Fe(CN)6−4-doped polypyrrole: a high-capacity and high-rate cathode material for sodium-ion batteries†
Min
Zhou
a,
Limin
Zhu
a,
Yuliang
Cao
*b,
Ruirui
Zhao
a,
Jianfeng
Qian
b,
Xinping
Ai
b and
Hanxi
Yang
*a
aCollege of Chemistry and Molecular Sciences, Wuhan University, Wuhan 430072, China. E-mail: hxyang@whu.edu.cn; Fax: +86-27-87884476; Tel: +86-27-68754526
bHubei Key Lab. of Electrochemical Power Sources, Department of Chemistry, Wuhan University, Wuhan 430072, China. E-mail: ylcao@whu.edu.cn
First published on 18th April 2012
Abstract
A redox-active Fe(CN)6−4-doped polypyrrole was synthesized and found to have a remarkable redox capacity of 135 mA h g−1 in Na+ electrolyte, an excellent rate capability of 1600 mA g−1 and a strong capacity retention of 85% over 100 cycles, showing great promise for enabling low-cost and environmentally benign Na-ion batteries for large-scale electric energy storage.
Rechargeable batteries with low cost and high energy density are in growing demand for future large-scale electric storage applications such as in electric vehicles and renewable power stations.1–3 At the present state of the art of electrochemical energy storage devices, lithium-ion batteries are now considered to be a promising technology for such applications due to their high electric storage densities. However, there is still a great concern for lithium availability in the world's reserves and the cost of lithium resources, even for the demand of electric vehicles.4 As an alternative to Li-ion batteries, sodium-ion batteries have recently attracted considerable interest because of the natural abundance and low cost of sodium resources, which can no doubt support worldwide electric storage applications.
Na-insertion compounds are very common in intercalation chemistry. However, few of the Na-insertion materials have been qualified as Na-storage materials for battery applications. Although a number of carbonaceous materials such as pyrolytic carbon5 and carbon fiber6 have demonstrated a reversible Na+ insertion, their storage capacity and cycling ability are much poorer than their lithium counterparts7 because larger Na+ ions are difficult to accommodate in graphitic hosts. In contrast, the host materials suitable for Na-storage cathodes are even more difficult to find, similarly due to the frustrated moving of Na+ ions in most metal oxide lattices. For example, layered xLi2MnO3·(1 − x)LiMn0.5Ni0.5O2 cathodes can deliver a reversible capacity of >250 mA h g−1 through Li+ insertion and extraction,8 but its sodium counterpart could realize a reversible capacity of only ∼100 mA h g−1.9 The single crystalline manganese oxide nanowires recently reported exhibit excellent reversible Na insertion behavior, but their capacities are merely ∼100 mA h g−1 at a normal discharge rate of 0.5C.10 In recent years, sodium fluorophosphates11–13 and fluorosulphate14 were developed as cathode materials for Na-ion batteries, however, their Na-storage capacities (70–100 mA h g−1) were not superior to those of transition metal oxides. Particularly, all the Na+-host cathodes developed so far have a severe drawback of poor rate capability, which adds an additional difficulty for electric storage applications.
In our recent work,15 we found that some electroactive polymers can be activated to deliver a greatly enhanced capacity by doping insoluble and redox-active ferricyanide anions in the polymer matrixes, which changes the redox mechanism of the conducting polymers from conventional p-doping/dedoping reactions of anions to the insertion/extraction reactions of small lithium cations, leading to an dramatically improved electrochemical utilization and cycling stability of the polymers. This method could in principle be adopted to create Na+-inserted polymers for achieving high capacity Na-host cathode materials, because polymer matrixes are flexible for reversible insertion and extraction of larger Na+ ions than rigid inorganic lattices.
Here, we report the redox-active Fe(CN)64−-doped polypyrrole with a high reversible capacity and excellent high-rate capability as a Na-storage cathode for ambient Na-ion batteries. As displayed in Fig. 1a, the PPy/FC composite appeared as well-dispersed particles with uniformly distributed size of ∼50 nm diameter, which is hardly soluble in commonly used organic electrolytes. XRD pattern of the samples (see Fig. S1, ESI†) showed a featureless and broad band at 2θ = 25°, suggesting an amorphous structure of the polymer. Fig. 1b compares the FTIR spectrum of the PPy/FC sample with that of pristine PPy polymer prepared at the same conditions. A main difference in the IR spectra is the appearance of a pair of strong bands at 1177 and 918 cm−1 and a single band at 2045 cm−1 in the PPy/FC sample, which are characteristic of doped state of PPy and the Fe(CN)64− dopant, respectively. Except for these IR features, all other IR modes in the PPy/FC sample are similar to those observed in pristine PPy polymer, implying that the Fe(CN)64− doping did not change the chemical structure of polypyrrole chains. Also, the main IR features of Fe(CN)64− are reflected in the PPy/FC polymer, indicative of the Fe(CN)64− moiety kept intact in the doped polymer (see Fig. S2, ESI†). By chemical and atomic emission spectroscopic analysis, the chemical composition of the PPy/FC composite was determined to be PPy17.14+·Fe(CN)64−.
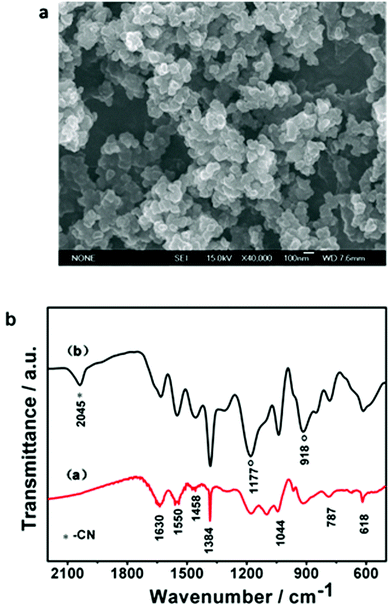 | ||
| Fig. 1 a. SEM image of the as-prepared PPy/FC particles. b. A comparison of the FT-IR spectra of pristine PPy (a) and Fe(CN)64−-doped PPy (b). | ||
The electrochemical redox properties of the PPy/FC composite were evaluated by cyclic voltammetry (CV) and galvanostatic charge/discharge cycling. Fig. 2a displays typical CV curves of the PPy and PPy/FC cathodes in Na+-containing electrolyte. The pristine PPy electrode shows a quasi-rectangular-shaped CV band, representing an electrochemical capacitive response, which is usually characteristic of a PPy electrode.16 Very differently, the CV curve of the PPy/FC electrode exhibits a pair of reproducible anodic and cathodic peaks at 2.75 V and 2.35 V (vs. Na+/Na), respectively, indicating a reversible redox reaction of the Fe(CN)64−-doped polymer matrix. In our recent work, Na4Fe(CN)6 nanoparticles were found to undergo a reversible Na+-insertion reaction with a pair of characteristic anodic and cathodic CV bands at 3.47 V and 3.27 V in 1.0 mol L−1 NaPF6 electrolyte.17 However, in the PPy/FC electrode, the redox bands due to the Fe(CN)64− doping were ∼400 mV shifted to negative region, probably resulting from a coordination of Fe+2/+3 ions to the electronegative N atoms in pyrrole rings. It can also be seen that the CV current from the PPy/FC electrode is greatly enhanced as compared with undoped PPy electrode, suggesting an electrochemical activation of the PPy chains by the redox-active Fe(CN)6−4 anions. This electrochemical activation can also be visualized from the charge/discharge profiles of the polymer electrodes as illustrated in Fig. 2b. Measured from coin-type Na-PPy and Na-PPy/FC cells, the pristine PPy electrode shows a typical capacitive charge-discharge behavior with a reversible capacity of only ∼35 mA h g−1, which is in agreement with frequently reported values (20∼60 mA h g−1).18 Nevertheless, the PPy/FC electrode exhibits a dramatic capacity enhancement with reversible capacity of 135 mA h g−1, which is one of the highest values ever reported for Na-storage cathodes, ∼80% higher than the radical polymer cathode recently reported for Na-storage,19 demonstrating an effective electrochemical activation of the PPy chains by Fe(CN)6−4 doping.
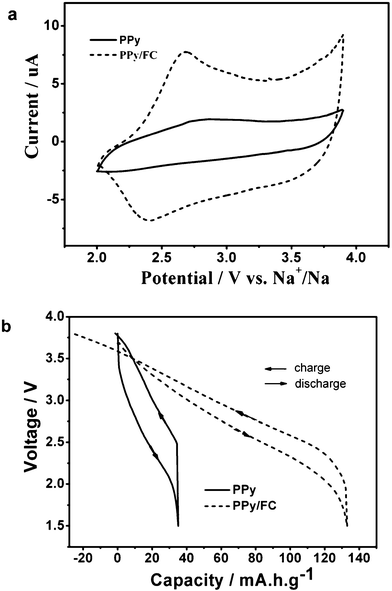 | ||
| Fig. 2 A comparison of electrochemical responses of pristine and Fe(CN)64−-doped polypyrrole in 1.0 mol L−1 NaPF6 + ethylene carbonate/diethyl carbonate (EC/DEC) electrolyte. a. Cyclic voltammograms measured by three-electrode cells using a Na reference at a slow scan of 0.1 mV s−1. b. Charge-discharge curves of coin-type cells measured at a constant current of 50 mA g−1. | ||
In addition to its high redox capacity, the PPy/FC material also exhibits superior high rate capability as a cathode of Na-batteries. Fig. 3a gives the charge and discharge curves of the PPy/FC cathode in a Na+ electrolyte at various current rates. As displayed in the figure, the PPy/FC electrode delivered a reversible capacity of 135 mA h g−1 at 50 mA g−1, 128 mA h g−1 at 100 mA g−1, and 105 mA h g−1 at 400 mA g−1. Even the current density was increased to a very high value of 1600 mA h g−1, the reversible capacity of the PPy/FC cathode can still realize ∼75 mA h g−1. This high rate performance is hardly seen from conventional metal oxide cathodes, apparently due to the flexible polymer matrix where the counter ions are allowed to move in and out with less restriction.
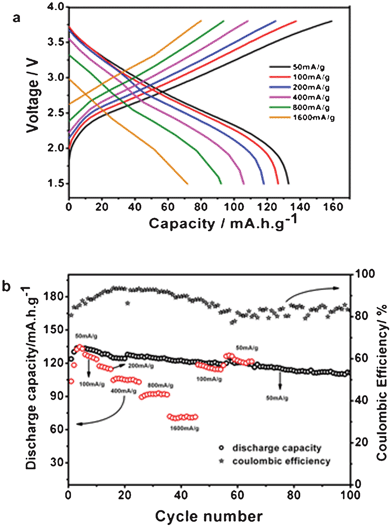 | ||
| Fig. 3 Rate capability and cycling performance of the PPy/FC polymer. a. Charge and discharge profiles of the PPy/FC cathode at various high rates as labeled. b. Reversible capacity of the PPy/FC cathode cycled at a constant rate of 50 mA g−1 and at various high rates. The cycling efficiency of the cells at 50 mA g−1 is also inserted in the figure. The data were derived from coin-type Na-PPy/FC cells using 1 mol L−1 NaPF6 +EC/DEC as electrolyte. | ||
The PPy/FC cathode has demonstrated not only high reversible capacity and high rate capability, but can also show good cyclability. Fig. 3b displays the cycling performances of the PPy/FC cathode, measured from 2032 coin-type cells with a Na disk anode. At a moderate rate of 50 mA g−1 charge/discharge, the reversible capacity of the PPy/FC cathode rose up from its initial value of 120 mA h g−1 rapidly to ∼135 mA h g−1 at fourth cycle and then remained ∼110 mA h g−1 after 100 cycles, corresponding to a capacity retention of 85% over 100 cycles. Even cycled at changing rates as shown in Fig. 3b, the PPy/FC electrode can still keep its capacity steadily without much degradation. After 70 cycles at various high rates from 50 to 1600 mA g−1, the polymer cathode can recover mostly its initial capacity of ∼125 mA h g−1, demonstrating an excellent cycling stability.
To reveal the underlying chemistry for the superior electrochemical performances of the polymer cathode, we characterized the changes in the bonding state of the PPy/FC polymer at different stages of charge and discharge, as shown in Fig. 4. When first discharged to 1.5 V, the PPy/FC polymer showed very similar IR features to its pristine state except for the disappearance of the strong 1170 and 918 cm−1 bands that are characteristic of the doped state of PPy chains, indicating an electrochemical reduction (or de-doping reaction) of the positively charged PPy skeleton to an intrinsic neutral state. Since the Fe(CN)6−4 anions are insoluble in the organic electrolyte and coordinated on the polymeric chains, the de-doping process could only be carried through by Na+ insertion for electric charge counterbalance rather than an extraction of large anions from the polymer chains. Once recharged to 3.4 V, the 1170 and 918 cm−1 bands suddenly re-emerged as a sign of p-doping of the polymer, while the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N vibration band shifted from 2042 cm−1 to 2058 cm−1, characterizing a concomitant oxidation of doped Fe(CN)6−4 ions to Fe(CN)6−3 ions. In addition, a new 867 cm−1 band featuring the P–F bond vibration in PF6− anions also appeared at the charging process, implying that the charging reaction involved the doping of PF6− anions into the polymer matrix. With continuous charging to a cutoff potential of +3.8 V, the IR bands at 1170, 918 and 867 cm−1 increased their intensities and the CN band gradually moved to a high wavenumber of 2071 cm−1. These changes reflect a continuous increase in the doping degree of the PPy chains accompanying Na+ extraction and PF6− insertion. X-ray photoelectron spectra of the Fe 2p3/2 electrons for the charged and discharged PPy/FC electrode (see Fig. S3, ESI†) confirm the redox reaction of the Fe+2/+3 ions in the polymer electrode. At the discharged state, the binding energy for the Fe 2p3/2 electrons appeared at 708.6 eV, reflecting the Fe ions existing in a valence state of +2 in the polymer matrix. When the electrode was charged, the binding energy for the Fe 2p3/2 electrons shifted to 708.9 eV at +3.4 V and then moved to 709.3 eV at complete charge to +3.8 V, demonstrating a progressive oxidation of the Fe+2 ions to Fe+3 ions in the Fe(CN)6−4 dopant during charge process and vice versa.
N vibration band shifted from 2042 cm−1 to 2058 cm−1, characterizing a concomitant oxidation of doped Fe(CN)6−4 ions to Fe(CN)6−3 ions. In addition, a new 867 cm−1 band featuring the P–F bond vibration in PF6− anions also appeared at the charging process, implying that the charging reaction involved the doping of PF6− anions into the polymer matrix. With continuous charging to a cutoff potential of +3.8 V, the IR bands at 1170, 918 and 867 cm−1 increased their intensities and the CN band gradually moved to a high wavenumber of 2071 cm−1. These changes reflect a continuous increase in the doping degree of the PPy chains accompanying Na+ extraction and PF6− insertion. X-ray photoelectron spectra of the Fe 2p3/2 electrons for the charged and discharged PPy/FC electrode (see Fig. S3, ESI†) confirm the redox reaction of the Fe+2/+3 ions in the polymer electrode. At the discharged state, the binding energy for the Fe 2p3/2 electrons appeared at 708.6 eV, reflecting the Fe ions existing in a valence state of +2 in the polymer matrix. When the electrode was charged, the binding energy for the Fe 2p3/2 electrons shifted to 708.9 eV at +3.4 V and then moved to 709.3 eV at complete charge to +3.8 V, demonstrating a progressive oxidation of the Fe+2 ions to Fe+3 ions in the Fe(CN)6−4 dopant during charge process and vice versa.
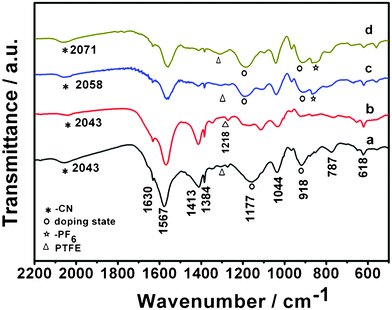 | ||
| Fig. 4 FT-IR spectra of the PPy/FC electrode. a. At open circuit. b. First discharged to 1.5 V. c. Charged to +3.4 V. d. Fully charged to +3.8 V. | ||
Based on the reaction mechanism stated above, the high discharge capacity observed from the PPy/FC electrode can be satisfactorily accounted for by a reversible Na-insertion/extraction reaction followed with a PF6− doping/dedoping process. At first discharge, the starting PPy17.14+Fe(CN)64− material must undergo an electroreduction reaction:
| PPy17.14+·Fe(CN)64− + 4Na+ + 4e ↔ PPy17.1·Fe(CN)64−·4Na+ | (1) |
According to this equation, the theoretical reversible capacity of the first discharge reaction could be determined to be ∼80 mA h g−1 upon 4 Na+ insertions. At reversed charging, the first step of the electrooxidation of the PPy/FC electrode must occur with 4 Na+ extractions, giving rise to a charging capacity of 80 mA h g−1. Direct quantitative analysis of the amount of cycled Na+ ions does give further evidence to support this charge-discharge mechanism (see Table 1, ESI†). The electrode samples taken from a fully discharged state showed a high Na/Fe ratio of 4.241, corresponding to 3.84 Na inserted into the polymer. When recharged to 3.8 V, the electrode samples gave a Na/Fe molar ratio of 1.199, demonstrating an extraction of the inserted Na ions from the polymer chains. Taking into account the experimental errors, the numbers of cycled Na+ ions agree very well with the theoretical value expected from eqn (1).
If the charging continues, successive oxidation of the Fe(CN)64− dopant should happen along with equal moles of PF6− anions inserted into the polymer matrix for charge counterbalance:
| PPy17.14+·Fe(CN)64− + PF6− − e ↔ PPy17.14+·Fe(CN)63−·PF6− | (2) |
In fact, the PPy chains can also undergo electrochemical redox or pseudocapacitive charge storage reaction at the same time in the oxidation of the Fe(CN)64− anions:
| PPy17.14+·Fe(CN)63−·PF6− + xPF6− − xe ↔ PPy 17.1(4+x)+·Fe(CN)63−·(1 + x)PF6− | (3) |
Taking into account the Fe(CN)64− content in the polymer, the theoretical reversible capacity given from eqn (2) is calculated to be ∼20 mA h g−1 because of only ∼5 wt% Fe(CN)64− doped in the PPy chains. The reversible capacity only due to the p-doping/dedoping reaction (eqn (3)) should be the same for pristine PPy and the PPy/FC electrode, which is ∼35 mA h g−1 as given from pristine PPy skeleton in Fig. 2, since the doping level of PF6− anions to the PPy chains should be at the same degree in both pristine PPy and in the PPy/FC electrode. Thus, we can thus sum up the stoichiometric capacities of Na-insertion/extraction (80 mA h g−1), Fe(CN)63−/4− redox (20 mA h g−1) and p-doping reaction (35 mA h g−1) as a totally realizable capacity of the PPy/FC electrode (135 mA h g−1). Obviously, this calculated redox capacity is in very close agreement with the experimental value (135 mA h g−1) for the PPy/FC electrode, suggesting that a major capacity enhancement by the redox-active Fe(CN)6−4 doping was contributed from the reversible Na+ insertion-extraction and Fe+2/+3 redox reactions. The chemical structure of the PPy/FC polymer and its reaction mechanism are schematically demonstrated in Scheme 1.
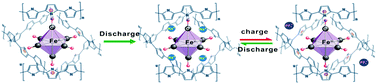 | ||
| Scheme 1 Chemical structure and redox mechanism of the PPy/FC polymer. | ||
In summary, we demonstrate that the composite polymer illustrated here exhibits a superior redox performances with a high reversible capacity of 135 mA h g−1, an excellent rate capability at 1600 mA g−1 and a strong capacity retention of 85% over 100 cycles, which exceeds all the inorganic Na-host materials ever reported and can possibly serve as a high-capacity and high-rate cathode materials for sodium-ion batteries. Particularly, the doping method described in this work is simple and easily extended to other conducting polymers, providing a new strategy for creating low cost and electrochemically active polymer materials for widespread electric storage applications.
Acknowledgements
We thank the 973 Program (2009CB220103) of China and the National Science Foundation of China (No. No. 2097132) for financial Support.References
- B. Dunn, H. Kamath and J. M. Tarascon, Science, 2011, 334, 928–934 CrossRef CAS.
- V. Palomares, P. Serras, I. Villaluenga, K. B. Hueso, J. Carretero-Gonzalez and T. Rojo, Energy Environ. Sci., 2012, 5, 5884 CAS.
- M. R. Palacin, Chem. Soc. Rev., 2009, 38, 2565–2575 RSC.
- J.-M. Tarascon, Nat. Chem., 2010, 2, 510 CrossRef CAS.
- R. Alcántara, P. Lavela, G. F. Ortiz and J. L. Tirado, Electrochem. Solid-State Lett., 2005, 8, A222–A225 CrossRef.
- P. Thomas and D. Billaud, Electrochim. Acta, 2002, 47, 3303–3307 CrossRef CAS.
- R. Alcántara, J. M. Mateos and J. L. Tirado, J. Electrochem. Soc., 2002, 149, A201–A205 CrossRef.
- M. M. Thackeray, C. S. Johnson, J. T. Vaughey, N. Li and S. Hackney, J. Mater. Chem., 2005, 15, 2257–2267 RSC.
- D. Kim, Adv. Energy Mater., 2011, 1, 333–336 CrossRef CAS.
- Y. Cao, Adv. Mater., 2011, 23, 3155–3160 CrossRef CAS.
- H. Zhuo, J. Power Sources, 2006, 160, 698–703 CrossRef CAS.
- J. Barker and M. Y. Saidi, Electrochem. Solid-State Lett., 2003, 6, A1 CrossRef CAS.
- N. Recham, J. Electrochem. Soc., 2009, 156, A993–A999 CrossRef CAS.
- P. Barpanda, Inorg. Chem., 2010, 49, 7401–7413 CrossRef CAS.
- M. Zhou, J. Qian, X. Ai and H. Yang, Adv. Mater., 2011, 23, 4913–4917 CrossRef CAS.
- C. Yang, P. Liu and T. Wang, ACS Appl. Mater. Interfaces, 2011, 3, 1109–1114 CAS.
- J. F. Qian, M. Zhou, Y. L. Cao, X. P. Ai and H. X. Yang, Adv. Energy Mater., 2012, 2, 410 CrossRef CAS.
- K. S. Park, S. B. Schougaard and J. B. Goodenough, Adv. Mater., 2007, 19, 848–851 CrossRef CAS.
- Y. Dai, Y. Zhang, L. Gao, G. Xu and J. Xie, Electrochem. Solid-State Lett., 2010, 13, A22–A24 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: experimental details. See DOI: 10.1039/c2ra20666h/ |
| This journal is © The Royal Society of Chemistry 2012 |