Redox processes in photochemistry of Pt(IV) hexahaloid complexes
Evgeni M.
Glebov
*ab,
Aleksandr V.
Kolomeets
a,
Ivan P.
Pozdnyakov
ab,
Victor F.
Plyusnin
ab,
Vjacheslav P.
Grivin
ab,
Nikolai V.
Tkachenko
c and
Helge
Lemmetyinen
c
aInstitute of Chemical Kinetics and Combustion, 3 Institutskaya St., 630090 Novosibirsk, Russian Federation. E-mail: glebov@kinetics.nsc.ru; Fax: +7 383 3307350; Tel: +7 383 3332385
bNovosibirsk State University, 2 Pirogova St., 630090 Novosibirsk, Russian Federation
cDepartment of Chemistry and Bioengineering, Tampere University of Technology, P.O. Box 541, 33101 Tampere, Finland. E-mail: nikolai.tkachenko@tut.fi
First published on 19th April 2012
Abstract
Ultrafast pump–probe spectroscopy (λpump = 405 nm) was applied to study the primary photochemical processes for PtCl62− and PtBr62− complexes in aqueous and alcohol solutions. For PtCl62−, an intermediate with a lifetime of ca. 200 ps was registered and identified as an Adamson radical pair [PtIIICl52−⋯Cl˙]. The transformations of the primary intermediate give rise to successive formation of different Pt(III) species. The reactions of Pt(III) results in chain photoaquation in aqueous solutions and reduction of Pt(IV) to Pt(II) in alcohol solutions. For PtBr62− complex, the previously reported (I. L. Zheldakov, M. N. Ryazantsev and A. N. Tarnovsky, J. Phys. Chem. Lett., 2011, 2, 1540; I. L. Zheldakov, PhD thesis, Bowling Green State University, 2010) formation of active 3PtBr5− intermediate is followed by very fast (15 ps) aquation of Pt(IV) in aqueous solutions and parallel reactions of solvation and reduction of Pt(IV) to Pt(II) in alcohol solutions. All the processes in alcohols are finished within 0.5 ns. The data of ultrafast experiments are supported by nanosecond laser flash photolysis and stationary photolysis.
1. Introduction
Photochemistry of Pt(IV) hexahaloid complexes is of current interest due to applications of PtCl62− complex as a surface modifier for TiO21,2 and CdS3 semiconductors. For a complete description of processes in these systems, photochemical reactions of complexes formed by desorbed platinum in the solution bulk should be taken into account.2 Another aspect of interest to the photochemistry of simple platinum complexes is caused by the formation of unstable Pt(III) intermediates. The quantitative information on the reactions of Pt(III) complexes could be useful for understanding mechanisms of action of platinum-containing anti-tumor drugs.4The photochemistry of platinum hexahaloid complexes is two hundred years old,5 but the photolysis mechanism is not finally established.6,7 Mechanisms put forward in the early age of photochemistry8,6 can now be examined directly. Generally, photochemistry of these complexes in aqueous and alcohol solutions is a combination of three types of reactions:
(i) photoaquation (photosolvation) occurring via heterolytic cleavage of metal–ligand (M–L) bond followed by an escape of a ligand to the solution bulk.
(ii) photoaquation (photosolvation) occurring via homolytic cleavage of M–L bond. An example is given by the classical mechanism of Adamson's radical pairs,8 which includes redox stages (1)–(4). In this case an inner-sphere electron transfer is the primary photochemical process, and two successive intermediates are formed, which are traditionally called primary and secondary radical pairs.
(iii) photoreduction occurring via outer-sphere electron transfer to the light-excited complex (e.g. from solvent).
| MIVL62− + hν → [MIIIL52−⋯L˙], primary radical pair | (1) |
| [MIIIL52−⋯L˙] + H2O → [MIIIL5(H2O)2−⋯L˙], secondary radical pair | (2) |
| [MIIIL5(H2O)2−⋯L˙] → MIVL5(H2O)− + L−, back electron transfer | (3) |
| [MIIIL5(H2O)2−⋯L˙] → MIIIL5(H2O)2− + L˙, radical escape to the bulk | (4) |
Different mechanisms of PtBr62− photolysis in aqueous solutions proposed in earlier works included inner-sphere electron transfer,8 two-electron reduction of Pt(IV) with an escape of Br2 molecule into the bulk,9 and one-stage exchange of two Br− anions to water molecules.10 The most reliable scenario was proposed by Balzani and co-workers.11–13 In their mechanism the heterolytic Pt–Br bond cleavage occurs:
| PtBr62− + hν + H2O → PtBr5(H2O)− + Br− | (5) |
The primary process (5) is followed by PtBr5(H2O)− photoaquation with the formation of PtBr4(H2O)2. As a result, the multistage photoaquation of PtBr62− occurs.12 The quantum yield of the primary stage (5) was estimated as 0.4, and no dependence on the exciting light wavelength (313–530 nm) was observed.12 The independence of the photoaquation quantum yield on wavelength was explained by a fast transition to the lower excited state, namely, triplet ligand field (LF), which was thought to be a precursor of the reaction products.13 This point of view was supported by means of nanosecond laser flash photolysis14 and ultrafast pump–probe spectroscopy15 experiments. Recently Zheldakov et al.16,17 have re-examined the data on PtBr62− photoaquation by means of ultrafast pump–probe spectroscopy (with better time resolution and signal-to-noise ratio than used in ref. 15) combined with CASPT2 and DFT/TDDFT calculations. As a result, a multistage pathway from the absorption of a light quantum by the parent PtBr62− complex to the formation of the final PtBr5(H2O)− photoaquation product was put forward. Two successive intermediates were discriminated in the initial stage of photolysis, which were identified as a nearly trigonal bipyramid 3PtBr5− complex and a square-pyramid 1PtBr5− complex. The last complex was identified as the direct precursor of PtBr5(H2O)−.16 Addition of free Br− anions to aqueous solutions containing PtBr62− results in changing of photolysis pathway from aquation to reduction of Pr(IV) to Pt(II).18 An electron transfer from a free Br− anion to the light-excited complex leads to the successive formation of a bromine atom and Br2˙− radical anion.18
In contrast to the case of PtBr62− photochemistry, the isoelectronic PtCl62− complex demonstrates redox photochemistry in aqueous solutions without addition of free Cl− anions. Starting with ref. 19, in most of the papers on the photochemistry of aqueous PtCl62− a homolytic cleavage of Pt–Cl bond with a chlorine atom escape into the solution bulk (eqn (6)) was considered as a primary photochemical process.
| PtCl62− + hν → PtCl52− + Cl˙ | (6) |
Nevertheless, the integral photochemical process is photoaquation with the formation of the PtCl5(H2O)− complex.20–24 When the primary process (6) is realized, photoaquation can follow a chain mechanism. A possible variant of chain development22 includes reactions (7)–(10).
| PtCl52− → PtCl4− + Cl−, chain propagation | (7) |
| PtCl4− + PtCl62− + H2O → PtCl5(H2O)− + PtCl4− + Cl−, chain development | (8) |
| 2PtCl4− → PtCl4(H2O)2 + PtCl42−, chain termination | (9) |
| PtCl4− + Cl˙ + H2O → PtCl5(H2O)−, chain termination | (10) |
In addition to the chain process, a direct (non-chain) mechanism can be related to the back electron transfer in the geminate pair, {Pt(III) ion − Cl atom}, like in reactions (1)–(3).21
The quantum yield of PtCl62− photoaquation (ϕaq) depends on the concentration of the initial complex, light wavelength and intensity.22 Depending on the experimental parameters, quantum yield could be both less19,24 and sufficiently higher than unity.19,22 When a chain process is predominant, ϕaq is linear versus PtCl62− concentration.22
Pt(III) intermediates probably involved in the chain processes were recorded by means of microsecond lamp flash photolysis,19 nanosecond24,25 and picosecond26 laser flash photolysis. Based on quantum chemical calculations of Goursot et al.24,26–29, the intermediate absorption was interpreted as belonging to different Pt(III) species. Two main intermediates occurring in laser flash photolysis experiments are short-living (a few microseconds) species with the maximum at 450 nm and long-living (several milliseconds) species with the maximum at 410 nm.25 The similar intermediates were recorded in pulse radiolysis experiments of Broszkiewich et al.30–32 Additional argument in favor of redox photoaquation was obtained by spin trapping of Cl atoms in the course of photolysis.33
Probably, the most intriguing question is the dramatic difference in photoaquation of isoelectronic PtCl62− and PtBr62− complexes.6,7 Ford et al.7 proposed that the different mechanisms of photoaquation are determined by different values of the rate constants of the reactions between chloride and bromide complexes of Pt(III) and Pt(IV), which were supposed to be responsible for chain propagation. However, in the experiments on laser flash photolysis of PtBr62− no intermediates were found in the time region > 50 ns.14
In the case of alcohol solutions, PtCl62− and PtBr62− also demonstrate different photochemical properties. For PtCl62− the primary photochemical process was reported to be an outer-sphere electron transfer from a solvent molecule to the light-excited complex34–36 with the formation of Pt(III) intermediates and hydroxyalkyl radicals:
| PtCl62− + CH3OH + hν → PtCl63− + ˙CH2OH + H+ | (11) |
This mechanism was supported by means of ESR registration of hydroxyalkyl radicals in frozen alcohol matrices both directly37 and by spin trapping.38 Specific intermediates characterized as complexes of Pt(III) with free radicals were reported to occur in a course of photolysis.35,36
In the case of PtBr62− photolysis in methanol solutions, the dependence of the reaction products on the content of dissolved oxygen was reported.39 In oxygen-free solutions an outer-sphere electron transfer similar to eqn (11) followed by reduction of Pt(III) to Pt(II) was proposed. In the presence of dissolved oxygen, photosolvation of Pt(IV) with the formation of PtBr5(CH3OH)− was reported in ref. 39. The effect of oxygen was explained by re-oxidation of Pt(III) intermediates by peroxide radicals. For support of this mechanism two types of intermediates interpreted as Pt(III) complexes were registered by means of lamp flash photolysis.39 The spectra of short-lived Pt(III) bromide complexes are known from pulse radiolysis.40 Photolysis of PtBr62− in a frozen methanol matrix (77 K) results in reduction of Pt(IV) to PtBr42−, and ˙CH2OH radicals are formed.41
Recently Zheldakov17 has applied femtosecond pump–probe spectroscopy to study primary processes in the photophysics of PtBr62− in methanol. As in the case of aqueous solutions, the formation of highly reactive 3PtBr5− complex of Pt(IV) was reported. The reactions of 3PtBr5− was reported to give rise to both solvation with the formation of PtBr5(CH3OH)− and reduction with the successive formation of intermediate PtBr52− and final PtBr42− complexes.
This work continues our efforts in application of ultrafast spectroscopy to the photochemistry of hexahaloid complexes of noble metals.15,42,43 Ultrafast processes for PtCl62− in aqueous and alcohol solutions and PtBr62− in alcohol solution with the excitation in the region of 405 nm were studied.
2. Materials and methods
A pump–probe spectroscopy was used to study transient absorption in femto- and picosecond time domains. The experimental setup was described in details in ref. 44. The samples were excited by ∼60 fs pulses at ∼405 nm (second harmonic of a Ti:sapphire generator-amplifier system, CDP Ltd., Moscow, Russia, ca. 20–25 μJ per pulse), the excitation pulse repetition rate was 10 Hz, excitation beam size was 0.4–0.5 mm in diameter and 200 pulses were used to record single time-resolved spectra. The samples were placed in 1 mm rotating cell (total volume of 1 ml) to provide the uniform irradiation of the sample and to avoid unwanted thermal effects from the heating of the sample by the pump pulse. The degradation of the sample after photochemical studies did not exceed 5%. Typically time-resolved spectra were collected with delay displacement of 100 fs in the first 3 ps after excitation and with exponentially increasing delay times at longer delays. Usually 60–70 spectra were collected for each sample with the longest delay of ∼100 ps. The experimental data were globally fitted, typically by a three-exponential model. The fitting program performed corrections of the group velocity dispersion and calculated the response time of the instrument. The overall time resolution was 150–200 fs.The absorption spectra were recorded using an HP-8453 spectrophotometer (Hewlett Packard). Stationary photolysis was performed using the irradiation of a high-pressure mercury lamp with a set of glass filters.
Solutions of PtCl62− complex were prepared from Na2PtCl6 salt (Aldrich). Solutions of PtBr62− complex were prepared from Na2PtBr6·H2O salt synthesized as described in ref. 45. Deionized water, methanol and ethanol (Aldrich) were used for the sample preparation. Methanol and ethanol were used without additional purification. When necessary, the samples were deaerated by bubbling with argon.
3. Results and discussion
3.1. UV spectra of PtCl62− and PtBr62− complexes
Platinum(IV), which has a d6 electronic configuration, forms stable and kinetically inert complexes with a variety of ligands. The UV absorption spectra of PtCl62− and PtBr62− complexes are shown in Fig. 1. The spectra of both complexes coincide with known from literature.20,13 Spectra slightly depend on solvent type.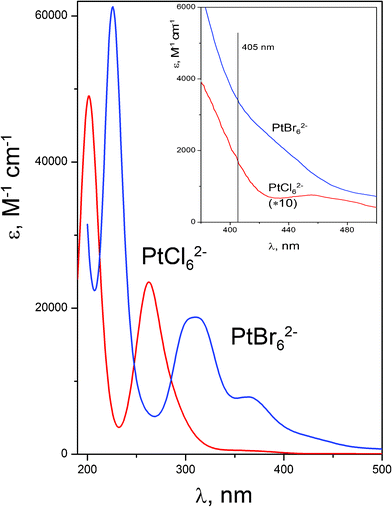 | ||
| Fig. 1 UV spectra of aqueous PtCl62− and PtBr62− solutions. Insert: Enlarged spectra in the visible region. | ||
The most intense ligand–metal charge transfer (LMCT) band of PtCl62− with the maximum at 202 nm corresponds to the electron density transfer from σ orbitals, predominantly located on ligands, to the vacant σ* orbitals, predominantly located on the metal ion.46,47 A less intense charge transfer band with maximum at 262 nm corresponds to the transitions from π orbitals of ligands. The shoulder in the region of 355 nm and low intensity broad band with maximum at 455 nm belongs to d–d transitions.46,47 According to the assignment of Goursot et al.,47 the laser excitation at 405 nm corresponds to the low-energy part of the 3T1g LF excited state.
The electronic absorption spectrum of PtBr62− is shifted to the red region in comparison with that of PtCl62−. The most intense LMCT band of PtBr62− is located at 226 nm. It corresponds to the electron density transfer from σ orbitals, predominantly located on ligands, to the vacant σ* orbitals, predominantly located on the metal ion.48 Less intense charge transfer bands (290–450 nm) correspond to the transitions from π orbitals of ligands. These bands are partially superimposed with the d–d bands.48 According to recent calculations of Zheldakov et al.16,17, transitions nearest to the excitation wavelength (405 nm) are a 1T1g→Eg* CT transition (401 nm) and a 1T2g→Eg* mixed LF/CT transition (413 nm). Both these transitions are forbidden.
3.2. Ultrafast pump–probe spectroscopy of PtCl62− in aqueous and alcohol solutions
Excitation of PtCl62− in aqueous solutions with a femtosecond laser pulse (405 nm) resulted in the formation of transient absorption which was almost completely decayed in 800 ps. Kinetic curves at several selected wavelengths are presented in Fig. 2. The global fit of the time profiles in the wavelength range 440–780 nm by iterative reconvolution shows that the use of a three-exponential function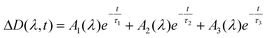 | (12) |
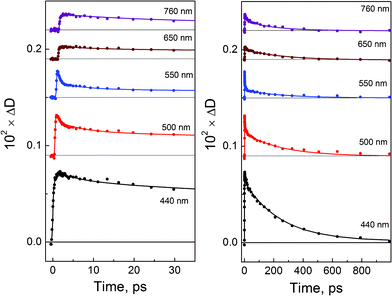 | ||
| Fig. 2 Femtosecond (λpump = 405 nm) photolysis of PtCl62− (0.03 M) in aqueous solutions. Cuvette, 1 mm. Kinetics of transient absorption at different wavelengths and time domains. Solid lines are the best three-exponential fits (eqn (12)) after reconvolution with the instrument response function. | ||
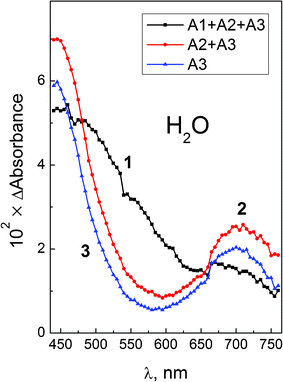 | ||
| Fig. 3 Femtosecond (λpump = 405 nm) photolysis of PtCl62− (0.03 M) in aqueous solutions. Intermediate absorption spectra at different times. 3-Exponential treatment of the data of Fig. 2. Curve 1 – zero time (sum of amplitudes A1(λ) + A2(λ) + A3(λ)); curve 2 – after the end of the first process (sum of amplitudes A2(λ) + A3(λ)); curve 3 – after the end of the second process – (amplitude A3(λ), eqn (12)). | ||
The total sum of amplitudes (curve 1) is the differential spectrum at zero time; the A2(λ) + A3(λ) sum (curve 2) is the differential spectrum at the end of first process (τ1 = 600 fs). After disappearance of A2(λ) (τ2 = 8.6 ps), the A3(λ) amplitude is the last differential spectrum before the complete decay of transient absorption (τ3 = 220 ps). Immediately after excitation the transient absorption spectrum appears as a wide band covering all the region of observation (440–760 nm). The structure of this band could not be resolved from the presented experimental data.
The first process with the characteristic time τ1 = 600 fs results in the formation of two absorption bands (curve 2 in Fig. 3).The maximum of the first band is in the region of 450 nm or probably shorter. The second band has a maximum at 700 nm. The second process with the characteristic time τ2 = 8.6 ps results in the narrowing of these bands without sufficient changes in their positions and relative intensities. The last process with the characteristic time τ3 = 220 ps results in the synchronous disappearance of intermediate absorption. The similar behavior of the two bands at ≤450 and 700 nm means that they belong to the same intermediate, further marked as intermediate A. The intermediate A seems to be the same species which was previously registered by Goursot et al.26 In ref. 26, the excitation of PtCl62− by 30 ps laser pulses at 355 nm resulted in the formation of the intermediate, which had two absorption bands at 440 and 640 nm decaying with the characteristic time of 210 ps. Because 640 nm was just the longest wavelength in which intermediate absorption was measured in ref. 26, we believe that intermediate A and intermediate observed in ref. 26 are exactly the same species.
Experiments on femtosecond laser excitation of PtCl62− in methanol and ethanol gave the results similar to those obtained in aqueous solutions. The differential intermediate absorption spectra for the case of methanol solutions are shown in Fig. 4. The formation of intermediate A with absorption bands at ≤450 and 700 nm is observed, similar to the case of aqueous solutions. The results of 3-exponential fit of the experimental kinetic curves for PtCl62− in different solvents are collected in Table 1. Therefore, both for aqueous and alcohol solutions of PtCl62− we observe fast (with the characteristic time τ1 = 600–700 fs) formation of the intermediate A from the initial Franck–Condon state (which is in the hot 3T1g state according to assignment in ref. 47). The second process with the characteristic time τ2 = 4–11 ps, in which no dramatic spectral changes occur, could be assigned to the vibrational cooling accompanied by the solvent relaxation. The thermalization time for transition metal complexes typically lies in a picosecond time domain (1–10 ps).49,50 The third characteristic time τ3 = 200–500 ps corresponds to the transition of thermalized intermediate A to the ground state of PtCl62− and/or to long-lived photolysis products.
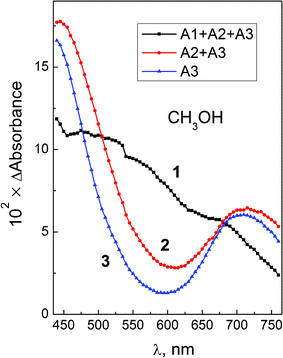 | ||
| Fig. 4 Femtosecond (λpump = 405 nm) photolysis of PtCl62− (0.052 M) in CH3OH. Intermediate absorption spectra at different times. 3-Exponential treatment of the kinetic curves. Curve 1 – zero time (sum of amplitudes A1(λ) + A2(λ) + A3(λ)); curve 2 – after the end of the first process (sum of amplitudes A2(λ) + A3(λ)); curve 3 – after the end of the second process (amplitude A3(λ), eqn (12)). | ||
| Solvent | τ 1, ps | Process | τ 2, ps | Process | τ 3, ps | Process |
|---|---|---|---|---|---|---|
| H2O | 0.6 ± 0.02 | 3T1g→A* | 8.6 ± 4.5 | A*→A | 220 ± 30 | Relaxation to ground state and products formation |
| CH3OH | 0.7 ± 0.1 | 4.3 ± 1.1 | 350 ± 20 | |||
| C2H5OH | 0.7 ± 0.2 | 11 ± 6 | 490 ± 50 |
3.3. Assignment of intermediates of PtCl62− photolysis
For the assignment of intermediate A the results of femtosecond experiments should be compared with the data of nanosecond laser flash photolysis. Fig. 5a shows the shapes of the spectra of intermediate A and successive Pt(III) intermediates followed by nanosecond laser excitation of aqueous PtCl62− solutions in the region of 308 nm.25 Two groups of intermediates, namely intermediate C with an absorption band at 440–450 nm (which characteristic lifetime is several microseconds) and intermediate D having an absorption band at 410 nm (with lifetime of more than one millisecond), are typically observed in the experiments on flash photolysis of PtCl62−![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 21,24,25 and pulse radiolysis of PtCl62− and PtCl42− complexes30–32 in aqueous solutions. Fig. 5b demonstrates the spectra of intermediate A (this work) and intermediate F (taken from ref. 34) followed by nanosecond laser flash photolysis of PtCl62− in methanol (308 nm). The spectra of intermediate F in ethanol and isopropanol are similar to that in methanol.36 The characteristic parameters of the intermediates obtained in different works together with the assignments proposed by their authors are collected in Table 2 (for aqueous solutions) and Table 3 (for methanol solutions).
21,24,25 and pulse radiolysis of PtCl62− and PtCl42− complexes30–32 in aqueous solutions. Fig. 5b demonstrates the spectra of intermediate A (this work) and intermediate F (taken from ref. 34) followed by nanosecond laser flash photolysis of PtCl62− in methanol (308 nm). The spectra of intermediate F in ethanol and isopropanol are similar to that in methanol.36 The characteristic parameters of the intermediates obtained in different works together with the assignments proposed by their authors are collected in Table 2 (for aqueous solutions) and Table 3 (for methanol solutions).
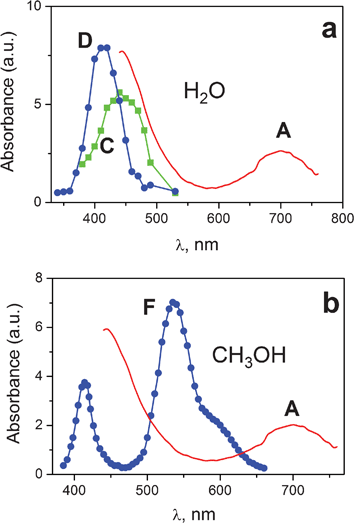 | ||
| Fig. 5 Spectra of intermediates occurred in the course of PtCl62− photolysis an water (a) and methanol (b). Intermediate A is a species obtained in ultrafast experiments (curve 3 in Fig. 3, 4, time delays 8.6 and 4.3 ps for water and methanol correspondingly). Intermediates C and D are successive Pt(III) species obtained in XeCl (308 nm, 15 ns) laser flash photolysis experiment (time delays 4 and 30 μs).25 Intermediate F is a Pt(III) species obtained in XeCl (308 nm, 15 ns) laser flash photolysis experiment (time delay 2 μs).34 Amplitudes of intermediate A are matched to the size of the picture. | ||
| Intermediate | λ max,a nm |
τ
forma |
τ
deca |
Assignment of PtIII species | ||
|---|---|---|---|---|---|---|
| ref. 26, 24 | ref. 25 | This work | ||||
| a λ max, τform and τdec are position of absorption band maxima, formation time and decay time of intermediates. b In ref. 26 intermediate absorption spectrum was interrupted at 650 nm. c From ref. 25. | ||||||
| A | 440, 700b | 600 fs | 220 ps | PtCl52− (C4v) or (PtCl62−)* | — | [PtCl52−⋯Cl˙] or (PtCl62−)* |
| B | Invisiblec | <100 nsc | 1.2 μsc | — | PtCl5(H2O)2− | PtCl5(H2O)2− |
| C | 440–450 | 1.2 μsc | 7 μsc | PtCl6−mXm (m = 0–2; X = OH, H2O) | PtCl4(OH)(H2O)2− | PtCl4X2 (X = OH, H2O) |
| D | 410 | 7 μsc | 1 msc | PtCl4−nXn (n = 1–3; X = OH, H2O) | PtIIICl4− | PtCl4−nXn (n = 1–3; X = OH, H2O) |
| Intermediate | λ max,a nm | τ form | τ dec | Assignment of PtIII species | |
|---|---|---|---|---|---|
| ref. 34 | This work | ||||
| a λ max, τform and τdec are position of absorption band maxima, formation time and decay time of intermediates. b From ref. 34. c Occurs in two-quantum process ref. 34. d From ref. 37. | |||||
| A | 440, 700b, | 700 fs | 350 ps | — | [PtCl52−⋯Cl˙] or (PtCl62−)* |
| E | Invisibleb | <100 nsb | 1 μsb | PtCl63− | PtCl63− |
| F | 414 535 | <50 nsb,c, 1.2 μsb | 2.6 msd | PtCl52− | PtCl52− (C4v) |
In the case of aqueous solutions, the yield of intermediate C observed in nanosecond laser flash photolysis experiments (308 nm) is linear versus laser intensity,25 corresponding to one-quantum processes only. In methanol solutions, intermediate F is formed both in one-quantum and in two-quantum processes.34
To identify the observed intermediates, the following points should be addressed:
1. Intermediates A in water and alcohols are the same species.
2. Long-lived Pt(III) intermediates in water and alcohols are different.
3. Decay of short-living intermediate A in the picosecond time domain does not result in occurrence of intermediates C and F, which are observed in the microsecond time domain. The decay of intermediates A should result in the formation of intermediates B (in water) and E (in alcohols), which are the precursors of intermediates C and F correspondingly. Intermediates B and E do not have a pronounced absorption in the visible spectral region.
4. Low relative yield of Pt(III) intermediates in aqueous solutions (ca. 10%21,25) should be explained.
5. Possibility of intermediate F formation both in photochemical and thermal reactions should be taken into account.
6. The assignment of intermediates should not contradict the Xα theoretical study of Pt(III) intermediates performed by Goursot et al.24,26–29 Numerous Pt(III) intermediates can exist. They have different composition, geometry and electron absorption spectra.
Different possibilities of intermediate A assignment are discussed below.
(i) Assume that A is PtIVCl5− complex, similar to the assignment of the short-lived intermediates of PtBr62− photolysis by Zheldakov et al.16,17. This possibility could be ruled out because of the impossibility of providing the formation of long-lived Pt(III) intermediates in aqueous solutions.
(ii) According to Goursot et al.,26 A is the PtIIICl52− (C4v) complex. In this case, it is hard to explain the difference between photolysis in aqueous and alcohol solutions. This difference is determined by the possibility of an electron transfer from alcohol to Pt complex, e.g. in the reaction
| PtIIICl52− + CH3OH → PtIICl53− + ˙CH2OH + H+ | (13) |
(iii) Assumption that A could be an excited state of the initial complex (PtCl62−)* was discussed in ref. 24. The authors24 have shown that the existence of an excited state with such a long lifetime is highly improbable but could not be completely ruled out.
(iv) The experimental results could be satisfactorily explained by the assumption that intermediate A is the Adamson's primary radical pair [PtIIICl52−⋯Cl˙]. The square-pyramidal PtCl52− (C4v) complex provides the two-band absorption in the visible spectral region,27 and the effect of apical Cl˙ atom on the spectral properties seems to be small in the case of sufficient elongation of PtIII–Cl˙ bond. The initial stages of photolysis (405 nm) in water and alcohols are assumed to be the same resulting in the same intermediate:
| PtCl62− + hν → [PtCl62−]*(3T1g) | (14) |
| [PtCl62−]*(3T1g) → [PtIIICl52− (C4v)⋯Cl˙], intermediate A (primary radical pair) | (15) |
The formation of the primary radical pair in water is followed by its relaxation to the ground state of PtCl62− (eqn (16)) and formation of the secondary radical pair (eqn (17)). Back electron transfer in the secondary radical pair (eqn (18)) results in photoaquation. Dissociation of the secondary pair (eqn (19)) results in the formation of PtIIICl5(H2O)2− complex. This intermediate, having one weakly coordinated chlorine, according to Xα calculations,28 absorbs in the near UV but not in the visible region. Therefore, it could be considered as “invisible” intermediate B. Then, the successive reactions of B results in formation of intermediates C and D absorbing in the visible spectral region, in accord with Xα calculations.29 The assignments of intermediates for PtCl62− photolysis in water are collected in Table 2. The structures of intermediates C and D may be not only [PtIIICl4(OH)(H2O)2−] and [PtIIICl3(OH) −], as shown in eqn (20) and (21), but another structures are possible as well. All the possible structures consistent with calculations29 are mentioned in Table 2.
| [PtIIICl52− (C4v)⋯Cl˙] → PtCl62− | (16) |
| [PtIIICl52− (C4v)⋯Cl˙] + H2O → [PtIIICl5(H2O)2−⋯Cl˙], secondary radical pair | (17) |
| [PtIIICl5(H2O)2−⋯Cl˙] → PtIVCl5(H2O)− + Cl−, back electron transfer | (18) |
| [PtIIICl5(H2O)2−⋯Cl˙] → PtIIICl5(H2O)2− + Cl˙, formation of B | (19) |
| PtIIICl5(H2O)2− + H2O → PtIIICl4(OH)(H2O)2− + Cl− + H+, formation of C | (20) |
| PtIIICl4(OH)(H2O)2− → PtIIICl3(OH) − + Cl− + H2O, formation of D | (21) |
The ratio of the rate constants of reactions (16) and (17) determines the primary quantum yield of photolysis. The ratio of the rate constants of the reactions (18) and (19) determines the relative yield of Pt(III) intermediates. Long-lived Pt(III) intermediates D can initiate chain reactions of photoaquation, similar to the mechanism shown in eqn (7)–(10).
In alcohols, intermediate A, which is formed in reaction (15) (in addition to backward reaction (16)), can oxidize a molecule of alcohol, which is situated in the second coordination sphere of platinum anion giving rise to PtCl63− complex. The reaction mechanism (for the case of methanol) is represented by the reactions (14)–(16) and (22)–(27) (note that reactions (23) and (24) occur under the conditions of XeCl laser flash photolysis experiment)
| [PtIIICl52− (C4v)⋯Cl˙] + CH3OH → PtIIICl63− + ˙CH2OH + H+, formation of E | (22) |
| PtIIICl63− + hν(308 nm)→ (PtIIICl63−)* | (23) |
| (PtIIICl63−)* → PtIIICl52− + Cl−, formation of F (photochemical) | (24) |
| PtIIICl63− → PtIIICl52− + Cl−, formation of F (thermal) | (25) |
| PtIIICl52− → PtIIICl4− + Cl− | (26) |
| PtIIICl4− + CH3OH → PtIICl42− + ˙CH2OH + H+ | (27) |
Reaction between the radical pair (intermediate A) and a solvent molecule gives rise to the Pt(III) complex PtCl63−. Omitting reactions occurring in the femto- and picosecond time domain, the formation of PtCl63− could be considered as an electron transfer from the solvent molecule to the light-excited complex (eqn (11)). PtCl63− complex distorted toward a D4h symmetry (with a moderate elongation of axial Pt–Cl bonds) exhibits LMCT transitions in the region of 308 nm, appearing as “dark” in the visible spectral region. Therefore, PtCl63− could be considered as intermediate E. Photochemical (when excited in the region of 308 nm, eqn (24)) and thermal (eqn (25)) reactions of PtCl63− provide two mechanisms of formation of PtCl52− complex (intermediate F).34,36 According to Xα calculations,27 the C4v structure of PtCl52− with a sufficient elongation of the axial Pt–Cl bond gives rise to two-peaks absorption spectrum in the visible region. The assignments of intermediates for the case of PtCl62− photolysis in alcohols are collected in Table 3. The final product of PtCl62− photolysis in alcohol solutions is the complex of bivalent platinum, PtCl42−.39,34–36
Finally, we believe that the proposed assignment of intermediates occurred upon photolysis of PtCl62− in aqueous and alcohol solution links the data of ultrafast spectroscopy, nanosecond and stationary laser flash photolysis.
3.4. Ultrafast pump–probe spectroscopy of PtBr62− in alcohol solutions
Kinetic curves at several selected wavelengths followed by ultrafast laser excitation (405 nm) of PtBr62− complex in methanol solutions are presented in Fig. 6. The global fit of the time profiles in the wavelength range 440–780 nm by iterative reconvolution shows that the use of a three-exponential function (12) with the instrument response function gives a good fitting with the time constants τ1 = 1.3 ps, τ2 = 8.7 ps and τ3 = 135 ps (solid lines in kinetic curves).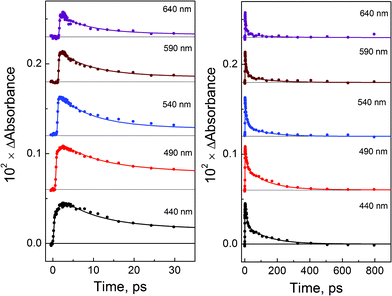 | ||
| Fig. 6 Femtosecond (λpump = 405 nm) photolysis of PtBr62− (1.9 × 10−3 M) in methanol solutions. Cuvette, 1 mm. Kinetics of transient absorption at different wavelengths and time domains. Solid lines are the best three-exponential fits (eqn (12)) after reconvolution with the instrument response function. | ||
The differential intermediate absorption spectra for the case of methanol solutions are shown in Fig. 7. For reference, Fig. 7 also shows the intermediate absorption spectra for the cases of ethanol (this work) and water (from ref. 15). Addition of 0.2–0.4 M NaBr did not affect both spectral and kinetic characteristics of intermediate absorption. The results of 3-exponential fit of the experimental kinetic curves for PtBr62− in different solvents are collected in Table 4. The main difference between the cases of PtBr62− photolysis in aqueous and alcohol solutions is that the longest time in alcohols (130–260 ps) is an order of magnitude larger that in water.
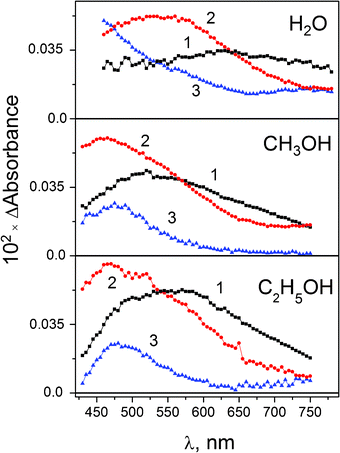 | ||
| Fig. 7 Photolysis of PtBr62− in aqueous (λpump = 420 nm, 3.8 × 10−3 M, data from ref. 15), methanol (λpump = 405 nm, 1.9 × 10−3 M) and ethanol (λpump = 405 nm, 1.9 × 10−3 M) solutions. Intermediate absorption spectra at different times (for methanol, 3-exponential treatment of the data from Fig. 6). Curve 1 – zero time (sum of amplitudes A1(λ) + A2(λ) + A3(λ)); curve 2 – after the end of the first process (sum of amplitudes A2(λ) + A3(λ)); curve 3 – after the end of the second process (amplitude A3(λ), eqn (12)). | ||
| Solvent | τ 1, ps | Process | τ 2, ps | Process | τ 3, ps | Process |
|---|---|---|---|---|---|---|
| a From ref. 15. b PtBr62− (1T1g, 1T2g); assignment of absorption bands corresponds to calculations of Zheldakov.17 c R = CH3; C2H5. | ||||||
| H2O | 0.4a | (PtBr62−)*b → 3PtBr5− + Br− |
2.2a | 3PtBr5− → 1PtBr5− | 15a | 1PtBr5− → PtBr62− and PtBr5(H2O)− |
| MeOH | 1.3 ± 0.3 |
3PtBr5− → 1PtBr5− and3PtBr5−c + ROH → 2PtBr52− |
8.7 ± 1.4 |
1PtBr5− + Br− → PtBr62− and1PtBr5−c + ROH → PtBr5(ROH)− |
130 ± 40 | 2PtBr52− + ROH → PtBr42− + products |
| MeOH + NaBr (0.3 M) | 1.5 ± 0.4 | 10.0 ± 1.6 | 140 ± 30 | |||
| EtOH | 1.3 ± 0.5 | 9.6 ± 2.0 | 260 ± 80 | |||
| EtOH + NaBr (0.3 M) | 2.0 ± 0.9 | 12.9 ± 2.4 | 260 ± 80 | |||
We now have to revise our views on the assignment of intermediates observed upon the photolysis of PtBr62− in aqueous solutions given in ref. 15. We considered the shorter time constant as attributed to the transition to the lower excited state (3T1g), which was in agreement with the independence of quantum yield of photoaquation on irradiation wavelength.12 Nevertheless, recently Zheldakov et al.16,17 have given conclusive evidence that 3T1g state is dissociative, and its lifetime is less than 150 fs. According to ref. 16, within 150 fs following 420 nm excitation of PtBr62− the molecule undergoes internal conversion and intersystem crossing into the dissociative lowest triplet excited 3T1g state, loses a ligand, and relaxes via the C4v conical intersection to the nearly trigonal bipyramid 3PtBr5− product in the triplet state. The next process is the intersystem crossing of 3PtBr5− to singlet 1PtBr5− state, which is the direct precursor of the reaction product PtBr5(H2O)− complex. Therefore, the intermediate absorption spectra 2 and 3 for aqueous solutions in Fig. 7 correspond to 3PtBr5− and 1PtBr5− (see Table 4). PtBr62− photolysis in the region of d–d bands in aqueous solutions is consistent with the mechanism in eqn (28)–(33).17
| PtBr62− (1A1g) + hν → PtBr62− (1T1g*, 1T2g*) | (28) |
| PtBr62− (1T1g*, 1T2g*) → PtBr62− (3T1g*) | (29) |
| PtBr62− (3T1g*) → {3PtBr5− + Br−}cage | (30) |
| {3PtBr5− + Br−}cage → {1PtBr5− + Br−}cage | (31) |
| 1PtBr5− + Br−}cage → PtBr62− (1A1g) | (32) |
| {1PtBr5− + Br−}cage + H2O → PtBr5(H2O) − | (33) |
In the case of PtBr62− photolysis in alcohol solutions, it is necessary to explain the formation of a relatively long-living (more that 100 ps) intermediates (Fig. 7, Table 4). The following assignment of intermediates is consistent with the reaction scheme proposed by Zheldakov.17
Initial bands (curves 1 in Fig. 7) with maxima at ca. 530 nm (methanol) and 570 nm (ethanol) look similar to the spectrum of the second intermediate in aqueous solution (curve 2 in Fig. 7). In 1.3–2 ps the spectrum with maximum at 465 nm is formed. Therefore, it could be proposed that the initial spectrum in alcohol solutions mainly belongs to the triplet 3PtBr5− complex. The spectral changes occurring in the first 200 fs include several processes, which are not resolved in our experiments. These processes are: (i) internal conversion and (ii) intersystem crossing into the dissociative lowest triplet excited 3T1g state of PtBr62− and (iii) loss of a ligand with the formation of the triplet 3PtBr5− product. Note that in the case of aqueous solutions we were able to resolve the transition of the initial hot Franck–Condon state to 3PtBr5−, which occurs at ca. 400 fs (curves 1 and 2 for H2O solutions in Fig. 7), but in the case of alcohol solutions it is not possible, probably because of higher reaction rates.
After the formation of the triplet 3PtBr5− complex it is a subject of (iv) transformation to the singlet state 1PtBr5−; and (v) the electron transfer from the solvent molecule to Pt(IV) with the formation of Pt(III) intermediate. Processes (i–iv) are the same as for aqueous solutions. Reaction (v) was monitored by Zheldakov17 in the spectral region 350–380 nm. Therefore, spectra 2 (Fig. 7, alcohols) correspond to the superposition of 1PtIVBr5− and 2PtIIIBr52− intermediates.
In ca. 10 ps, 1PtBr5− complex recombines in the solvent cage with Br− anion and is solvated with the formation of photosolvation product PtIVBr5(ROH) −. Spectra 3 (Fig. 7, alcohols) correspond to pure Pt(III) intermediate, 2PtBr52− complex. The last one transfers an electron from the solvent molecule giving rise to the final product of photoreduction, which is PtBr42− complex.14 Therefore, parallel reactions of photosolvation and photoreduction occur in the case of PtBr62− photolysis in alcohol solutions. The mechanism of photolysis includes reactions (28)–(32), like in aqueous solutions, and reactions (34)–(36), which are written for the case of methanol solution.
| {1PtBr5− + Br−}cage + CH3OH → PtBr5(CH3OH) − | (34) |
| 3PtBr5− + CH3OH → PtIIIBr52− + ˙CH2OH + H+ + Br− | (35) |
| PtIIIBr52− + CH3OH → PtIIBr42− + ˙CH2OH + H+ + Br− | (36) |
Eqn (35) and (36) reflect the fact that the hydroxyalkyl radical ˙CH2OH was the only free radical registered in the course of PtBr62− photolysis in frozen methanol matrix (77 K).14 In the case of MCl62− (M = Pt, Pd, Ir) photolysis in frozen ethanol, the formation of both CH3˙CHOH and CH3CH2O˙ radicals was reported,38 and the lack of experimental data for PtBr62− does not allow one to select a proper variant.
In addition to the data obtained by ultrafast spectroscopy, the results of stationary photolysis and nanosecond laser flash photolysis should be taken into account.
PtBr62− complex is not stable in alcohol solutions (Fig. 8). The observed spectral changes are caused by solvation reaction, because the addition of free bromide anions completely stops the reaction. An isosbestic point at 215 nm is exactly conserved. The conservation of isosbestic points at 260 and 280 nm is not exact because of successive solvation with the formation of complexes PtBr5(CH3OH)−, PtBr4(CH3OH)2, etc.
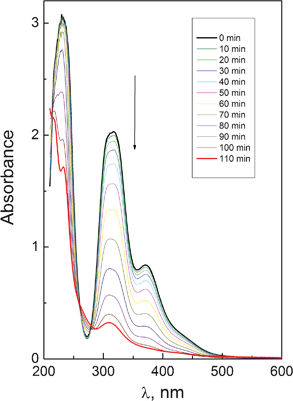 | ||
| Fig. 8 Spectral changes caused by thermal reaction of PtBr62− complex (1.8 × 10−4 M in a 1 cm cell) in methanol solution. | ||
Fig. 9 shows the changes in optical density of PtBr62− absorption caused by irradiation in methanol with natural content of oxygen. At the initial stage of photolysis (Fig. 9a) three isosbestic points at 216, 250 and 286 nm are conserved, which are close to those observed in the case of thermal solvation, but not the same. It could be assumed that photosolvation of PtBr62− in alcohols is accompanied by another reaction. The conservation of isosbestic points is violated in the course of prolonged irradiation, resulting in the formation of an absorption band with maximum at 270 nm (curve 8 at Fig. 9b). The appearance of the maximum in the region of 270 nm is a characteristic feature of PtBr42− complex,51,52 which is the product of Pt(IV) photoreduction to Pt(II).41 Taking the absorption coefficient of PtBr42− in methanol equal to that obtained in water (9100 M−1 cm−141), it is possible to perform a rough estimation that the relative yield of Pt(II) at the end of photolysis (Fig. 9b) is about 40% of the disappeared PtBr62− complexes. Therefore, the observed spectral changes caused by stationary photolysis of PtBr62− in alcohols could be explained by parallel reactions of multistage photosolvation and photoreduction. It supports the data of ultrafast measurements.
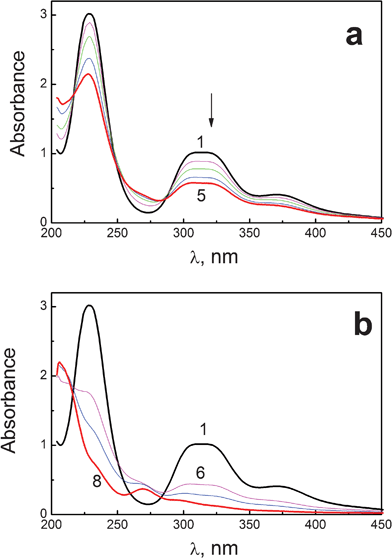 | ||
| Fig. 9 Spectral changes caused by irradiation (313 nm) reaction of PtBr62− complex (6 × 10−5 M in a 1 cm cell) in methanol solution. Natural content of oxygen. a – initial stage of photolysis; b – prolonged photolysis. Curves 1–8 correspond to 0, 3, 6, 9, 12, 17, 27, 46 s of irradiation. | ||
It should be noted that the removal of oxygen from methanol solutions did not affect the photolysis, which is in contradiction with the results of previous work.39
The experiments on nanosecond laser flash photolysis (XeCl laser, 308 nm, 10 ns) of PtBr62− in methanol solutions did not show the formation of any intermediates. Only instant changes in absorption corresponding to the disappearance of the initial complex were observed both in oxygen-free and in oxygen-saturated solutions. Pt(III) intermediates were registered in the experiments on pulse radiolysis of PtBr62− in aqueous solutions,40 and their lifetimes were about tens of microseconds. In the case of alcohol solutions the lifetime of Pt(III) intermediates is exactly less than 50 ns, which is in accord with the mechanism of Pt(II) formation in picosecond time domain proposed in ref. 17.
Therefore, the mechanism of fast (with a total time much less than 0.5 ns) parallel reactions of photosolvation and photoreduction of PtBr62− in alcohols is consistent with ultrafast pump–probe spectroscopy measurements, nanosecond flash photolysis and stationary photolysis.
The results on the photochemistry of PtBr62− and PtCl62− complexes demonstrate that the predominantly LF excitation of the chloro complex leads to Pt–Cl bond homolysis, whereas mixed LF-LMCT excitation of the bromo complex results in Br− ligand dissociation without a redox process. It means that the simple intuitive rule “d–d band excitation results in the heterolytic bond cleavage while charge transfer excitation leads to the homolytic bond cleavage” does not always work. Although charge-transfer states are most commonly associated with excited-state electron transfer, LF states can also be engaged in photoredox chemistry.53 An example of reductive behavior followed by ligand field excitation is given by Cr(III) photochemistry. The ligand field 2Eg excited state of [Cr(bpy)3]3+ complex is a strong one-electron photooxidant (see ref. 54 and references therein). In the case of hexahaloid complexes of platinum metals, similar examples could be taken from IrCl62− photochemistry.55 The LMCT bands of this complex in the visible spectral region are not photoactive, while excitation to the LF bands in the near UV region results in either photosubstitution for aqueous and acetonitrile solutions or photoreduction for alcohol solutions.55 High density of states and extensive mixing among the various excited states (both ligand-field and charge transfer) is the feature of transition metal complexes.53 It could result, for example, in redox reactions caused by the LF excitation.
Conclusions
In conclusion we stress the reasons for differences in photochemistry of isoelectronic PtBr62− and PtCl62− complexes both in aqueous and in alcohol solutions. There are two main points:(1) The first point is the difference in the nature of the initial reactive intermediates. For PtBr62− the absorption of a light quantum results in ultrafast (less than 150 fs) formation of the active Pt(IV) intermediate, triplet 3PtBr5− complex, which is a subject of very fast (15 ps) aquation or solvation. For the case of PtCl62−, the initial Pt(III) intermediate assigned as the Adamson radical pair is sufficiently more stable, which leads (in aqueous solutions) to the partial escape of Pt(III) complexes from the solvent cage. It makes possible a chain mechanism of photoaquation. The existence of long-living intermediates in the case of PtCl62− photolysis in aqueous solutions is beyond question. However, the nature of these intermediates is a question still open for discussion. To examine the radical character of these intermediates, the experiments in low-temperature matrices with the ESR registration seems to be prospective. Another possible way of solving the problem is to perform the quantum chemical calculations of the radical complexes.
(2) The second point is a great difference in reactivities of bromide and chloride intermediates of Pt(III) (as was proposed in ref. 7). This is a key factor in the case of alcohol solutions. For bromide complexes, very fast electron transfer from the solvent molecules to Pt(III) is accompanied by still faster Pt(IV) solvation. As a result, all the reactions are completed within 0.5 ns. For chloride complexes, relative stability of Pt(III) intermediates gives rise to a number of secondary reactions.
Acknowledgements
The financial support of the Russian Foundation of Basic Research (grants no. 11-03-00268, 11-03-90406-UKR, 11-03-92605-KO) is gratefully acknowledged.References
- (a) L. Zang, W. Macyk, C. Lange, W. F. Mayer, C. Antonius, D. Meissner and H. Kish, Chem.–Eur. J., 2000, 6, 379 CrossRef CAS; (b) H. Kish and W. Macyk, ChemPhysChem, 2002, 3, 399 CrossRef; (c) W. Macyk, G. Burgeth and H. Kish, Photochem. Photobiol. Sci., 2003, 2, 322 RSC; (d) W. Macyk, G. Burgeth and H. Kish, Adv. Inorg. Chem., 2004, 56, 241 CrossRef; (e) A. Janczyk, A. Wolnicka-Glubisz, H. Kish, G. Stochel and W. Macyk, Free Radical Biol. Med., 2008, 44, 1120 CrossRef CAS; (f) H. Kish, Adv. Inorg. Chem., 2011, 63, 371 CrossRef.
- W. Macyk and H. Kish, Chem.–Eur. J., 2001, 7, 1862 CrossRef CAS.
- (a) Q. Li, Zh. Chen, X. Zheng and Zh. Jin, J. Phys. Chem., 1992, 96, 5959 CrossRef CAS; (b) Zh. Jin, Zh. Chen, Q. Li, Ch. Xi and X. Zheng, J. Photochem. Photobiol., A, 1994, 81, 177 CrossRef CAS.
- K. L. Swancutt, S. P. Mezyk and J. J. Kiddle, Radiat. Res., 2007, 168, 423 CrossRef CAS.
- J. Hershel, Phil. Mag., 1832, 1, 58 Search PubMed.
- V. Balzani and V. Carassiti, Photochemistry of coordination compounds, Acad. Press, New York, 1970, pp. 258–262 Search PubMed.
- P. G. Ford, J. D. Petersen and R. E. Hintze, Coord. Chem. Rev., 1974, 14, 67 CrossRef CAS.
- A. W. Adamson and A. H. Sporer, J. Am. Chem. Soc., 1958, 80, 3865 CrossRef CAS.
- S. A. Penkett and A. W. Adamson, J. Am. Chem. Soc., 1965, 87, 2514 CrossRef CAS.
- V. V. Vasilyev, K. P. Balashev and G. A. Shagisultanova, Koord. Khim. (Russ. J. Coord. Chem.), 1982, 8, 1235 Search PubMed (in Russian).
- V. Balzani, V. Carassiti and F. Scandola, Gazz. Chim. Ital., 1966, 96, 1213 CAS.
- V. Balzani, M. F. Manfrin and L. Moggi, Inorg. Chem., 1967, 6, 354 CrossRef CAS.
- V. Balzani and V. Carassiti, J. Phys. Chem., 1968, 72, 383 CrossRef CAS.
- E. M. Glebov, V. F. Plyusnin, V. P. Grivin, A. B. Venediktov and S. V. Korenev, Russ. Chem. Bull., 2007, 56, 2357 CrossRef CAS.
- I. P. Pozdnyakov, E. M. Glebov, V. F. Plyusnin, N. V. Tkachenko and H. Lemmetyinen, Chem. Phys. Lett., 2007, 442, 78 CrossRef CAS.
- I. L. Zheldakov, M. N. Ryazantsev and A. N. Tarnovsky, J. Phys. Chem. Lett., 2011, 2, 1540 CrossRef CAS.
- I. L. Zheldakov, Ultrafast Photophysics and Photochemistry of Hexacoordinated Bromides of Pt(IV), Os(IV), and Ir(IV) in the Condensed Phase Studied by Femtosecond Pump–Probe Spectroscopy, PhD Thesis, Bowling Green State University, 2010 Search PubMed.
- E. M. Glebov, V. P. Grivin, V. F. Plyusnin, A. B. Venediktov and S. V. Korenev, J. Photochem. Photobiol., A, 2010, 214, 181 CrossRef CAS.
- R. L. Rich and H. Taube, J. Am. Chem. Soc., 1954, 76, 2608 CrossRef CAS.
- L. E. Cox, D. G. Peters and E. L. Wehry, J. Inorg. Nucl. Chem., 1972, 34, 297 CrossRef CAS.
- R. C. Wright and G. S. Laurence, J. Chem. Soc., Chem. Commun., 1972, 132 Search PubMed.
- K. P. Balashev, V. V. Vasilyev, A. M. Zimnyakov and G. A. Shagisultanova, Koord. Khim. (Russ. J. Coord. Chem.), 1984, 10, 976 CAS (in Russian).
- K. P. Balashev, I. I. Blinov and G. A. Shagisultanova, Zh. Neorg. Khim. (Russ. J. Inorg. Chem.), 1987, 32, 2470 CAS (in Russian).
- W. L. Waltz, J. Lillie, A. Goursot and H. Chermette, Inorg. Chem., 1989, 28, 2247 CrossRef CAS.
- I. V. Znakovskaya, Yu. A. Sosedova, E. M. Glebov, V. P. Grivin and V. F. Plyusnin, Photochem. Photobiol. Sci., 2005, 4, 897 CAS.
- A. Goursot, A. D. Kirk, W. L. Waltz, G. B. Porter and D. K. Sharma, Inorg. Chem., 1987, 26, 14 CrossRef CAS.
- A. Goursot, H. Chermette, E. Peigault, M. Chanon and W. L. Waltz, Inorg. Chem., 1984, 23, 3618 CrossRef CAS.
- A. Goursot, H. Chermette, E. Peigault, M. Chanon and W. L. Waltz, Inorg. Chem., 1985, 24, 1042 CrossRef CAS.
- A. Goursot, H. Chermette, W. L. Waltz and J. Lillie, Inorg. Chem., 1989, 28, 2241 CrossRef CAS.
- G. E. Adams, R. K. Broszkiewicz and B. D. Michael, Trans. Faraday Soc., 1968, 64, 1256 RSC.
- R. K. Broszkiewicz and J. Grodkowski, Int. J. Radiat. Phys. Chem., 1976, 8, 359 CrossRef CAS.
- E. Bothe and R. K. Broszkiewicz, Inorg. Chem., 1989, 28, 2988 CrossRef CAS.
- D. Rehorek, C. M. Dubose and E. S. Jansen, Inorg. Chim. Acta, 1984, 83, L7 CrossRef CAS.
- V. P. Grivin, I. V. Khmelinski, V. F. Plyusnin, I. I. Blinov and K. P. Balashev, J. Photochem. Photobiol., A, 1990, 51, 167 CrossRef CAS.
- V. P. Grivin, I. V. Khmelinski and V. F. Plyusnin, J. Photochem. Photobiol., A, 1990, 51, 379 CrossRef CAS.
- V. P. Grivin, I. V. Khmelinski and V. F. Plyusnin, J. Photochem. Photobiol., A, 1991, 59, 153 CrossRef CAS.
- G. A. Shagisultanova, Koord. Khim. (Russ. J. Coord. Chem.), 1981, 7, 1527 CAS (in Russian).
- A. G. Fadnis and T. J. Kemp, J. Chem. Soc., Dalton Trans., 1989, 1237 RSC.
- K. P. Balashev, I. I. Blinov and G. A. Shagisultanova, Koord. Khim. (Russ. J. Coord. Chem.), 1987, 13, 1674 CAS (in Russian).
- R. K. Broszkiewicz and B. Voinovic, Radiat. Phys. Chem., 1992, 40, 11 CrossRef CAS.
- E. M. Glebov, V. F. Plyusnin, A. B. Venediktov and S. V. Korenev, Russ. Chem. Bull., 2003, 52, 1305 CrossRef CAS.
- A. V. Litke, I. P. Pozdnyakov, E. M. Glebov, V. F. Plyusnin, N. V. Tkachenko and H. Lemmetyinen, Chem. Phys. Lett., 2009, 477, 304 CrossRef CAS.
- E. M. Glebov, A. V. Kolomeets, I. P. Pozdnyakov, V. F. Plyusnin, N. V. Tkachenko and H. Lemmetyinen, Photochem. Photobiol. Sci., 2011, 10, 1709 CAS.
- N.V. Tkachenko, L. Rantala, A. Y. Tauber, J. Helaja, P. H. Hynninen and H. Lemmetyinen, J. Am. Chem. Soc., 1999, 121, 9378 CrossRef CAS.
- I. I. Chernyaev, Sintez Kompleksnyh Soedinenii Metallov Platinovoi Gruppy (Synthesis of the Coordination Compounds of the Platinum Group Metals), Nauka, Moscow, 1964, p. 339 (in Russian) Search PubMed.
- D. L. Swihart and W. R. Mason, Inorg. Chem., 1970, 9, 1749 CrossRef CAS.
- A. Goursot, E. Penigault and H. Chermette, Chem. Phys. Lett., 1983, 97, 215 CrossRef CAS.
- C. K. Jorgensen, Mol. Phys., 1959, 2, 309 CrossRef CAS.
- P. J. Reid, C. Silva, P. F. Barbara, L. Karki and J. T. Hupp, J. Phys. Chem., 1995, 99, 2609 CrossRef CAS.
- E. A. Juban and J. K. McCusker, J. Am. Chem. Soc., 2005, 127, 6857 CrossRef CAS.
- H. Ito, J. Fujita and K. Saito, Bull. Chem. Soc. Jpn., 1967, 40, 2584 CrossRef CAS.
- W. R. Mason and H. B. Gray, J. Am. Chem. Soc., 1968, 90, 5721 CrossRef CAS.
- E. A. Juban, A. L. Smeigh, J. E. Monat and J. K. McCusker, Coord. Chem. Rev., 2006, 250, 1783 CrossRef CAS.
- N. A. P. Cane-Maguire, Photochemistry and Photophysics of Coordination Compounds: Chromium, in Photochemistry and Photophysics of Coordination Compounds I, V. Balzani and S. Campagna (ed.), Springer, 2007, pp. 37–68 Search PubMed.
- (a) E. M. Glebov, V. F. Plyusnin, N. I. Sorokin, V. P. Grivin, A. B. Venediktov and H. Lemmetyinen, J. Photochem. Photobiol., A, 1995, 90, 31 CrossRef CAS; (b) E. M. Glebov, V. F. Plyusnin, V. P. Grivin, Yu. V. Ivanov, N. V. Tkachenko and H. Lemmetyinen, Int. J. Chem. Kinet., 1998, 30, 711 CrossRef CAS; (c) E. M. Glebov and V. F. Plyusnin, Russ. J. Coord. Chem., 1998, 24, 507 CAS; (d) E. M. Glebov, V. F. Plyusnin, N. V. Tkachenko and H. Lemmetyinen, Chem. Phys., 2000, 257, 79 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |