Application of a multi-SO3H Brønsted acidic ionic liquid in water: a highly efficient and reusable catalyst for the regioselective and scaled-up synthesis of pyrazoles under mild conditions†
Shirin
Safaei
a,
Iraj
Mohammadpoor-Baltork
*a,
Ahmad Reza
Khosropour
*a,
Majid
Moghadam
a,
Shahram
Tangestaninejad
a,
Valiollah
Mirkhani
a and
Reza
Kia
b
aCatalysis Division, Department of Chemistry, University of Isfahan, Isfahan, 81746-73441, Iran. E-mail: imbaltork@sci.ui.ac.ir; khosropour@chem.ui.ac.ir; Fax: +98(311)6689732; Tel: +98(311)7932705
bDepartment of Chemistry, Science and Research Branch, Islamic Azad University, Tehran, Iran
First published on 11th April 2012
Abstract
An elegant and efficient procedure with exceptionally mild conditions for the regioselective synthesis of pyrazoles by the reaction of various 1,3-diketones and hydrazines/hydrazides using a multi-SO3H Brønsted acidic room temperature ionic liquid as a powerful catalyst in aqueous media has been developed. The ionic liquid was easily separated from the reaction mixture and was recycled and used for at least six consecutive runs without any loss of activity.
Introduction
Since a great amount of waste in the environment is attributed to the use of organic solvents,1 there is a growing demand for the development of organic reactions in environmentally-friendly media.2 In recent years, water as a nontoxic, nonflammable, cheap, renewable and environmentally friendly reaction medium has gained particular interest in organic synthesis.3 In addition, the remarkable properties seen in water as a result of its chemical and physical properties are very useful for selectivity/reactivity that cannot be attained in organic solvents.4 Consequently, the development of highly efficient and environmentally-benign procedures using reusable catalysts in water as the reaction medium is highly desirable.Recently, ionic liquids have attracted increasing interest in the context of green synthesis.5 Although ionic liquids were initially introduced as an alternative green reaction medium, today they have progressed far beyond this, having a significant role in controlling reactions as catalysts.6 However, moisture sensitivity and the decomposition of some ionic liquids under normal atmospheric conditions are two major drawbacks to their practical use.7 Furthermore, selecting ionic liquids with the appropriate attributes for a technological application is a daunting task, due to available structural possibilities.8 It is clear from the published data that the type of cation and its substituents, and the anion can improve or suppress the catalytic properties of the ionic liquids for a particular reaction. A specific category of ionic liquids, namely Brønsted acidic ionic liquids (BAILs) with SO3H-functionalized cations have been reported to act as active and green catalysts for various acid-catalyzed reactions.9 So, the design of organic syntheses in the presence of these BAIL catalysts, typically in water, at room temperature as an environmentally-benign and economic fashion is of practical importance.
Over the years, pyrazole derivatives have emerged as a significant class of nitrogenated heterocycles that have attracted much synthetic interest due to their broad spectrum of pharmacological and therapeutic properties such as anti-anxiety,10 antipyretic,11 analgesic,12 anti-inflammatory,13 antimicrobial,14 antiviral15 and anticancer16 activities. Celecoxib (Fig. 1) is a drug containing the pyrazole core that has been used for the treatment of osteoarthritis, acute pain, rheumatoid arthritis, painful menstruation and menstrual symptoms, and for reducing the number of colon and rectum polyps in patients with familial adenomatous polyposis.17,18
 | ||
| Fig. 1 Structure of Celecoxib. | ||
Also, the pyrazole nucleus appears in a number of naturally occurring products like 1-pyrazolylalanine that has been found in the seeds of watermelons.19 In addition, some of these derivatives have attracted attention due to their potential as herbicides, fungicides and insecticides.20 These pharmacological and biological properties of pyrazole skeletons encourage the development of convenient and mild methods for their synthesis. A number of synthetic methods for these heterocycles have been developed.21
Traditionally the synthesis of these heterocyclic compounds is performed by the condensation reaction of hydrazines and their derivatives with 1,3-diketones.22 Such reactions are generally carried out in the presence of Brønsted or Lewis acids such as Al2O3/montmorillonite,23 PSSA,24 K2CO3/[Cp*IrCl2]2,25 zirconium sulfophenyl phosphonate26 or H3PW12O40.27 Very recently, this transformation was modified by Kidwai et al. using Zn[L-proline]2.28 Moreover, several examples have been reported of the synthesis of trifluoromethyl substituted pyrazoles due to their specific applications in medicine and agriculture.29 One of the best methods to introduce a trifluoromethyl group into these heterocycles is based on the trifluoromethylated building block approach.30 Recently, Bonacorso et al. described the synthesis of trifluoromethyl substituted pyrazoles from the reaction of trifluoroacetylated enol ethers with hydrazines employing diethylaminosulfur trifluoride (DAST) in dichloromethane as the solvent at room temperature for 24 h.31
However, many aforementioned methods suffer from serious drawbacks including unsatisfactory yields, long reaction times, tedious experimental procedures, high temperature, low selectivity, expensive reagents, and hazardous solvents, as well as large amount of catalysts and their non-recyclability which lead to difficulty in product separation. Therefore, the development of a reusable and more convenient catalyst for the synthesis of pyrazoles without the use of any hazardous solvents would extend the scope of this transformation.
In 2002, Cole et al. reported a functional ionic liquid with strong Brønsted acidity as a task specific ionic liquid (TSIL).32 After that, sulfonic alkyl group functionalized ionic liquids were reported as efficient and convenient acidic catalysts due to the combined advantages of both liquid and solid acids, e.g., easy separation, reusability and uniform acid sites.33 So, the research and application of various –SO3H functionalized ionic liquids have received more and more attention.34
These results in combination with our recent work on designing new synthetic methodologies,35 especially in Brønsted acidic ionic liquids,36 led us to present our recyclable multi-SO3H Brønsted acidic room temperature ionic liquid A in water as a powerful catalyst for the green and efficient synthesis of pyrazoles under mild conditions (Scheme 1).
 | ||
| Scheme 1 Regioselective synthesis of pyrazoles catalyzed by A. | ||
Results and discussion
The ionic liquid A, was prepared by modification of the method reported by Liang et al.34a as shown in Scheme 2. | ||
| Scheme 2 Synthesis of A, a Brønsted acidic ionic liquid. | ||
Hexamethylenetetramine was allowed to react with four equimolar amount of 1,3-propanesultone in THF solution at room temperature to produce the zwitterionic hexamethylenetetrammonium salt. Treatment of this salt with sulfuric acid at 120 °C for 10 h gave A in quantitative yield.37
Initially we selected benzohydrazide and acetylacetone for screening the reaction conditions (Table 1). Using 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratios of benzohydrazide and acetylacetone and 15 mol% of A as the catalyst in an aqueous medium at 25 °C for 5 min, the desired (3,5-dimethyl-1H-pyrazol-1-yl)(phenyl)methanone was obtained in 96% yield (Table 1, entry 2). With respect to the quantity of the catalyst, there was a significant decline in the yield when the amount of catalyst was decreased (Table 1, entry 1). It is noteworthy that the desired product was obtained in a very low yield in the absence of the catalyst even after 24 h (Table 1, entry 3).
:1 molar ratios of benzohydrazide and acetylacetone and 15 mol% of A as the catalyst in an aqueous medium at 25 °C for 5 min, the desired (3,5-dimethyl-1H-pyrazol-1-yl)(phenyl)methanone was obtained in 96% yield (Table 1, entry 2). With respect to the quantity of the catalyst, there was a significant decline in the yield when the amount of catalyst was decreased (Table 1, entry 1). It is noteworthy that the desired product was obtained in a very low yield in the absence of the catalyst even after 24 h (Table 1, entry 3).
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Amount (mol%) | Time (min) | Yield (%)b |
| a Acetylacetone (1 mmol), phenylhydrazide (1 mmol) in H2O (3 ml) at room temperature. b Isolated yield. | ||||
| 1 | A | 10 | 5 | 82 |
| 2 | A | 15 | 5 | 96 |
| 3 | — | — | 1440 | 19 |
| 4 | p-TSA | 15 | 5 | 73 |
| 5 | [Hmim]HSO4− | 15 | 5 | 30 |
| 6 | [Mspim]HSO4 | 15 | 5 | 45 |
| 7 | [Bmim]BF4− | 15 | 5 | 10 |
| 8 | [Bmim]PF6− | 15 | 5 | — |
| 9 | [Mspim]HSO4 | 60 | 5 | 70 |

Comparison of the catalytic activity of A with other Brønsted or Lewis acidic ionic liquids under the optimal reaction conditions, showed that it had the highest catalytic activity of the present catalyst (Table 1, compare entries 2 and 4–8). It is surprising that in the presence of commercially available ionic liquids such as [Bmim]BF4 or [Bmim]PF6 or even Brønsted acidic ionic liquids such as 1-methylimidazolium hydrogen sulfate ([Hmim]HSO4) or 1-methyl-3-(3-sulfopropyl)-1H-imidazol-3-ium hydrogen sulfate ([Mspim]HSO4), the corresponding product was obtained in low yields (Table 1). It is also noteworthy that, even in the presence of 60 mol% of monofunctional ionic liquid Brønsted acidic, [Mspim]HSO4 (equal to 15 mol% of A), the corresponding product was obtained in 70% yield (Table 1, entry 9).
After determining the optimum reaction conditions, we turned our attention towards studying the scope of this method. So, we attempted a series of reactions with a variety of 1,3-diketones and hydrazines or hydrazides under mild reaction conditions.38 The results are summarized in Table 2.
| Entry | 1,3-Diketone | Hydrazine/hydrazide | Product | Time (min) | Yield (%)b | TOF (h−1) | |
|---|---|---|---|---|---|---|---|
| a Diketone (1 mmol), hydrazine/hydrazide (1 mmol) and catalyst (15 mol%) in H2O (3 ml) at room temperature. b Isolated yield. c Reaction was performed in the presence of 15 mol% of p-TSA. d Reaction was performed in the presence of 60 mol% of p-TSA. | |||||||
| 1 |

|

|

|
3a | 2 | 98 | 196.0 |
| 2 |

|

|
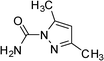
|
3b | 5 | 97 | 77.6 |
| 3 |

|

|
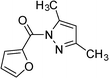
|
3c | 5 | 95 | 76.0 |
| 4 |

|

|

|
3d | 5 | 96 | 76.8 |
| 5 |

|

|

|
3e | 2 | 91 | 182.0 |
| 6 |

|

|
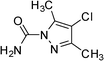
|
3f | 5 | 95 | 76.0 |
| 7 |
|

|

|
3g | 15 | 74 | 19.7 |
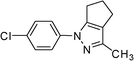
| 8 |

|

|
|
3h | 5 | 78 | 62.4 |
| 9 |

|

|

|
3i | 2 | 97 | 194.0 |
| 10 |

|

|

|
3j | 5 (5c,d) | 96 (61c, 82d) | 76.8 (48.8c, 16.4d) |
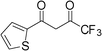
| 11 |
|

|

|
3k | 2 | 94 | 188.0 |
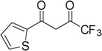
| 12 |
|

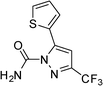
|
|
3l | 5 | 96 | 76.8 |
A wide range of structurally diverse arylhydrazines or arylhydrazides and 1,3-diketones underwent condensation by this reaction to provide pyrazole derivatives in high to excellent yields. Phenyl hydrazine and its derivatives with both activating and deactivating groups such as Me or Cl, reacted rapidly to afford the corresponding products in high to excellent yields (Table 2, entries 1, 5 and 8–11).
The present method is also equally effective with acyclic 1,3-diketones such as pentane-2,4-dione, 3-chloropentane-2,4-dione, 1,1,1-trifluoropentane-2,4-dione or cyclic 1,3-diketone such as 2-acetylcyclopentanone, producing the desired products in high yields (Table 2). This catalytic system also worked well with 4,4,4-trifluoro-1-(thiophen-2-yl)butan-1,3-dione as an acid-sensitive heteroaromatic 1,3-diketone to afford the corresponding products in 94–96% yields without accompanying polymerization (Table 2, entries 10–12). Semicarbazide was also afforded excellent yields (Table 2, entries 2, 6 and 12). The success of our efficient synthesis of pyrazole derivatives also encouraged us to explore this methodology for trifluoromethyl substituted pyrazoles. Interestingly, the 5-(trifluoromethyl) pyrazole derivatives were obtained from the reaction of 1,1,1-trifluoromethyl β-diketones with aryl hydrazines or semicarbazides by this methodology (Table 2, entries 9–12).
In general, this method was clean, and the conversions were almost complete within 2–15 min, affording the products in high to excellent yields (74–98%).
In order to compare the catalytic activity of p-TSA with A in the conversion of unsymmetrical 1,3-diketones to their corresponding pyrazoles, the reaction of 4,4,4-trifluoro-1-(thiophen-2-yl)butan-1,3-dione with 4-methylphenylhydrazine (Table 2, entry 10) in the presence of 15 mol% and 60 mol% (equal to 15 mmol of A) of p-TSA was examined. Under these conditions, the desired product (3j) was obtained in only 61% and 82% yields, respectively, and the regioselectivity was as same as that of A.
Moreover, to the best of our knowledge, previous works reported in the formation of mixtures of regioisomeric pyrazoles.39 Recently, this regioselectivity was modified by Müller et al. through a harsh method via Sonogashira coupling/Michael addition/cyclocondensation reaction.40 Surprisingly, our procedure has very high regional selectivity and provides excellent pathway for the synthesis of only one regioisomer of unsymmetrical pyrazoles (Table 2, entries 7–12).
As a result, we obtained the target product 3 in the form of compounds 3g–3l exclusively. To verify the selectivity, 3h and 3k were selected as representative compounds, and the position of the N heteroatoms in the corresponding products was proved by X-ray crystallography emphasized excellent regioselectivity (Fig. 2, CCDC 850136 for 3h and Fig. 3, CCDC 850135 for 3k). This regioselectivity could be attributed to the nucleophilic attack of hydrazine/hydrazide to the more active carbonyl group of 1,3-diketones.
 | ||
| Fig. 2 ORTEP diagram of 3h. | ||
 | ||
| Fig. 3 ORTEP diagram of 3k. | ||
The proposed mechanism for the formation of the products can be explained by the pathway presented in Scheme 3. Indeed, due to better enolizeability of one of the carbonyl group relative to the other one, the terminal nitrogen of hydrazine/hydrazide initially attacks the more electrophilic carbon. So, the first step involves facile enolization/imination reaction activated by the catalyst A which results in intermediate 4. intramolecular nucleophilic addition in 4 catalyzed by A gives 5. Finally, dehydration of 5 in the presence of the catalyst produces the corresponding pyrazole 3 and releases the catalyst for the next catalytic cycle.
 | ||
| Scheme 3 Proposed reaction pathway. | ||
Since, the first step of the proposed mechanism involves enolization/imination, a multivalent acid can speed up the reaction by protonation of both carbonyl groups simultaneously in a single step. Simultaneous protonation of the two carbonyl groups of a 1,3-dicarbonyl compound by monovalent acids such as p-TSA and [Mspim]HSO4 has higher activation entropy than that by the four-valent acids such as A.41 Thus, the synthesis of pyrazoles in the presence of A is expected to be faster with higher yields. In order to p-TSA and [Mspim]HSO4 play their acidic role in the early stage of the reaction, it is required that two molecules approach a small space around the 1,3-dicarbonyl entity which is unfavorable because of intermolecular electrostatic repulsion. This problem is already solved in the formation of A which contains four acidic groups in the same molecule.
However, when the reaction of thiosemicarbazide or furan-2-carbohydrazide with 1,1,1-trifluoropentane-2,4-dione was carried out under the optimized reaction conditions, interestingly the corresponding 4,5-dihydro-1H-pyrazoles 4 were obtained by simple filtration in 98% and 97% yields respectively (Scheme 4, 4a and 4b). The structure of 4a has also been confirmed by X-ray crystallographic analysis (Fig. 4, CCDC 850137). The stability of these compounds (4a and 4b) can be attributed to: (i) the destabilizing effect of the CF3 group on carbocation at C-5; (ii) intramolecular hydrogen bonding between OH and thiocarbonyl/carbonyl groups (based on X-ray crystal structure of 4a); and (iii) resonance between N1 and thiocarbonyl/carbonyl group, which prevents its participation in stabilizing carbocation at C-5 resulting from elimination of a hydroxy group.
 | ||
| Scheme 4 Synthesis of 4,5-dihydro-1H-pyrazoles catalyzed by A. | ||
 | ||
| Fig. 4 ORTEP diagram of 4a. | ||
The catalyst (due to excellent solubility in water), can be easily separated by washing with distilled water. It was easily recyclable after evaporation at 80 °C, reaching its initial volume. The results illustrated that the catalyst retained its activity even after six consecutive runs (Table 3).
Finally, the scale-up synthesis of the reaction was investigated in the case of 3d as a model reaction. Encouraged by the above results, we increased the scale of the reaction to 50.0 mmol, keeping the reaction stoichiometry unchanged. The reaction was found to proceed successfully and the corresponding product was obtained in 93% yield.
Conclusions
In summary, we have demonstrated an efficient, scale-up and facile synthesis of pyrazoles in the presence of catalytic amounts of a multi-SO3H Brønsted acidic room temperature ionic liquid, which acts as a powerful catalyst in aqueous media. Using water as a solvent make the process simple, cheap and clean. This new acidic ionic liquid has shown high catalytic activity for the generation of a diverse range of pyrazoles in high to excellent yields and excellent regioselectivity. The experimental simplicity, ease of product isolation, reusability of the catalyst, low cost, and ready availability of the reagents make this process very useful for the large scale synthesis of pyrazole derivatives. Further exploration of this ionic liquid is under way.Acknowledgements
The authors are grateful to the Center of Excellence of Chemistry and also the Research Council of the University of Isfahan for financial support of this work.References
- (a) K. H. Shaughnessy and R. B. DeVasher, Curr. Org. Chem., 2005, 9, 585 CrossRef CAS; (b) J.-F. Wei, J. Jiao, J.-J. Feng, J. Lv, X.-R. Zhang, X.-Y. Shi and Z.-G. Chen, J. Org. Chem., 2009, 74, 6283 CrossRef CAS.
- P. G. Jessop, Green Chem., 2011, 13, 1391 RSC.
- (a) R. A. Breslow, Fifty-Year Perspective on Chemistry in Water. In Organic Reactions in Water, ed. U. M. Lindström, Blackewll, Oxford, U.K., 2007; pp 1–28 Search PubMed; (b) S. Minakata and M. Komatsu, Chem. Rev., 2009, 109, 711 CrossRef CAS; (c) A. Chanda and V. V. Fokin, Chem. Rev., 2009, 109, 725 CrossRef CAS; (d) R. N. Butler and A. G. Coyne, Chem. Rev., 2010, 110, 6302 CrossRef CAS; (e) X. Zhang, Y. Zhou, H. Wang, D. Guo, D. Ye, Y. Xu, H. Jiang and H. Liu, Green Chem., 2011, 13, 397 RSC; (f) V. K. Rai, P. K. Rai, S. Bajaj and A. Kumar, Green Chem., 2011, 13, 1217 RSC; (g) X. Fan, Y. He, L. Cui, X. Zhang and J. Wang, Green Chem., 2011, 13, 3218 RSC; (h) P. Gunasekaran, K. Balamurugan, S. Sivakumar, S. Perumal, J. C. Menéndez and A. I. Almansour, Green Chem., 2012, 14, 750 RSC; (i) Y.-Y. Peng, J. Liu, X. Lei and Z. Yin, Green Chem., 2010, 12, 1072 RSC.
- (a) M. B. Gawande and P. S. Branco, Green Chem., 2011, 13, 3355 RSC; (b) P. Das and J. McNulty, Tetrahedron Lett., 2010, 51, 3197 CrossRef CAS.
- (a) P. Wasserscheid and P. Welton, Ionic Liquids in Synthesis, Wiley-VCH, Weinheim, 2003 Search PubMed; (b) T. Welton, Chem. Rev., 1999, 99, 2071 CrossRef CAS; (c) J. Dupont, R. F. Souza and P. A. Z. Suarez, Chem. Rev., 2002, 102, 3667 CrossRef CAS; (d) F. Rantwijk and R. A. Sheldon, Chem. Rev., 2007, 107, 2757 CrossRef; (e) M. A. P. Martins, C. P. Frizzo, D. N. Moreira, N. Zanatta and H. G. Bonacorso, Chem. Rev., 2008, 108, 2015 CrossRef CAS; (f) H. Olivier-Bourbigou, L. Magna and D. Morvan, Appl. Catal., A, 2010, 373, 1 CrossRef CAS.
- Q. Zhang, S. Zhang and Y. Deng, Green Chem., 2011, 13, 2619 RSC.
- (a) T. M. Potewar, S. A. Siddiqui, R. J. Lahoti and K. V. Srinivasan, Tetrahedron Lett., 2007, 48, 1721 CrossRef CAS; (b) A. R. Khosropour, Can. J. Chem., 2008, 86, 264 CrossRef CAS.
- (a) H. Tokuda, K. Hayamizu, K. Ishii, M. A. B. H. Susan and M. Watanabe, J. Phys. Chem. B, 2004, 108, 16593 CrossRef CAS; (b) R. E. Baltus, R. M. Counce, B. H. Culbertson, H. Luo, D. W. DePaoli, S. Dai and D. C. Duckworth, Sep. Sci. Technol., 2005, 40, 525 CrossRef CAS; (c) H. Tokuda, K. Hayamizu, K. Ishii, M. A. B. H. Susan and M. Watanabe, J. Phys. Chem. B, 2005, 109, 6103 CrossRef CAS; (d) W. Shi, D. C. Sorescu, D. R. Luebke, M. J. Keller and S. Wickramanayake, J. Phys. Chem. B, 2010, 114, 6531 CrossRef CAS; (e) H. B. Nulwala, C. N. Tang, B. W. Kail, K. Damodaran, P. Kaur, S. Wickramanayake, W. Shic and D. R. Luebke, Green Chem., 2011, 13, 3345 RSC.
- (a) P. A. Ganeshpure, G. George and J. Das, J. Mol. Catal. A: Chem., 2008, 279, 182 CrossRef CAS; (b) S. Zhang, H. Lefebvre, M. Tessier and A. Fradet, Green Chem., 2011, 13, 2786 RSC.
- M. Abdel-Aziz, G. E.-D. A. Abuo-Rahma and A. A. Hassan, Eur. J. Med. Chem., 2009, 44, 3480 CrossRef CAS.
- F. R. Souza, V. T. Souza, V. Ratzlaff, L. P. Borges, M. R. Oliveira, H. G. Bonacorso, N. Zanatta, M. A. P. Martins and C. F. Mello, Eur. J. Pharmacol., 2002, 451, 141 CrossRef CAS.
- M. M. M. El-Din, A. M. Senbel, A. A. Bistawroos, A. El-Mallah, N. A. N. El-Din, A. A. Bekhit and H. A. Abd El Razik, Basic Clin. Pharmacol. Toxicol., 2011, 108, 263 CrossRef CAS.
- A. K. Tewari, P. Srivastava, V. P. Singh, A. Singh, R. K. Goel and C. G. Mohan, Chem. Pharm. Bull., 2010, 58, 634 CrossRef CAS.
- D. Boschi, S. Guglielmo, S. Aiello, G. Morace, E. Borghi and R. Fruttero, Bioorg. Med. Chem. Lett., 2011, 21, 3431 CrossRef CAS.
- O. I. El-Sabbagh, M. M. Baraka, S. M. Ibrahim, G. Andrei, R. Snoeck, J. Balzarini and A. A. Rashad, Eur. J. Med. Chem., 2009, 44, 3746 CrossRef CAS.
- (a) G. M. Nitulescu, C. Draghici and A. V. Missir, Eur. J. Med. Chem., 2010, 45, 4914 CrossRef CAS; (b) P. C. Lv, H. Q. Li, J. A. Sun, Y. Zhou and H. L. Zhu, Bioorg. Med. Chem., 2010, 18, 4606 CrossRef CAS.
- (a) R. Yatsyshyn, O. Shtefiuk, Y. Sandurska, T. Shtefiuk and L. Kulba, Osteoporos. Int., 2011, 22, 267 Search PubMed; (b) D. Clemett and K. L. Goa, Drugs, 2000, 59, 957 CrossRef CAS.
- W. P. Zhao, Z. G. Zhang, X. D. Li, D. Yu, X. F. Rui, G. H. Li and G. Q. Ding, Braz. J. Med. Biol. Res., 2009, 42, 963 CrossRef CAS.
- T. Eicher and S. Hauptmann, The Chemistry of Heterocycles: Structure, Reactions, Syntheses, and Applications, Wiley-VCH, New York, 2003 Search PubMed.
- (a) A. Ali, J. K. Nayar and W. D. Gu, J. Am. Mosq. Control Assoc., 1998, 14, 216 CAS; (b) M. Ogawa, H. Miyake and E. Maeda, Plant Prod. Sci., 2001, 4, 291 CrossRef CAS; (c) A. K. Culbreath, T. B. Brenneman, R. C. Kemerait and G. G. Hammes, Pest Manage. Sci., 2009, 65, 66 CrossRef CAS.
- (a) P. Simunek, M. Svobodova, V. Bertolasi and V. Machacek, Synthesis, 2008, 1761 CrossRef CAS; (b) K. Alex, A. Tillack, N. Schwarz and M. Beller, Org. Lett., 2008, 10, 2377 CrossRef CAS; (c) J. J. Neumann, M. Suri and F. Glorius, Angew. Chem., Int. Ed., 2010, 49, 7790 CrossRef CAS; (d) S. Safaei, I. Mohammadpoor-Baltork, A. R. Khosropour, M. Moghadam, S. Tangestaninejad and V. Mirkhani, Synlett, 2011, 2214 CAS; (e) H. G. Bonacorso, L. M. F. Porte, G. R. Paim, F. M. Luz, M. A. P. Martins and N. Zanatta, Tetrahedron Lett., 2010, 51, 3759 CrossRef CAS; (f) S. Fustero, M. Sánchez-Roselló, P. Barrio and A. Simón-Fuentes, Chem. Rev., 2011, 111, 6984 CrossRef CAS; (g) L. Buriol, C. P. Frizzo, D. N. Moreira, L. D. T. Prola, M. R. B. Marzari, T. S. München, N. Zantta, H. G. Bonacorso and M. A. P. Martins, Monatsh. Chem., 2011, 142, 515 CrossRef CAS; (h) A. Corradi, C. Leonelli, A. Rizzuti, R. Rosa, P. Veronesi, R. Grandi, S. Baldassari and C. Villa, Molecules, 2007, 12, 1482 CrossRef CAS.
- Z. Wang and H. Qin, Green Chem., 2004, 6, 90 RSC.
- F. Texier-Boullet, B. Klein and J. Hamelin, Synthesis, 1986, 409 CrossRef CAS.
- V. Polsheettiwar and S. R. Varma, Tetrahedron Lett., 2008, 49, 397 CrossRef.
- C. T. Eary and D. Clausen, Tetrahedron Lett., 2006, 47, 6899 CrossRef.
- M. Curini, O. Rosati, V. Campagna, F. Montanari, G. Cravatto and M. Boclini, Synlett, 2005, 2927 CrossRef CAS.
- X. Chen, J. She, Z. Shang, J. Wu, H. Wu and P. Zhang, Synthesis, 2008, 3478 CAS.
- M. Kidwai, A. Jain and R. Poddar, J. Organomet. Chem., 2011, 696, 1939 CrossRef CAS.
- J. Prabhakaran, M. D. Underwood, R. V. Parsey, V. Arango, V. J. Majo, N. R. Simpson, R.V. Heertum, J. J. Mann and J. S. Dileep Kumar, Bioorg. Med. Chem., 2007, 15, 1802 CrossRef CAS.
- S. V. Druzhinin, E. S. Balenkova and V. G. Nenajdenko, Tetrahedron, 2007, 63, 7753 CrossRef CAS.
- H. G. Bonacorso, L. M. F. Porte, G. R. Paim, F. M. Luz, M. A. P. Martins and N. Zanatta, Tetrahedron Lett., 2010, 51, 3759 CrossRef CAS.
- A. C. Cole, J. L. Jensen, I. Ntai, K. L. T. Tran, K. J. Weaver, D. C. Forbes and J. H. Davis, J. Am. Chem. Soc., 2002, 124, 5962 CrossRef CAS.
- (a) N. S. Gupta, G. L. Kad and J. Singh, Catal. Commun., 2007, 8, 1323 CrossRef CAS; (b) F. Dong, L. Jun, Z. Xinli, Y. Zhiwen and L. Zuliang, J. Mol. Catal. A: Chem., 2007, 274, 208 CrossRef CAS; (c) F. Dong, F. Zhenghao and L. Zuliang, Catal. Commun., 2009, 10, 1267 CrossRef CAS; (d) A. H. Kategaonkar, S. S. Sonar, K. F. Shelke, B. B. Shingate and M. S. Shingare, Org. Commun., 2010, 3, 1 CAS; (e) Q. Zhang, H. Su, J. Luo and Y. Wei, Green Chem., 2012, 14, 201 RSC.
- (a) X. Liang and J. Yang, Green Chem., 2010, 12, 201 RSC; (b) J. Yang, H. Zhou, X. Lu and Y. Yuan, Catal. Commun., 2010, 11, 1200 CrossRef CAS; (c) A. S. Amarasekara and O. S. Owereh, Catal. Commun., 2010, 11, 1072 CrossRef CAS; (d) X. Liang and C. Qi, Catal. Commun., 2011, 12, 808 CrossRef CAS and references therein.
- (a) M. Rostami, A. R. Khosropour, V. Mirkhani, M. Moghadam, S. Tangestaninejad and I. Mohammadpoor-Baltork, Appl. Catal., A, 2011, 397, 27 CrossRef CAS; (b) Z. Khorsandi, A. R. Khosropour, V. Mirkhani, I. Mohammadpoor-Baltork, M. Moghadam and S. Tangestaninejad, Tetrahedron Lett., 2011, 52, 1213 CrossRef CAS.
- A. R. Khosropour, I. Mohammadpoor-Baltork and F. Kiani, C. R. Chim., 2011, 14, 441 CrossRef CAS.
- Synthesis of multi-SO 3 H Brønsted acidic ionic liquid A : the catalyst was prepared according to known process.34a Hexamethylentetramine (10 mmol) and 1,3-propanesultone (40 mmol) was stirred in dry THF (40 ml) for 72 h at room temperature to form a white solid zwitterion. After washing the salt with Et2O to remove any unreacted starting materials, the solid was derided in vacuo. Then, sulfuric acid (40 mmol) was added and the mixture was stirred for 10 h at 120 °C during which time the solid zwitterion liquefied.
- General procedure for regioselective synthesis of pyrazoles: a mixture of 1,3-diketone (1 mmol), arylhydrazine/hydrazide (1 mmol) and catalyst (15 mol%) was stirred at room temperature for the appropriate time according to Table 2. After completion of the reaction, in the case of solid products, the mixture was filtered and dried. In the case of liquid products, the mixture was extracted with Et2O and the organic phase was dried over MgSO4 and evaporated. In all cases, the product was pure enough for identification. If necessary, the crude product was purified by recrystallization from ethanol. All products were characterized by IR, mass, 1H NMR and 13C NMR spectral data and elemental analysis.
- (a) G. Coispeau and J. Elguero, Bull. Soc. Chim. Fr., 1970, 2717 Search PubMed; (b) L.-P. Song and S.-Z. Zhu, J. Fluorine Chem., 2001, 111, 201 Search PubMed; (c) F. Gosselin, P. D. O'Shea, R. A. Webster, R. A. Reamer and R. D. Tillyer, Synlett, 2006, 3267 Search PubMed; (d) S. Fustero, R. Román, J. F. Sanz-Cervera, A. Simón-Fuentes, J. Bueno and S. Villanova, J. Org. Chem., 2008, 73, 8545 Search PubMed.
- B. Willy and T. J. J. Müller, Eur. J. Org. Chem., 2008, 4157 Search PubMed.
- J. E. House, Principles of Chemical Kinetics, 2nd Ed., Elsevier, Amsterdam, 2007 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC reference numbers 850135, 850136 and 850137. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2ra20624b |
| This journal is © The Royal Society of Chemistry 2012 |