Functionalisation of polybutylene succinate nanocomposites: from structure to reinforcement of UV-absorbing and mechanical properties†
Christian
Coelho
*ab,
Mohammed
Hennous
b,
Vincent
Verney
cd and
Fabrice
Leroux
*cd
aClermont Université, ENSCCF, Institut de Chimie de Clermont-Ferrand, BP 10448, F-63000 CLERMONT-FERRAND, France. E-mail: christian.coelho@univ-bpclermont.fr
bUniversité des Sciences et de la Technologie d'Oran, Mohamed Boudiaf Faculté des sciences, Département de chimie industrielle, ORAN, Algeria
cClermont Université, Université Blaise Pascal, Institut de Chimie de Clermont-Ferrand, BP 10448, F-63000 CLERMONT-FERRAND, France
dCNRS, UMR 6296, ICCF, BP 80026, F-63171 AUBIERE, France. E-mail: fabrice.leroux@univ-bpclermont.fr
First published on 3rd April 2012
Abstract
Cinnamic derivatives intercalated into ZnAl layered double hydroxides by coprecipitation are subsequently dispersed into a biodegradable polymer, polybutylene succinate (PBS). The structure and composition of the associated organic–inorganic hybrid assemblies are first characterized by X-ray diffraction (XRD) and UV spectroscopy, and then PBS extruded nanocomposite derivatives are evaluated against UV exposure. Using a combination of the PBS nanocomposite structures and the UV and rheological properties relationship, the effect of radiation time on the macromolecular changes undergone by the polymer chains, i.e. chain scissions and cross-linking, as well as on the UV-barrier evolution, is also scrutinized by means of transmission and emission spectroscopies. The combination of organically modified hydrotalcites with PBS could be used as an innovative route for sustainable development of UV protected materials and reduce the environmental impact of UV absorbing chemicals.
1 Introduction
The solar radiation that reaches Earth consists of UV-B (280–315 nm), UV-A (315–400 nm), visible (400–750 nm) and infrared (>750 nm) radiation. To avoid or reduce the photodegradation resulting mostly from UV-B and UV-A radiation, UV absorbers should be integrated into the polymer matrix and give a UV screening effect by blocking the H-abstraction of the polymer chromophore, or by stopping the homolytic breaking of hydroperoxides. Generally, the efficiency can be raised only at high UV absorber amounts, which is economically viable neither for industries nor for sustainable development. Moreover, the presence of additives free to migrate inside the polymer medium may cause two main drawbacks: firstly, a concentration gradient of diffuse additives and their subsequent leaching out of the polymer, and secondly a deleterious effect on the polymer backbone inherent to the additive photoreactivity. In the first case, the expelled molecules may have a negative environmental impact, while the stability of the material is a concern in the second case.An elegant strategy consists of inserting the organic functionalized molecule into an inorganic host filler to avoid diffusion, as well as to limit as much as possible their action against the polymer chains. Interest here is focused on the cinnamic-type organic backbone since trans-cinnamic acid derivatives are known as UV blockers and antioxidants. Tested substituents are hydroxyl and methoxy groups. In para-hydroxycinnamic acid, one hydroxyl group is in the para position, while two hydroxyl groups are in the meta and para positions for ferulic acid, and cafeic acid has one hydroxyl group in the para position and one methoxy group in the meta position. UV-shielding properties were investigated with these four cinnamic derivatives separately, confined in a polymer matrix. The organic blends were compared to the organic–inorganic dispersions. These molecules have been chosen in order to check their UV-absorbing properties, particularly ferulic acid, which was formerly studied and successfully intercalated in hydrotalcite-like anionic clays.1,2
Indeed, organic–inorganic layered double hydroxide (LDH) sandwich structures have been extensively evaluated for their properties. Their photochemical and photophysical characterizations have been largely investigated by Costantino et al.,3–6 but to our knowledge only a few were studied as an organic–inorganic filler acting as a UV-absorber (patent literature).
In comparison, layered double hydroxide nanofillers were synthesized and tested for their UV absorption properties. Most of these studies concerned sulfonic acid derivatives1,7–9 and cinnamic acid derivatives, and the associated nanostructures were evaluated for cosmetic use as UV sunscreens and UV stabilizers. As far as filler dispersion is concerned, it may be preceded by melt-intercalation inside a polymer matrix, as exemplified by polypropylene10 and low density polyethylene,11 leading to partially or totally exfoliated structures. It has been shown that totally exfoliated LDH–polymer nanocomposites have enhanced thermostability and UV-stability.11 Moreover, incorporating nanosized fillers into polymers presents the advantage of increasing film barrier properties, enhancing mechanical properties or even increasing antimicrobial activity.12,13 Such negatively charged organic absorbers may be incorporated between positively charge platelets such as a layered double hydroxide. Layered double hydroxides (LDHs) of the general formula 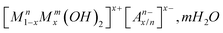 consist of cationic hydrotalcite-like layers and an exchangeable interlayer anion. The resulting 2D-confinement supplied by the inorganic structure prevents the UV absorber from migrating. Therefore, assembled hybrid materials can act as nanocontainers for UV screening molecules. Using a combination of inorganic hosts and organic chromophores offers a multiple functionalisation of materials. Stabilization and mechanical improvement is obtained by LDHs, and organic molecules endow them with optical properties such as color, UV absorption and fluorescence.3–6,14
consist of cationic hydrotalcite-like layers and an exchangeable interlayer anion. The resulting 2D-confinement supplied by the inorganic structure prevents the UV absorber from migrating. Therefore, assembled hybrid materials can act as nanocontainers for UV screening molecules. Using a combination of inorganic hosts and organic chromophores offers a multiple functionalisation of materials. Stabilization and mechanical improvement is obtained by LDHs, and organic molecules endow them with optical properties such as color, UV absorption and fluorescence.3–6,14
Polybutylene succinate (PBS) polymer, an aliphatic polyester, is selected here since it is a good candidate for biodegradable and compatible materials in the agricultural and sanitary fields, as well as in packaging applications. For such purposes, the polymer is potentially exposed to sunlight, and therefore photoaging becomes of greatest concern. Concerning PBS, its photooxidation mechanism pathways were recently proposed and found to be based on oxidation of hydroxyl end groups, H-abstraction decomposition and Norrish I photocleavage, leading to a scission of its chain.15
Therefore, the nanocomposite approach in association with the use of cinnamic acid derivatives, due to their antioxidant properties derived from its free radical scavenging ability, is appealing.16
They have been intercalated inside layered double hydroxides and tested for their UV shielding properties when melted inside PBS.
Molecular changes have been assayed by means of UV-visible absorption spectroscopy and fluorescence spectroscopy. Melt rheological measurements were done to assess the mechanical properties associated with microstructural changes in the molten state.
The originality here is to understand the relationships using functional nanocontainers incorporated in a polymer in conjunction with characterization of the hybrid filler and polymer nanocomposite structure, and to evaluate the associated UV-absorbing properties. It is also crucial to relate the subsequent mechanical changes undergone by the whole thing as a function of the photoaging time.
2 Experimental
2.1 Materials
Starting reagent grade materials ZnCl2, AlCl3·6H20, NaOH and the cinnamic-based organic molecule, all commercially available, were used as received: trans-cinnamic acid C6H5–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–COOH (97%), p-hydroxycinnamic acid OH–C6H4–CHCH–COOH (98%), ferulic acid (OH)(CH3) –C6H3–CHCH–COOH (99%) and cafeic acid (OH)2–C6H3–CHCH–COOH (98%) were purchased from Aldrich. pKa dissociation constants of the carboxylic function at 25 °C are, respectively, 4.44, 4.64, 4.58 and 4.62. PBS, EnPol G-4560 was provided by Ire Chemicals Co., Korea.
CH–COOH (97%), p-hydroxycinnamic acid OH–C6H4–CHCH–COOH (98%), ferulic acid (OH)(CH3) –C6H3–CHCH–COOH (99%) and cafeic acid (OH)2–C6H3–CHCH–COOH (98%) were purchased from Aldrich. pKa dissociation constants of the carboxylic function at 25 °C are, respectively, 4.44, 4.64, 4.58 and 4.62. PBS, EnPol G-4560 was provided by Ire Chemicals Co., Korea.
2.2 Synthesis of LDH nanofillers
For the preparation of the layered LDH hybrids, coprecipitation was achieved by keeping pH constant at 10.0 (pH > pKa), to simultaneously precipitate both cations under their hydroxide form, yielding LDH platelets, and to immobilize the cinnamic anion derivatives.2.3 Preparation of polybutylene succinate blends
PBS with 5% (w/w) LDH nanofiller was mixed and melt-extruded using a Hakke MINILAB microcompounder (Thermo Electron Corporation, Germany) operated at 120 °C with a rotating speed of 100 rpm for 5 min. This operation led to PBS nanocomposites.PBS organic blends were obtained by the same protocol just by changing the weight ratio of the organic molecules to 2% (w/w). This ratio takes into account the relative content of molecules interleaved in LDH filler and dispersed in PBS nanocomposites.
PBS blend films were prepared by compression molding between two Teflon sheets during one minute under 200 bars at 120 °C. The thickness of the films was about 50 to 60 μm.
2.4 Characterization methods
X-ray powder diffraction (XRD) patterns obtained on a Philips X-Pert Pro diffractometer using Cu-Kα radiation were collected in a step scan mode between 2.0 and 70° (2θ) and with a step size of 0.03 (2θ) and a counting time of 10 s/step. An identical procedure was applied to PBS nanocomposite films. Additionally, a lower angle domain, down to 2θ = 0.7°, was recorded to scrutinize the larger intercalated structure.The UV-visible spectra were recorded in reflection mode for powders, and in transmission mode for films, with a Shimadzu UV-2101 PC spectrophotometer equipped with an integrating sphere.
Molecular emission on films was performed on a Perkin-Elmer LS-55 luminescence spectrophotometer equipped with a front surface accessory and a pulsed xenon excitation source. Excitation was set at 280 nm and the emission signal was collected by the monochromator from 250 to 500 nm at a scan speed of 120 nm min−1.
The melt rheological properties of the polymer blends were measured at 120 °C using a rotational spectrometer (ARES, TA, USA) equipped with a parallel plate geometry (plate diameter 8 mm, gap 1 mm). The imposed oscillatory shear stress amplitude was tested to validate the measurements inside the linear viscoelastic domain. This strain was fixed and used for measuring the dynamic stress against the oscillatory shearing frequency (from 0.1 to 100 rad s−1). G′ (storage modulus), G′′ (loss modulus) and tan δ (ratio of G′′ to G′) were monitored automatically against frequency. The curve representing storage viscosity (η′′) against loss viscosity (η′) is described by the Cole–Cole plot. It was used to determine the Newtonian zero-shear viscosity η0 of PBS blends, which corresponds to the extrapolation from the convex downward semi-circle for η → 0. This value is proportional to the molecular weight Mw according to the α power-law relationship η0 = K · 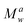 .
.
2.5 Photoageing experiments
PBS blend films were irradiated in a photoaging device equipped with a mercury-vapour lamp (Novalamp RVC 400 W) which emits spectral rays above 295 nm. This device was designed to accelerate the photodegradation of the polymer in artificial conditions that were still representative of natural ageing. PBS nanocomposite films were introduced inside a closed pyrex reactor with a volume of 335 mL, equipped with a pressure and temperature MKS Instrument®, a CO2 Vaisala® probe and a O2 Mettler-Toledo® probe. The temperature inside the reactor was set constant at 40 °C.The amount of expelled gas calculated by the Henry's law was normalized against the weight of the polymer film in mol kg−1.
3 Results and Discussion
3.1 Organic charges and nanofiller characterization
![Powder XRD diffraction patterns of cinnamic acids derivatives with LDH compared to [(Zn,Al)CO3]–LDH](/image/article/2012/RA/c2ra20579c/c2ra20579c-f1.gif) | ||
| Fig. 1 Powder XRD diffraction patterns of cinnamic acids derivatives with LDH compared to [(Zn,Al)CO3]–LDH | ||
The [(Zn,Al)CO3]–LDH shows a basal 001 reflection peak at d = 6.2 Å (2θ = 11.6°) and corresponds to the interlayer distance, i.e. the gallery size between two consecutive inorganic brucite-type slabs.
This value is largely increased when the organic unit is intercalated: 18.2 Å (2θ = 4.84°), 17.8 Å (2θ = 4.96°), 16.9 Å (2θ = 5.24°) and 12.2 Å (2θ = 7.24°), for LDH t-cinnamic, LDH p-hydroxycinnamic, LDH ferulic and LDH cafeic, respectively. These values are close to those obtained by co-precipitation in the zinc–aluminium layered double hydroxide,2 except for the LDH cafeic interlayer distance which is relatively small. A recent study reports a value of 22.5 Å for the (Mg,Al)–LDH intercalated with cafeic acid but the synthesis was quite different and based on an exchange process at 100 °C.17
Therefore, the presence of cinnamic acid derivative chromophores inside LDH is confirmed by XRD since the interlayer distance is higher than 6.2 Å and there are no more diffraction peaks due to carbonate LDH.
Counter-intuitively, the basal spacings of the resulting hybrid LDH materials follow the order t-cinnamic acid > p-hydroxycinnamic acid > ferulic acid > cafeic acid, this order somehow being in the opposite trend to the molecular size of the guests with R and R′ substituent groups, thus suggesting a particular interleaved accommodation.
Indeed, considering the thickness of a double hydroxide layer of 4.7 Å,18 the interlayer region of the hybrid LDH materials has a gallery height of 13.5, 13.1, 12.2 and 7.5 Å for t-cinnamic acid, p-hydroxycinnamic acid, ferulic acid and cafeic acid, respectively.
With an estimated length of these cinnamic derivatives of approximately 9.5 to 10 Å, cafeic acid cannot fit between the layers in a perpendicular orientation. The only possibility is an arrangement of the guest molecules in a tilted position with the molecules inclined at 45° with regard to the hydroxide sheet. This inorganic–organic arrangement is typical of J-aggregates,19 and should cause a bathochromic shift when compared to the corresponding absorption band of the organic chromophore alone, as previously observed with inserted perylene compounds.
Concerning t-cinnamic acid, the hypothesis of H-aggregates is not excluded. In this case the t-cinnamic acid chromophore should be held by two hydrogen bonds with a length of 2 Å in the host–guest complex.
3.1.2 UV shielding properties of nanofillers
UV-visible reflectance spectra are measured for the organic molecules and associated hybrid LDH materials (Fig. 2). The reflectance spectra recorded in the solid state should be considered to be derived from the absorption spectra of the aqueous solution.19 This was experimentally validated.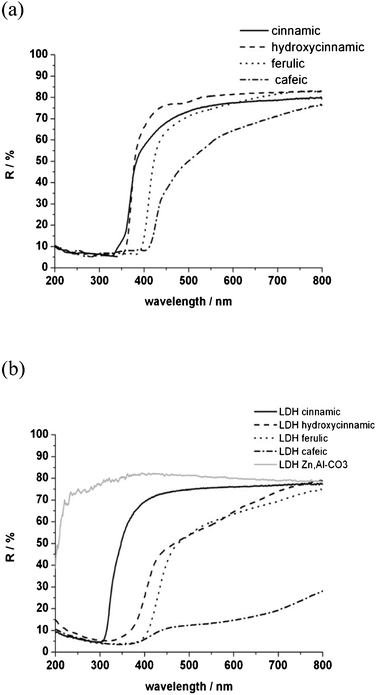 | ||
| Fig. 2 UV-visible reflectance curves of organic charges (a) and nanofillers (b). | ||
The four derivatives of cinnamic acid are rather transparent in the visible part and absorbing in the UV-A (200 to 280 nm) and UV-B (280 to 320 nm) wavelength domains. Cinnamic and hydroxycinnamic acids are partially UV-C (320 to 400 nm) absorbers in the shorter wavelengths, while ferulic and cafeic acids bearing both R and R′ substituent groups absorb in the entire UV region.
When incorporated into the LDH gallery, the optical reflectance in the visible part is mostly kept, except for the interleaved cafeic acid. Besides, the UV absorbing properties are unchanged, except for trans-cinnamic acid which is hypsochromically shifted from 350 to 300 nm. Such a shift is typical of H-type aggregates of trans-cinnamic acid chromophores.19
3.2 Structure and characterization of polybutylene succinate blends and nanocomposites
PBS organic blends (on the left) and nanocomposites (on the right) presented in the graphical abstract, exhibit a large variety of colour. For p-hydroxycinnamic acid, ferulic acid and cafeic acid, a browning of the PBS nanocomposites is noted. However, the visible properties are unchanged for the PBS nanocomposite adopting a t-cinnamic hybrid filler, and it remained transparent.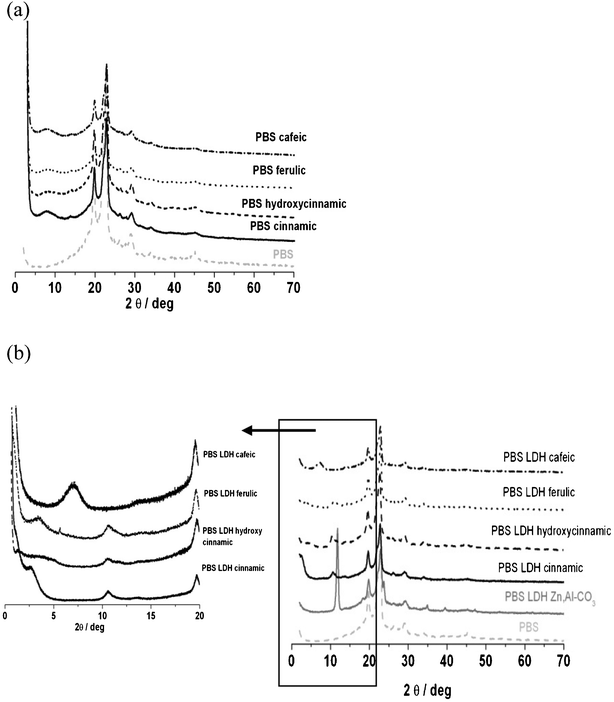
At least six observed diffraction peaks, at 2θ = 19.7° (d = 4.51 Å), 22.8° (d = 3.9 Å), 26.2° (d = 3.4 Å), 28.9° (d = 3.1 Å), 34.0° (d = 2.6 Å) and 45.2 (d = 2.0 Å) are characteristic of PBS. Two of them, 19.7° and 22.8°, are typical for a PBS crystal in the monoclinic unit cell and ascribed to the (020) and (110) diffraction planes, respectively.20 The associated full width at half maximum, as well as the diffraction peak positions, after incorporation of the hybrid LDH filler, remain unchanged (Fig. 3a). This means that this dispersion has an impact neither on the crystallinity of PBS nor on its coherence length, i.e. size of the crystallized domain.
In some cases, the interlamellar distance of the hybrid LDH filler is propped apart when dispersed into PBS as for t-cinnamic and ferulic acid, with an increase from 1.82 nm to 2.96 nm (Δd = 1.64 nm) and from 1.69 nm to 2.60 nm (Δd = 0.91 nm), respectively, for LDH t-cinnamic and LDH ferulic. This increase should be interpreted as being due to diffusion of the polymer chain between the layers, thus resulting in an intercalated polymer nanocomposite structure. The greater basal expansion is obtained with LDH t-cinnamic.
However, the PBS nanocomposite with LDH cafeic and LDH p-hydroxycinnamic leads to the same diffraction peak in comparison to the pristine LDH assembly, meaning that the PBS polymer chain is not diffusing inside the filler gallery, resulting in a “non miscible” structure.
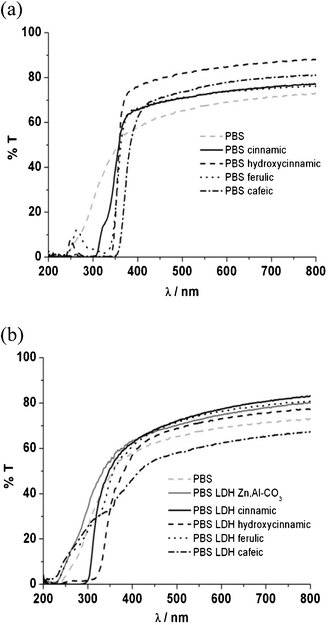 | ||
| Fig. 4 UV-visible spectra of PBS organic blend films (a) and PBS nanocomposite films (b). | ||
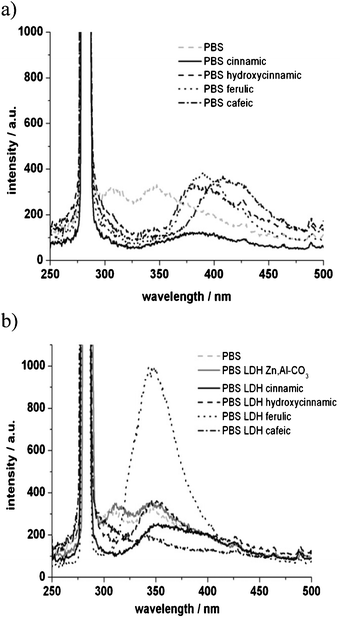 | ||
| Fig. 5 Emission spectra of PBS organic blends (a) and nanocomposites (b), excitation wavelength was set at 280 nm. | ||
From the comparison between spectra in Fig. 5a and b, it can be noted that the emission wavelengths are different for the molecules because of different intermolecular accommodations when blended with PBS, while this effect is levelled out when the organic molecules are well ensconced within the host filler.
PBS emission presents two maximum peaks centered at 310 and 350 nm. PBS blends with the cinnamic derivatives present an emission wavelength which varies from 380 nm for p-hydroxycinnamic to 410 nm for cafeic acid. These emission wavelengths for the organic molecule are in agreement with the literature.21
When the derivatives are intercalated into LDH, the emission of all PBS nanocomposites is shifted down to 350 nm. This unique maximum centered at 350 nm still originates from the fluorescence of cinnamic derivatives which exist in organized systems inside the PBS nanocomposites. These fluorophores present the same steric restrictions due to LDH, or the same chemical interactions with PBS.
The emission wavelength shift may be tentatively explained by an energy transfer between LDH and the organic chromophore, as previously described22 by a bathochromic shift of rhodamine derivatives when immobilized in AlPO4-5. To unravel the possible role of LDH platelets alone, the fluorescence spectrum of the LDH carbonate phase is compared with PBS, and it is found that both fluorescence spectra are similar. Therefore, we propose the hypothesis of an energy transfer between the LDH nanofiller and PBS to explain the observed hypsochromic shift.
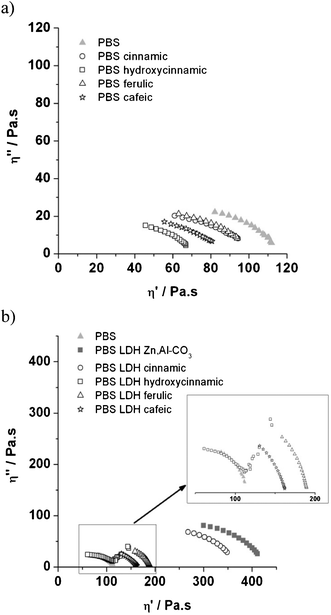
On the other hand, when LDH nanofillers are incorporated into a PBS matrix, the values of viscosity are increased and arranged in the order LDH p-hydroxycinnamic acid < LDH cafeic acid < LDH ferulic acid < LDH t-cinnamic acid. Once again, but in opposition to organic PBS blends, this may be explained by an increase in molecular weight with the presence of a LDH nanofiller,23,25–27 even if a total conversion in a gel-like structure is not observed. The lack of strong reinforcement may be due to the rather low loading of LDH–cinnamic derivatives of 5% (w/w) in the PBS nanocomposite, and the weak attritive interactions developed between the LDH nano-assembly and PBS chains.
However, PBS with LDH p-hydroxycinnamic acid presents two repartitions of molecular motions. One part of the macromolecules behaves like a Newtonian liquid, quite similar to PBS itself, whereas the other part presents a gel-like structure, as illustrated by the straight line at low frequency (Fig. 6).
It is interesting to notice that a slight reinforcement is observed, even in the case of a non miscible structure as previously reported.23
One can compare the effect of the functional groups susceptible to interaction with the polymer chain on the rheological behaviour of the blends and nanocomposites. Regardless of the fact that the polymer and the filler are non miscible and that they do mix and yield an intercalated structure, Cole–Cole circles still follow the same order: p-hydroxycinnamic < cafeic < ferulic < t-cinnamic. This means that the functional groups in the UV absorbers are of crucial importance to the mobility of the polymer chains. However, the apparent molecular weight remains quite similar, while a pronounced increase in η0 is observed for both expanded structures, i.e. adopting t-cinnamic and ferulic LDH filler. For non miscible structures, and comparing between p-hydroxycinnamic and cafeic molecules, it is interesting to note that an additional hydroxyl reinforces the nanostructure more, probably due to the multiplicity of the reaction with PBS chains.
3.3 Polybutylene succinate blends and nanocomposite photodegradation
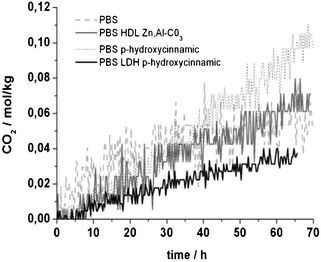 | ||
| Fig. 7 CO2 emission rate evolution during PBS blend photodegradation concerning the hydroxycinnamic acid UV absorber. | ||
A trend concerning CO2 emission upon irradiation may be drawn, with the most CO2-emitting composite being PBS hydroxycinnamic, followed by PBS and PBS LDH carbonate. Indeed, PBS LDH p-hydroxycinnamic is the lowest emitting nanocomposite.
For the studied cinnamic derivatives, PBS nanocomposites emit less CO2 compared to PBS organic blends (ESI,† Fig. S.I.1.) and as much as for p-hydroxycinnamic and LDH cafeic (two times less than for PBS composites).
Admitting that carbon dioxide is the last photoproduct of polymer photodegradation,24 the resulting UV stability of these materials may be arranged as a function of the incorporation of these hybrid fillers. However, some kinetics studies should be undertaken to confirm this quantitatively but are beyond the purpose of this work.
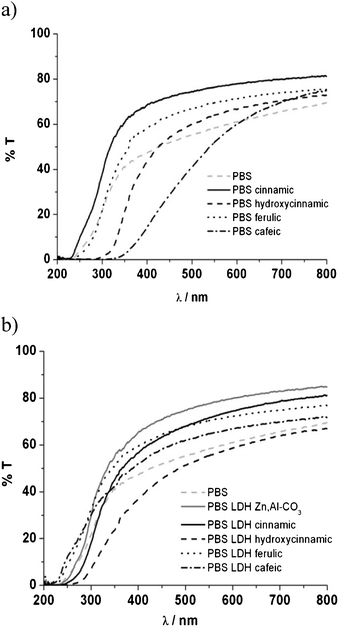 | ||
| Fig. 8 UV-visible spectra of PBS organic blends (a) and PBS nanocomposites (b) after 70 h of irradiation. | ||
PBS itself presents very few changes in terms of transmittance of UV and visible light. Without the presence of any additive, PBS is stable upon irradiation, while PBS blends loaded with t-cinnamic acid, ferulic acid and p-hydroxycinnamic acid exhibit a shift 50 nm down after irradiation, meaning that some molecular changes are occurring, probably due to interactions between PBS and additives and/or additive stability. A more pronounced shift of more than 100 nm towards longer wavelengths was observed for PBS filled with cafeic acid. The photochemical reactions occurring from the cinnamic derivatives affect PBS differently.
Concerning PBS nanocomposites, UV radiation also damages the molecular arrangement, and a blue shift of 20 nm in the UV-B region is noticed. Even if not strongly pronounced, a subtle change in the UV-visible spectra occurs for the polymer nanocomposites, thus leading to a shielding property that is truncated when compared to the initial time. The domains of the UV region are not so well defined compared to the same samples before irradiation. In other words, the LDH container acts locally as an efficient local UV absorber, but in a full extent an optimized concentration should be adjusted in association with an intercalated polymer nanocomposite structure.
Interestingly, this affirmation could also be observed with the fluorescence spectra (ESI,† Fig. S.I.2). Three of the four cinnamic derivatives present the same emission properties in terms of localized peaks and intensity. However, the PBS nanocomposite with LDH ferulic acid, with a high intense peak centered at 350 nm before irradiation, decreases greatly after irradiation. This means that the expanded nanostructure was more fragile to the aggressive UV conditions. In our case, the non miscible structure obtained with the adapted UV nanofiller seems to better protect the PBS nanocomposite from UV irradiation. This result is in agreement with the smaller amount of CO2 observed for these nanomaterials.
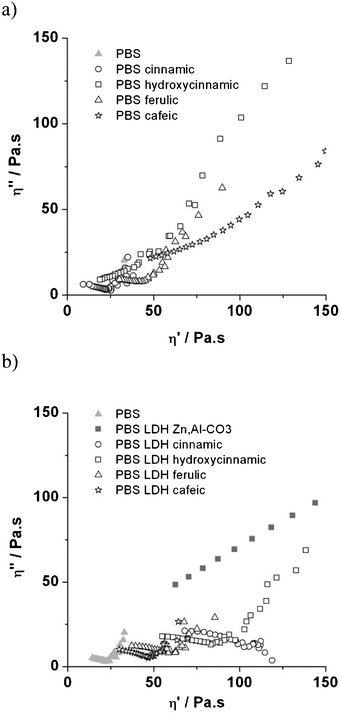 | ||
| Fig. 9 Melt Cole–Cole plots of 70 h irradiated PBS organic blends (a) and PBS nanocomposites (b). | ||
From the partial depressed circle, the Newtonian zero-shear viscosity η0 has largely decreased, both for PBS unfilled and PBS organic blended with t-cinnamic acid and ferulic acid, unless the PBS organic blend with p-hydroxycinnamic acid and cafeic acid presented a completely reticulated structure after irradiation.
Values of η0 for PBS nanocomposites are much lower after 70 h of irradiation and are arranged in the following order: LDH cafeic > LDH ferulic > LDH p-hydroxycinnamic > LDH t-cinnamic. This is typical of chain scission occurring in the polymer matrix. The circles describing the viscous part of the PBS nanocomposite with LDH t-cinnamic and LDH ferulic appeared less depressed, compared to the PBS organic blend. This means that the molecular weight distribution in the PBS nanocomposite is more homogenous with the use of expanded nanomaterials. This represents a lesser degradation extent in the bulk material.
There are two antagonistic effects during the photodegradation of PBS nanocomposites: chain scission of PBS and cross-linking, depending on whether the LDH nanofiller presents a “non miscible” or an expanded structure towards the polymer.
Conclusions
PBS nanocomposites were here successfully characterized with cinnamic derivatives intercalated in layered double hydroxides. The associated dispersion corresponds to either a non miscible or an intercalated polymer structure. Adding organic–inorganic fillers to the polymer enhances mechanical properties in the molten state and produces less carbon dioxide during photodegradation compared to the blends obtained from the direct incorporation of the UV-absorber. Contrary to the general idea that a benefit could only be obtained from the exfoliated polymer nanocomposite structure, the non miscible structure is here found to be of interest for UV-stability as well as for the macromolecular cross-linking upon irradiation, the latter being more pronounced, with hydroxyl substitution on the aromatic cinnamic-based molecule. Better UV stability was not fully demonstrated because the kinetics were not realized. A photophysical mechanism based on an energy transfer from the LDH nanofiller and PBS is surmised to improve UV stability. This approach helps to understand the crucial parameters for UV properties, as well as their evolution in use, and may be adapted to other functionalities such as dyeing and luminescent properties, gas barrier properties, thermal stability and flame retardancy.Acknowledgements
This work was supported by a grant from a strategic bilateral cooperation (Algerian France PROFAS) as well as by financial support of Ecole Nationale Supérieure de Chimie de Clermont-Ferrand.References
- Q. He, S. Yin and T. Sato, J. Phys. Chem. Solids, 2004, 65, 395–402 CrossRef CAS.
- C. Rossi, A. Schoubben, M. Ricci, L. Perioli, V. Ambrogi, L. Latterini, G.G. Aloisi and A. Rossi, Int. J. Pharm., 2005, 295, 47–55 CrossRef CAS.
- G. Aloisi, U. Costantino, F. Elisei, L. Latterini, C. Natali and M. Nocchetti, J. Mater. Chem., 2002, 12, 3316–3323 RSC.
- L. Latterini, F. Elisei, G.G. Aloisi, U. Costantino and M. Nocchetti, Phys. Chem. Chem. Phys., 2002, 4, 2792–2792 RSC.
- U. Costantino, N. Coletti, M. Nocchetti, G. Gian, F. Elisei and L. Latterini, Langmuir, 2000, 16, 10351–10358 CrossRef CAS.
- U. Costantino, N. Coletti, M. Nocchetti, G.G. Aloisi and F. Elisei, Langmuir, 1999, 15, 4454–4460 CrossRef CAS.
- L. Perioli, M. Nocchetti, V. Ambrogi, L. Latterini, C. Rossi and U. Costantino, Microporous Mesoporous Mater., 2007, 107, 180–189 CrossRef.
- Y. Feng, D. Li, Y. Wang, D.G. Evans and X. Duan, Polym. Degrad. Stab., 2006, 91, 789–794 CrossRef CAS.
- H. Chai, Y. Lin, D.G. Evans and D. Li, Ind. Eng. Chem. Res., 2008, 47, 2855–2860 CrossRef CAS.
- D. Li, Z. Tuo, D.G. Evans and X. Duan, J. Solid State Chem., 2006, 179, 3114–3120 CrossRef CAS.
- P. Ding and B. Qu, Polymer Engineering and Science, 2006, 46, 1153–1159 CAS.
- U. Costantino, M. Nocchetti, M. Sisani and R. Vivani, Z. Kristallogr., 2009, 224, 273–281 CrossRef CAS.
- U. Costantino, V. Bugatti, G. Gorrasi, F. Montanari, M. Nocchetti, L. Tammaro and V. Vittoria, ACS Appl. Mater. Interfaces, 2009, 1, 668–677 CAS.
- G. Schulz-Ekloff, D. Wöhrle, B. van Duffel and R. A. Schoonhevdt, Microporous Mesoporous Mater., 2002, 51, 91–138 CrossRef CAS.
- S. Carroccio, P. Rizzarelli and C. Puglisi, Macromolecules, 2004, 37, 6576–6586 CrossRef CAS.
- E. Graf, Free Radical Biol. Med., 1992, 13, 435–448 CrossRef CAS.
- Y. Wei, Y. Gao, K. Zhang and Y. Ito, J. Liq. Chromatogr. Relat. Technol., 2010, 33, 837–845 CrossRef CAS.
- J. Bauer, P. Behrens, M. Speckbacher and H. Langhals, Adv. Funct. Mater., 2003, 13, 241–248 CrossRef CAS.
- S. Kim, T.K. An, J. Chen, I. Kang, S.H. Kang, D.D. Chung, C.E. Park, Y.H. Kim and S.K. Kwon, Adv. Funct. Mater., 2011, 21, 1616–1623 CrossRef CAS.
- E.S. Yoo and S.S. Im, J. Polym. Sci., Part B: Polym. Phys., 1999, 37, 1357–1366 CrossRef CAS.
- J. Min, X. Meng-Xia, Z. Dong, L. Yuan, L. Xiao-Yu and C. Xing, J. Mol. Struct., 2004, 692, 71–80 CrossRef CAS.
- M. Bockstette, D. Wohrle, I. Braune and G. Schulz-Ekloff, Microporous Mesoporous Mater., 1998, 23, 83–96 CrossRef CAS.
- Q. Zhou, V. Verney, S. Commereuc, I.J. Chin and F. Leroux, J. Colloid Interface Sci., 2010, 349, 127–133 CrossRef CAS.
- S. S. Fernando, P. A. Christensen, T. A. Egerton and J. R. White, Polym. Degrad. Stab., 2007, 92, 2163–2172 CrossRef CAS.
- H. H. Winter, Gel point in encyclopedia of polymer science and engineering, John Wiley & Sons, New York, 1989 Search PubMed.
- J. R. Costa, M. Abdel-Goad, U. Wagenknecht and G. Heinrich, Polymer, 2005, 46, 4447–4453 CrossRef.
- R. Krishnamoorti and E. P. Giannelis, Macromolecules, 1997, 30, 4097–4102 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/c2ra20579c/ |
| This journal is © The Royal Society of Chemistry 2012 |