DOI:
10.1039/C2RA20518A
(Paper)
RSC Adv., 2012,
2, 4891-4903
Enhanced hydrogen storage performance for MgH2–NaAlH4 system—the effects of stoichiometry and Nb2O5 nanoparticles on cycling behaviour†
Received
21st March 2012
, Accepted 21st March 2012
First published on 22nd March 2012
Abstract
Nowadays, the technological utilization of reactive hydride composites (RHC) as promising hydrogen storage materials is hampered by their reaction kinetics. In the present work, effects of reactant stoichiometry on ensuing hydrogen sorption properties and pathway of the MgH2–NaAlH4 (mole ratios 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, 1:1 and 2:1) system, both undoped and doped with Nb2O5 nanoparticles, were investigated. It was found that the as-prepared reactant stoichiometry of MgH2/NaAlH4 system had a profound impact on its dehydrogenation kinetics and reaction mechanism. Variable temperature dehydrogenation data revealed that undoped binary composites possessed enhanced hydrogen desorption properties compared to that of pristine NaAlH4 and MgH2. The use of Nb2O5 displayed superior catalytic effects in terms of enhancing dehydriding/rehydriding kinetics and reducing the dehydrogenation temperature of MgH2–NaAlH4 system. Isothermal volumetric measurements at 300 °C revealed that enhancements arising upon adding Nb2O5 were almost double that of undoped MgH2–NaAlH4 composites. The apparent activation energies for NaAlH4, Na3AlH6, MgH2, and NaH relevant decompositions in doped composite were found to be much lower than that for the undoped one. Moreover, Nb2O5 doping also markedly enhanced the reversible capacity of MgH2–NaAlH4 composites under moderate conditions, persisting well during three de/rehydrogenation cycles. XRD, XPS, and FESEM-EDS analyses demonstrated that reduction of Nb2O5 during first desorption was coupled to the migration of reduced niobium oxide species from the bulk to the surface of the material. It was suggested that these finely dispersed oxygen-deficient niobium species might contribute to kinetic improvement by serving as the active sites to facilitate hydrogen diffusion through the diffusion barriers both during dehydrogenation and rehydrogenation.
:2, 1:1 and 2:1) system, both undoped and doped with Nb2O5 nanoparticles, were investigated. It was found that the as-prepared reactant stoichiometry of MgH2/NaAlH4 system had a profound impact on its dehydrogenation kinetics and reaction mechanism. Variable temperature dehydrogenation data revealed that undoped binary composites possessed enhanced hydrogen desorption properties compared to that of pristine NaAlH4 and MgH2. The use of Nb2O5 displayed superior catalytic effects in terms of enhancing dehydriding/rehydriding kinetics and reducing the dehydrogenation temperature of MgH2–NaAlH4 system. Isothermal volumetric measurements at 300 °C revealed that enhancements arising upon adding Nb2O5 were almost double that of undoped MgH2–NaAlH4 composites. The apparent activation energies for NaAlH4, Na3AlH6, MgH2, and NaH relevant decompositions in doped composite were found to be much lower than that for the undoped one. Moreover, Nb2O5 doping also markedly enhanced the reversible capacity of MgH2–NaAlH4 composites under moderate conditions, persisting well during three de/rehydrogenation cycles. XRD, XPS, and FESEM-EDS analyses demonstrated that reduction of Nb2O5 during first desorption was coupled to the migration of reduced niobium oxide species from the bulk to the surface of the material. It was suggested that these finely dispersed oxygen-deficient niobium species might contribute to kinetic improvement by serving as the active sites to facilitate hydrogen diffusion through the diffusion barriers both during dehydrogenation and rehydrogenation.
Introduction
The development of new practical hydrogen storage materials with high volumetric and gravimetric hydrogen densities is a key technical challenge to implement the fuel cell technology for transportation applications. According to the U.S. DOE's 2015 targets, a viable hydrogen storage system for fuel cell applications has necessitated a system gravimetric capacity of more than 5.5 wt% with fast desorption kinetics (1.5 wt% min−1). Solid-state hydrogen storage is attractive because it offers a volumetric hydrogen density greater than that of either compressed gas or liquid hydrogen storage, without high pressure containment or cryogenic tanks.1–5 During the past decade, MgH26–8 and light-element complex hydrides such as alanates,9–14 amides,15,16 borohydrides,17–19 and ammonia borane20,21 are extensively investigated due to their higher gravimetric and volumetric hydrogen densities. Among the solid-state hydrogen storage materials, magnesium hydride is a good candidate possessing large gravimetric density (7.6 wt% H2), abundant resources, low cost, and superior reversibility compared to that of conventional transition metal-based hydrides. However, its use is unfortunately hampered by its high thermodynamic stability and its slow kinetics. During the recent years, these disadvantages have been overcome by preparing nanocrystalline MgH2 produced by high energy milling,22,23 or doping with various catalysts such as metals,24–26 metal oxides,27–29 and metal halides,30,31etc. Nonetheless, all these efforts have only ameliorated its kinetic performances. The thermodynamic characteristics of the interaction between Mg and hydrogen are hardly modified. During the past decade, another pioneer work in the hydrogen storage field is the use of a mixture of single and complex hydrides with amides based systems.32–34 Later this approach has been extended to borohydrides systems,35–37 which are known as Reactive Hydride Composites (RHCs). This approach is aimed at modifying the thermodynamics and kinetics of the hydrogen sorption reaction based on mutual interactions between different hydrides by reducing the enthalpy of dehydrogenation and rehydrogenation reactions. Recent studies have demonstrated that it is possible to reduce the activation energy and decomposition enthalpy of magnesium hydride by mixing it with a second hydride specie, such as LiAlH4, indicating an effective way to improve the hydrogen storage properties of LiAlH4 and MgH2.38–40 Analogous to LiAlH4, NaAlH4 has also been used to modify the de/rehydrogenation thermodynamics of MgH2, albeit up to now there is limited investigation on sorption reactions of this RHC system by experimental means. Recently, Ismail et al.41 have investigated the kinetic sorption properties and reaction pathway of a binary MgH2–NaAlH4 mixture with 4:1 molar stoichiometry prepared by ball milling. Unlike the results obtained for the mixture LiAlH4/MgH2, where the destabilization could be caused by the formation of either Al12Mg17 or LixMgy alloys, only the intermetallic Al12Mg17 is ascribed as a destabilizing agent in the system NaAlH4/MgH2. The present work intends to contribute to the clarification of the sorption properties, more precisely, indicating the most probable dehydrogenation pathway for each of the various stoichiometries of this system. It is important to properly understand the effect of stoichiometry on such multicomponent system as well as the effect of the dehydrogenation conditions on the reaction paths and sorption properties of the system. Moreover, in our recent studies on LiAlH4,10 we have found that Nb2O5 nanoparticles are superior to its analogue Cr2O3 in enhancing the kinetic and thermodynamic performance of LiAlH4. Previous studies29,42 have also indicated that the additive Nb2O5 is known to substantially improve the decomposition thermodynamics and kinetics of MgH2. Motivated by these experimental findings, we have further extended the usage of Nb2O5 nanoparticles in the MgH2–NaAlH4 system. The addition of Nb2O5 is found to result in significantly improved de/rehydrogenation kinetics in the MgH2–NaAlH4 system.
To clarify the impact of reactant stoichiometry on the properties of the undoped and doped MgH2–NaAlH4 composites, herein we report the synthesis, characterization, hydrogen storage characteristics, and hydrogen desorption pathway for composites with stoichiometric ratios of 1:2, 1:1, and 2:1. Thermogravimetry (TG), differential scanning calorimetry (DSC), and isothermal sorption measurements have been conducted to investigate its thermodynamic and kinetic behavior. On the basis of XRD, XPS, and FESEM-EDS analyses, we suggest a set of consecutive reactions that is consistent with the observed phase evolution and hydrogen desorption behavior. On the basis of these data, we discuss factors that may explain differences between these three sets of related compositions. Furthermore, we have performed a full analysis of the surface and bulk regions in order to investigate the evolution, oxidation state, and the local structure of the Nb species during the different steps of milling, dehydrogenation, and rehydrogenation.
Experimental section
NaAlH4 (hydrogen storage grade, ≥93% purity) and MgH2 (hydrogen storage grade) were purchased from Sigma–Aldrich Co. The high purity nano-oxide Nb2O5 was provided by SINONANO Co., Ltd (China). All the materials were used as received without any further purification. All material handling (including weighing and loading) was performed in a high purity argon filled glove box, with low oxygen and water vapour content. A hydride mixture was prepared by mixing the as-received MgH2 and NaAlH4 together in the mole ratios of 1:2, 1:1, and 2:1. Subsequently, the mixtures were ball milled for 30 min by a high energy Spex mill. All the samples were loaded into the hardened steel vial under an argon atmosphere in a glove box. Steel balls (1 g and 3 g) were added with a ball to powder weight ratio of 15:1. Air-cooling of the vial was employed to prevent its heating during the ball-milling process. 2 mol% of Nb2O5 nanopowders was also ball milled with 1MgH2–2NaAlH4, 1MgH2–1NaAlH4, and 2MgH2–1NaAlH4 under the same conditions to investigate their catalytic effects. MgH2 and NaAlH4 doped with 2 mol% of Nb2O5were also prepared under the same conditions for comparison purposes.
Non-isothermal dehydrogenation performances were investigated by thermogravimetry (TG) and differential scanning calorimetry (DSC). The DSC and TG analyses were conducted using NETZSCH STA 449C. All measurements were carried out under a flow (50 ml min−1) of high purity argon (99.999%). Sample mass was typically 5 mg. Heating runs were performed at different rates (4, 7 and 10 °C min−1) from 35 to 500 °C.
The isothermal de/re-hydrogenation kinetics were measured using a pressure composition–temperature (PCT) apparatus. The details of the apparatus are given in our previous reports.9,10,43 The apparatus can be operated up to a maximum pressure of 10 MPa and 600 °C. About 0.5 g of sample was loaded into the sample vessel. The isothermal dehydrogenation measurements for the undoped and doped samples were performed at 280 °C and 300 °C under a controlled vacuum atmosphere. Following the first complete dehydrogenation, the samples were subjected to rehydrogenation studies at 300 °C under 9.5 MPa for 1 h. The pressure drop with time in the closed system testified the rehydrogenation of the samples. Subsequently, the rehydrogenated samples were dehydrogenated at similar temperature.
The phase structure of the sample following the ball milling, dehydrogenation, and rehydrogenation was determined by a MXP21VAHF X-ray diffractometer (XRD with Cu-Kα radiation) at room temperature. XRD was done at a tube voltage of 40 kV and a tube current of 200 mA. The samples were covered with paraffin film to prevent the oxidation during the XRD test. X-ray photoelectron spectroscopy (XPS) was performed with a PHI-5300 XPS spectrometer.
The as-received, doped, dehydrogenated, and rehydrogenated samples were examined by a field emission scanning electron microscope (FESEM-6301F) coupled with energy dispersive spectroscopy (EDS). Sample preparation for the FESEM measurement was carried out inside the glove box and, moreover, the samples were transferred to the SEM chamber by means of a device maintaining an Ar overpressure.
Results and discussion
The TG profiles in Fig. 1 depict the non-isothermal dehydrogenation performance of the milled MgH2–NaAlH4 composites with different mole ratios (1:2, 1:1 and 2:1) undoped and doped with 2 mol% Nb2O5. The manometric desorption profiles recorded on the unary components NaAlH4 and MgH2, with and without 2 mol% Nb2O5, are also reported in Fig. 1 for comparison. In the case of pristine NaAlH4, the hydrogen desorption starts at 178 °C. After heating to 450 °C, this material has released a total hydrogen capacity of 6.3 wt%. For the bare MgH2, the hydrogen release starts at 345 °C, and the weight loss is about 7.2 wt% after heating to 450 °C. It is evident that both the undoped and doped 1MgH2–2NaAlH4 and 1MgH2–1NaAlH4 systems display three significant stages of dehydrogenation occurring during the heating process. On the contrary, the undoped 2MgH2–1NaAlH4 composite has exhibited four-stage hydrogen release. The relative proportion of hydrogen evolved during these three or four stages is of course dependent upon the ratio of the as prepared samples. The first stage for all the undoped and doped composite samples seems to be the two-step decomposition of NaAlH4 as indicated in eqn (1) and (2), because both the dehydrogenation temperatures and the hydrogen liberation amounts correspond to the decomposition of NaAlH4.| | | 3NaAlH4 → Na3AlH6 + 2Al + 3H2 | (1) |
| | | Na3AlH6 → 3NaH + Al + 3/2H2 | (2) |
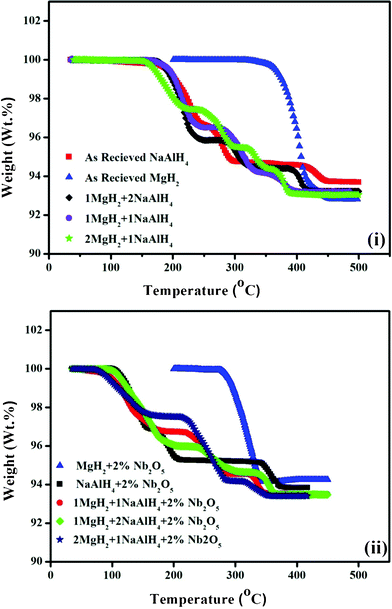 |
| | Fig. 1 Comparison of the TG profiles of neat MgH2 and NaAlH4, and MgH2–NaAlH4 composites (in mole ratios of 1:2, 1:1 and 2:1), (i) without Nb2O5 nanoparticles and (ii) with Nb2O5 nanoparticles. The ramping rate is 4 °C min−1. | |
Although the first step dehydrogenation temperature (175 °C and 170 °C) in the MgH2–NaAlH4 (molar ratios, 1:2 and 1:1) samples has not exhibited the significant reduction compared to that of the pristine NaAlH4 sample, faster dehydrogenation kinetics have been observed for both samples. The first stage of the decomposition of 1MgH2–2NaAlH4 and 1MgH2–1NaAlH4 samples terminates at 255 and 248 °C, respectively, which are 45 °C and 52 °C lower than that for the pure NaAlH4 sample. The 1MgH2–2NaAlH4 composite starts to decompose at 278 °C and terminates at 326 °C for the second stage, whereas the 1MgH2–1NaAlH4 sample starts the second decomposition at 275 °C and concludes at 335 °C. Obviously, The MgH2 decomposition temperature in 1MgH2–1NaAlH4 composite has reduced by 70 °C compared to that of the pristine MgH2 sample. Further heating of the 1MgH2–1NaAlH4 sample leads to the third decomposition, starting at about 360 °C, corresponding to the decomposition of NaH according to eqn (3), which also occurs at 50 °C lower than that of the pure NaAlH4 sample.
For the undoped 2MgH2–1NaAlH4 system, the first and second steps (Fig. 1(i)) occur in the temperature ranges of 152–225 °C and 240–300 °C while the onset temperatures of the third and fourth desorption steps are at 320 °C and 360 °C, respectively. The lowered dehydrogenation temperatures of 2MgH2–1NaAlH4 system compared to that for 1MgH2–2NaAlH4 and 1MgH2–1NaAlH4 systems suggest that higher amount of MgH2 enhances the decomposition of NaAlH4. After heating to 390 °C the total desorption capacity from the 2MgH2–1NaAlH4 mixture is about 6.9 wt%, which is higher than that of both MgH2 and NaAlH4 alone at the same temperature. These results indicate that the mixing between NaAlH4 and MgH2 can decrease the onset desorption temperature compared to that of NaAlH4 (178 °C) and MgH2 (345 °C). Therefore, the dehydrogenation properties of the MgH2–NaAlH4 system are improved compared to those of its unary components (NaAlH4 and MgH2), suggesting that a mutual destabilization has occurred in the binary MgH2–NaAlH4 system. Obviously, a greater reduction in the decomposition temperatures has occurred for the composites with higher concentrations of MgH2. This kinetic enhancement in the undoped 2:1 system can be partially ascribed to the more refinement of powders in these composites compared to that in other mixtures (Fig. 14).
In order to further clarify the mechanistic effects, three undoped binary composites have been also heated at much lower heating rate of 1 °C min−1. The results are displayed in Fig. S1 of the ESI.† Evidently, all the decomposition steps still exist, albeit the dehydrogenation temperatures have been further reduced.
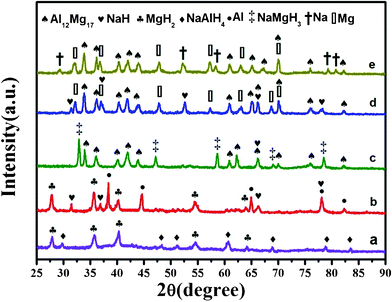
Fig. 2 and 3 present the evolution of the XRD patterns of the 1MgH2–1NaAlH4 and 2MgH2–1NaAlH4 samples upon ball-milling and heating to different temperatures, successively. The as-milled samples are detected as the physical mixtures of MgH2 and NaAlH4. Fig. 2(c) and 3(b) characterize the NaH, Al and MgH2 phases in the XRD patterns of the 1MgH2–1NaAlH4 and 2MgH2–1NaAlH4 systems after dehydrogenation at 250 °C and 230 °C, respectively. It excludes the possibility of MgH2-relevant reactions, demonstrating that only self-decomposition of NaAlH4 dominates this stage. For the 1:1 composite, after dehydrogenation at 350 °C, the Al phase has disappeared, and the peaks pertaining to Al12Mg17 and Mg are observed clearly from the XRD pattern in Fig. 2(d). These results indicate that the hydrogen release in the temperature range of 250–350 °C is mainly from the reaction of Al with MgH2 and the decomposition of MgH2 in accordance with the following reactions:
| | | 12Al + 17MgH2 → Al12Mg17 + 17H2 | (4) |
After further heating to 400 °C, the NaH phase has disappeared with the evolution of Na phase, suggesting that the hydrogen release in the temperature range of 350–400 °C is due to the decomposition of NaH as indicated in eqn(3). These results demonstrate that the three stages of dehydrogenation in the 1MgH2–1NaAlH4 composite correspond to the sequential decomposition of the NaAlH4, MgH2 and NaH phases (NaH resulting from the decomposition of NaAlH4).
The XRD pattern of 2MgH2–1NaAlH4 sample, heated to 310 C, shows the formation of the NaMgH3 phase together with Al12Mg17 in the temperature range of 240–300 °C (Fig.3c). The presence of NaMgH3, in agreement with the TG analysis of Fig. 1 (the third decomposition at about 320 °C), strongly suggests that apart from the reaction between Al and MgH2, a partial reaction between Mg/MgH2 and NaH has also taken place in the temperature range of 240–300 °C. The formation of NaMgH3 during the 2MgH2–1NaAlH4 decomposition reaction demonstrates that the desorption reaction occurs via the formation of an intermediate phase. On the contrary, the 1MgH2–1NaAlH4 sample has not exhibited the formation of NaMgH3. This corroborates that NaMgH3 can be formed while the ratio of MgH2/NaAH4 is relatively high. Otherwise, NaH and Mg are formed preferentially without the formation of NaMgH3. Ismail et al.41 have also identified the formation of NaMgH3 for 4MgH2–1NaAlH4 composite. XRD measurements of 2MgH2–1NaAlH4 composite at 360 °C indicate that the decomposition of NaMgH3 has accomplished below that temperature according to the following reaction.
| | | NaMgH3 → NaH + Mg + H2 | (6) |
Moreover, the XRD pattern in Fig. 3(e) confirms that the hydrogen release in the temperature range of 360–390 °C is induced by the decomposition of NaH. These experimental results, described above, point out the fact that various stoichiometric ratios of the MgH2/NaAlH4 system can lead to significant variations of the reaction mechanism and the sorption properties, even if the same final decomposition products (Al12Mg17, Mg and Na) are obtained. It can be surmised that the more MgH2-rich compositions facilitate the favorable formation of NaMgH3 phase. It has already been hypothesized that the formation of a mixed compound such as NaMgH3 is strongly associated with the microstructure of the starting materials, which should contain interfaces between the reacting phases.36 It is believed that higher concentrations of MgH2 favour the formation of NaMgH3 by creating large amounts of such interfaces. Moreover, the binary NaAlH4–MgH2 systems displayed superior hydrogen storage properties compared to that of the unary components NaAlH4 and MgH2. This phenomenon may be attributed to the mutual interactions among the two hydrides.
The desorption curves in Fig.1(ii) clearly depict that the use of nanometric Nb2O5 additions has rendered quite striking effects not only on the dehydrogenation characteristics of the binary composites but also on the unary components. In order to comprehend the catalytic role of Nb2O5 on the binary system, the samples of NaAlH4 and MgH2 doped with 2 mol% Nb2O5 nanopowders are prepared and investigated in comparison with the undoped binary samples as well as the undoped and doped unary components. It can be seen that nanosized Nb2O5 is effective in ameliorating the dehydrogenation properties of the unary components NaAlH4 and MgH2 as well. In the case of the Nb2O5-doped NaAlH4 and MgH2, the onset dehydrogenation temperatures are decreased to 100 °C and 275 °C, which are 78 °C and 70 °C lower than that of pure NaAlH4 (178 °C) and MgH2 (345 °C) samples. In addition, the dehydrogenation temperatures of the NaAlH4 and MgH2 in all the MgH2–NaAlH4 systems are also lower than that of the pure and doped NaAlH4 and MgH2 samples. The onset temperatures of hydrogen desorption for the milled 2 mol% Nb2O5 added MgH2–NaAlH4 with molar ratios of 2:1, 1:1, and 1:2 composites appear around 70, 80, and 95 °C, respectively, which are 82, 90, and 80 °C lower than the corresponding decomposition temperatures of the milled MgH2–NaAlH4 composite without Nb2O5 addition. Further heating leads to the decomposition of MgH2 near 213, 220 and 235 °C (Fig. 1(ii)), which are also lower than the corresponding decomposition temperatures at 240, 275 and 278 °C in Fig. 1(i). In particular for the doped samples with molar ratios of 1:1 and 2:1, the dehydrogenation temperature ranges have lowered to 80–355 °C and 70–350 °C due to the additive Nb2O5. Obviously, the addition of Nb2O5 nanoparticles plays a key role in decreasing the decomposition temperature of the MgH2–NaAlH4 system. Moreover, the nanometric Nb2O5-doped MgH2–NaAlH4 samples have shown faster desorption rates compared to undoped ones during the heating process. For example, the hydrogen desorption capacities of 6.6, 6.4 and 5.9 wt% are reached on heating the doped MgH2–NaAlH4 (molar ratios of 2:1, 1:1 and 1:2) samples to 350 °C. In contrast, the MgH2–NaAlH4 composites (molar ratios of 2:1, 1:1 and 1:2) without additive have exhibited hydrogen release capacities of 5.7, 5.8 and 5.6 wt% hydrogen, respectively, by 350 °C. In addition, the total hydrogen release contents from 2MgH2–1NaAlH4 doped samples are around 6.6 wt%. It indicates that a significant reduction in the decomposition temperature arising from the addition of a Nb2O5 catalyst is achieved without much penalty in the practical capacity of the materials. The above results clearly indicate that the dehydrogenation properties of the NaAlH4–MgH2 composites have been enhanced by the addition of Nb2O5 nanopowders.
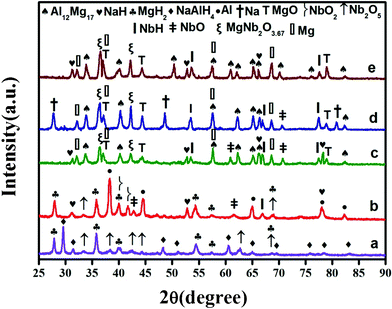
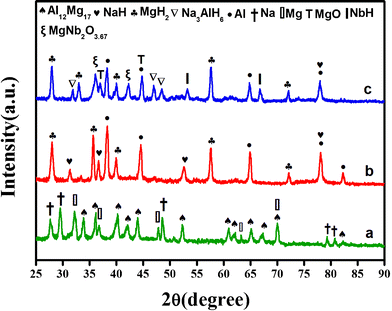
With the aim of structurally elucidating the catalytic mechanism of Nb2O5 nanoparticles, XRD measurements of the doped composites, before and after being subjected to the dehydrogenation at different temperatures, are displayed in Fig. 4. It is clear that the Nb2O5 phase can be detected in the XRD pattern of the as milled materials. It suggests that the Nb2O5 nanocrystalline particles remain stable with the composite matrix during ball-milling under the high-energy impact mode. Although, the dehydrogenation products of the doped 1:1 composite are analogous to that observed for undoped one, nevertheless, some by-products due to the decomposition/reaction of the dopant can be revealed in the discharged mixture of the doped sample. Fig. 4a clearly depicts that the doped 1:1 sample, at the beginning, consists mainly of Nb2O5 and changes significantly during the heating and desorption process. It is evident that the heating of the sample up to 200 °C has induced the disappearance of the crystalline Nb2O5, coupled to a parallel growth of the newly reduced niobium species with different oxidation states similar to NbO2 (+4) and NbO/NbH (+2, +1). During further heating, the Mg liberated from the hydrogen induces a fast reduction of the niobium oxide due to its very low redox potential, leading to the evolution of new peaks corresponding to pure MgO. Obviously, the reaction between the reduced niobium oxide species and the Mg/MgH2 has also yielded the formation of a ternary oxide phase MgNb2O3.67. It has been previously documented42 that NbO and MgO exhibit similar crystal structure. Moreover, both have similar lattice parameters with comparable bond lengths between metal and oxygen atoms. Therefore, a reaction between the niobium oxide and the Mg/MgH2 with a formation of a ternary oxide MgxNbyO is possible. Hence, the evolution of the additive to the real active species has taken place in the form of mixed MgNb2O3.67 phase. In the literature, the formation of ternary MgxNbyO has already been reported for the MgH2 doped with Nb2O5. It has been documented that these ternary Mg–Nb oxides facilitate the hydrogen diffusion by acting as pathways by the formation of metastable niobium hydrides.42 When the desorption process is finished, a steady state is reached consisting mainly of phases with the oxidation states rich in +1(NbH) and +2(NbO).
Fig. 4e presents the evolution of the XRD pattern of the Nb2O5-doped 2MgH2–1NaAlH4 composite upon heating to 300 °C. Interestingly, no diffraction peaks corresponding to the NaMgH3 phase have been detected for the 2:1 doped mixtures (in the sensitivity limits of the XRD), suggesting that the presence of Nb2O5 alters the desorption pathway by inhibiting the formation of NaMgH3. These results coincide well with those obtained in Fig. 1, indicating that the four-stage decomposition for the undoped 2MgH2–1NaAlH4 sample has transformed to three- stage owing to the additive Nb2O5. Moreover, these results are also in contrast with that reported by Milanese et al.,44 pointing to the fact that the dopants hamper the formation of NaMgH3 phase.
The thermal decomposition behavior of the undoped and doped MgH2–NaAlH4 (molar ratios of 2:1, 1:1 and 1:2) composites, as well as of undoped unary components NaAlH4 and MgH2, is further investigated by DSC, as presented in Fig. 5, 6, and 7. Fig. 5 illustrates the DSC results of the as-received MgH2 and alanate samples at various heating rates (4 °C min−1, 7 °C min−1 and 10 °C min−1, respectively). the DSC results of Nb2O5-doped NaAlH4 at various heating rates (4 °C min−1, 7 °C min−1 and 10 °C min−1, respectively) have been reported in Fig. S2 of the ESI.† The calorimetric profiles, recorded on the binary 1:1, 1:2 and 2:1 mixtures, are reported in Fig. 6. Five endothermic events in the 1:1 and 1:2 mixtures are assigned to the melting of NaAlH4, the decomposition of molten NaAlH4 to Na3AlH6, the decomposition of Na3AlH6 to NaH and Al, the decomposition of MgH2, and the decomposition of NaH, respectively, which is comparable with the three-step dehydrogenation in the TG results. Concomitant with the aforementioned XRD and TG results, the first four endothermic processes in 2:1 sample are similar to that in other composites, while the fifth and sixth events are attributed to the decompositions of NaMgH3 and NaH, respectively. The resulting peak temperatures, measured in Fig. 6(i), are small compared to that of pure NaAlH4 and MgH2 samples. For instance, the peak temperatures for 2:1 composite for the first and second dehydrogenation steps of NaAlH4 are 191 °C and 213 °C, respectively, which are 49 °C and 58 °C lower than those of the bare alanate sample. It is also evident that the peak temperature for pure MgH2 is 406 °C, which is 97 °C higher than that for the MgH2-relevant decomposition in 1MgH2–1NaAlH4 sample. These results further testify the mutual destabilization between NaAlH4 and MgH2. In contrast with the calorimetric profiles of the undoped MgH2/NaAlH4 composites, the features of all the doped samples are strikingly different, displaying only four endothermic peaks [Fig. 7]. The results indicate that NaAlH4 decomposes at a much lower temperatures without melting with Nb2O5 catalysis. Obviously, the addition of Nb2O5 nanopowders has rendered a significant reduction in the decomposition temperatures associated with the MgH2–NaAlH4 system.
 |
| | Fig. 5 The DSC profiles at various heating rates (4 °C min−1, 7 °C min−1 and 10 °C min−1) for pristine (i) MgH2 and (ii) NaAlH4. | |
 |
| | Fig. 6 The DSC profiles for (i) MgH2–NaAlH4 composites (in mole ratio of 1:2, 1:1 and 2:1) at heating rate of 4 °C min−1 and (ii) MgH2–NaAlH4 composite (in mole ratio of 1:1) at various heating rates (4 °C min−1, 7 °C min−1 and 10 °C min−1). | |
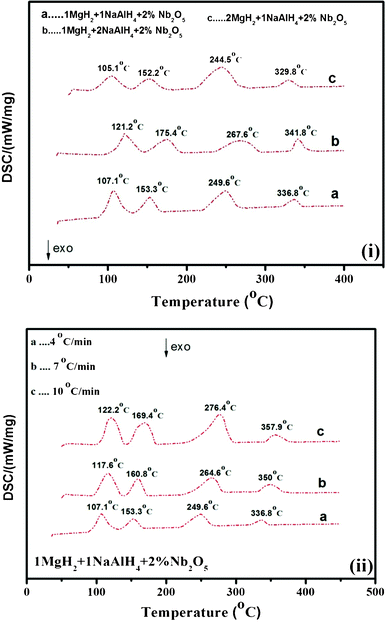 |
| | Fig. 7 The DSC profiles for (i) Nb2O5-doped MgH2–NaAlH4 composites (in mole ratios of 1:2, 1:1 and 2:1) at heating rate of 4 °C min−1 and (ii) Nb2O5-doped MgH2–NaAlH4 composite (in mole ratio of 1:1) at various heating rates (4 °C min−1, 7 °C min−1 and 10 °C min−1). | |
To acquire detailed information about the kinetics of the reactions, the apparent activation energy related to the NaAlH4, Na3AlH4, MgH2, and NaH decompositions in both the undoped and doped 1MgH2–1NaAlH4 composites, are calculated by using the nonisothermal Kissinger method45 which is based on the shift in peak temperatures with the heating rates of 4, 7, and 10 °C min−1. The activation energies of pristine NaAlH4 and MgH2, and Nb2O5-doped NaAlH4, for hydrogen desorption, are also reported. The derived values of the activation energies are listed in Tables 1 and 2. For comparison, the activation energies of NaAlH4 and MgH2, undoped and doped with other additives, are also recapitulated in Tables 1 and 2.6,25–27,46–49,50–55 The calculated apparent activation energies for the first, second, and third decomposition steps for the pure alanate are 116 kJ mol−1, 149 kJ mol−1, and 180 kJ mol−1, respectively. It is evident in Table 1 that the activation energy values for the first and second stages obtained in our work, agrees very well with that reported by Fan et al.47 The apparent activation energy calculated is about 153 kJ mol−1 for pure MgH2, which is consistent with the values of 150, 156, 158, and 160 kJ mol−1 H2 for un-milled MgH2 in Table 2. With the mixing of two hydrides, the activation energy has lowered to 109, 124, 120, and 164 kJ mol−1 for NaAlH4, Na3AlH6, MgH2, and NaH relevant decompositions, respectively, which exhibits an enhancement in kinetics. This result clearly indicates that the mixing of two hydrides reduces the energy barriers for all the four stages but is more efficient for the last three stages, i.e., the dehydrogenation of Na3AlH6, MgH2, and NaH, respectively. For the uncatalyzed MgH2–NaAlH4 composite, the reduction of activation energy may be attributed to the particle size reduction and the occurrence of mutual interactions between two species. The derived apparent activation energies corresponding to NaAlH4, Na3AlH6, MgH2, and NaH decompositions for the MgH2–NaAlH4 composite, catalyzed with Nb2O5 nanoparticles, are 68, 81, 75, and 130 KJ mol−1, respectively. It implies that the activation energies of four stages are reduced by around 41, 43, 44, and 34 kJ mol−1, compared with the activation energies associated with the undoped 1MgH2–1NaAlH4 sample, respectively. Table 1 and 2 clearly demonstrate that these values are slightly higher than those reported for TiO2-doped NaAlH4 and MgH2 doped with TiH2, but smaller than that reported for NaAlH4 and MgH2 doped with all other additives. Thereby, it is surmised that the presence of the Nb2O5 nanoparticles has essentially rendered the diminution of activation energy barrier for the dehydrogenation of the MgH2–NaAlH4 samples.
Table 1 Comparison of Activation energies (KJ mole−1) of pure and doped NaAlH4
| Sample |
Determination method |
NaAlH4 |
NaAlH3 |
References |
| Pristine NaAlH4 |
Kissinger Method |
114.2,132 |
156.8,268 |
47,46
|
| Pristine NaAlH4 |
Isothermal |
118.1 |
120.7 |
48
|
| Pristine NaAlH4 |
Kissinger Method |
116 |
149 |
Present work |
| NaAlH4 + MgH2 |
Kissinger Method |
109 |
124 |
Present work |
| NaAlH4 + MgH2 + Nb2O5 |
Kissinger Method |
68 |
81 |
Present work |
| Nb2O5-doped |
Kissinger Method |
65 |
86 |
Present work |
| CeCl3-doped |
Kissinger Method |
80.8 |
97.2 |
47
|
| CeAl4-doped |
Kissinger Method |
80.9 |
98.9 |
47
|
| TiCl3-doped |
Isothermal |
80 |
97.5 |
48
|
| Ti-doped |
Kissinger Method |
77 |
|
49
|
| TiO2-doped |
Kissinger Method |
67 |
|
49
|
| Ti/Til3•6THF-doped |
Kissinger Method |
96.2 |
197.2 |
50
|
| Ti/TiCl3-doped |
Kissinger Method |
139.5 |
|
50
|
| CeAl-doped |
Kissinger Method |
72.3 |
98.9 |
51
|
| LaCl3-doped |
Kissinger Method |
86.4 |
96.1 |
52
|
| La3Al11-doped |
Kissinger Method |
92.9 |
99.2 |
52
|
| SmCl3-doped |
Kissinger Method |
89 |
96.7 |
52
|
| SmAl3-doped |
Kissinger Method |
91.9 |
98.9 |
52
|
| TiF3-doped |
Kissinger Method |
98 |
130 |
46
|
| SiO2-doped |
Kissinger Method |
127 |
138 |
46
|
| TiF3 + SiO2-doped |
Kissinger Method |
99 |
122 |
46
|
Table 2 Comparison of Activation energies (KJ mole−1) of pure and doped MgH2
| Sample |
Determination method |
MgH2 |
References |
| Pristine MgH2 |
Kissinger Method |
120,135,150,191,191.3 |
53,6,27,26,54
|
| Pristine MgH2 |
Isothermal |
158.5,156,160 |
25
|
| Pristine MgH2 |
Kissinger Method |
153.5 |
Present work |
| NaAlH4 + MgH2 |
Kissinger Method |
120 |
Present work |
| NaAlH4 + MgH2 + Nb2O5 |
Kissinger Method |
75 |
Present work |
| TiC-doped |
Kissinger Method |
144.6 |
54
|
| NbF5-doped |
Kissinger Method |
90,88 |
53
|
| Cr2O3-doped |
Kissinger Method |
84 |
27
|
| Fe2O3-doped |
Kissinger Method |
124 |
27
|
| Ni-doped |
Kissinger Method |
81 |
26
|
| TiO2-doped |
Kissinger Method |
94 |
27
|
| Fe3O4-doped |
Kissinger Method |
115 |
27
|
| In2O3-doped |
Kissinger Method |
122 |
27
|
| ZnO-doped |
Kissinger Method |
147 |
27
|
| TiH2-doped |
Kissinger Method |
58.4 |
6
|
| Nb2O5-doped |
Kissinger Method |
71 |
55
|
| CoCl2-doped |
Kissinger Method |
121.3 |
25
|
| NiCl2-doped |
Kissinger Method |
102.6 |
25
|
The pronounced effect of Nb2O5 nanopowders in promoting dehydrogenation reactions of the Na–Mg–Al–H system is further demonstrated in the isothermal volumetric measurements. Fig. 8 presents the dehydriding kinetics at 280 °C and 300 °C for the samples with and without the Nb2O5 additive, respectively. For comparison, the dehydrogenation kinetics of the pure MgH2, decomposed at 280 °C and 300 °C, are also included. The desorption rate is much slower for the pristine MgH2 both at 280 °C and 300 °C. The desorption kinetics exhibit an enhancement after mixing with NaAlH4. However, the enhancements arising upon the addition of Nb2O5 to the MgH2/NaAlH4 sample are much more significant. Within 60 min, the pure MgH2 has only desorbed 0.7 wt% and 1.1 wt% hydrogen at 280 °C and 300 °C, respectively. The 1MgH2–1NaAlH4 composite can release 4.5 wt% and 5.2 wt% hydrogen in 60 min at 280 °C and 300 °C, respectively. Under the same conditions, the doped sample has yielded 5.9 wt% and 6.2 wt% of hydrogen. Moreover, the MgH2–NaAlH4–Nb2O5 sample has desorbed about 5.2 wt% hydrogen after 25 min at 300 °C, which is higher than for the MgH2–NaAlH4 (3.2 wt%) and much higher than for the pure MgH2 (0.5 wt%) at same temperature. In contrast, 60 min is required for the undoped MgH2–NaAlH4 sample to release 5.2 wt% hydrogen at 300 °C. These results indicate that the dehydrogenation kinetics of MgH2 is significantly improved by combining with NaAlH4, and also further enhanced by the addition of Nb2O5. In conclusion, the Nb2O5-doped MgH2/NaAlH4 mixture exhibits superior dehydrogenation properties than that of the pure MgH2 as well as the uncatalyzed MgH2/NaAlH4 samples.
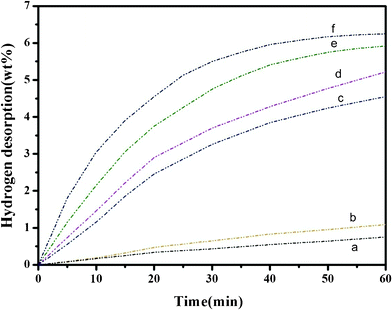 |
| | Fig. 8 Comparison of the isothermal dehydriding curves for neat MgH2 at (a) 280 °C and (b) at 300 °C, undoped MgH2–NaAlH4 composite (in mole ratio of 1:1) at (c) 280 °C and (d) at 300 °C, and Nb2O5-doped MgH2–NaAlH4 composite (in mole ratio of 1:1) at (e) 280 °C and (f) at 300 °C. | |
In order to further analyze the catalytic efficiency of Nb2O5 nanoparticles for the rehydrogenation/dehydrogenation cycles, the reversibility and cyclic properties of the Nb2O5-doped binary composite system have been investigated. The rehydrogenation of the samples has been conducted under 9.5 MPa of H2 at 300 °C after complete dehydrogenation. The undoped binary system has also been examined for comparison. Fig. 9 displays the isothermal rehydrogenation kinetics for the first three hydrogenation cycles of the Nb2O5-doped 1:1 system and the first rehydrogenation cycle of the undoped 1:1 sample. It is evident that the rehydriding capacity for the undoped sample (3.3 wt%) is smaller than that of the Nb2O5-doped sample (4.4 wt%), indicating that the reversibility is facilitated by the Nb2O5 dopant. Moreover, the recycled MgH2–NaAlH4–Nb2O5 sample exhibits well maintained kinetics and some capacity loss compared to the performance in the first cycle. It decreases from 4.4 wt% (first rehydrogenation) to 4.2 wt% (second rehydrogenation) and 4 wt% (third rehydrogenation). The XRD profiles of both the undoped and doped samples following the rehydrogenation, shown in Fig. 10b and c, indicate the reformation of MgH2 and NaH. This suggests that reaction (3) is reversible. However, no Al3Mg2 alloy is found in the rehydrogenated samples, indicating that full recovery of MgH2 from Al12Mg17 alloy has been acquired according to the following equations.
| | | Mg17 Al12 + (17 − 2y)H2 → yMg2Al3 + (17 − 2y)MgH2 + (12 − 3y)Al | (7) |
| | | Mg2Al3 + 2H2 → 2MgH2 + 3Al | (8) |
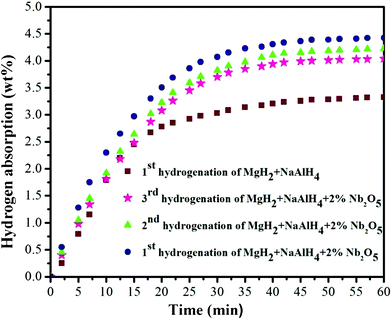 |
| | Fig. 9 Comparison of the rehydriding curves for the undoped MgH2–NaAlH4 composite (in mole ratio of 1:1) and Nb2O5-doped MgH2–NaAlH4 composite (in mole ratio of 1:1) at 300 °C under 9.5 MPa. | |
In addition, the Na3AlH6 phase has been detected for the rehydrogenated doped sample (Fig. 10c), which is not found for the rehydrogenated undoped sample (Fig. 10b). The formation of Na3AlH6 may be accounted for the enhanced rehydriding capacity of the doped sample engendered by the Nb2O5 dopant, indicating that the Nb2O5 component in the MgH2–NaAlH4–Nb2O5 sample plays a catalytic role through the formation of Nb-containing catalytic species.
 |
| | Fig. 10 XRD spectra for (a) undoped MgH2–NaAlH4 (1:1) composite after complete dehydrogenation, (b) undoped MgH2–NaAlH4 (1:1) composite after rehydrogenation, and (c) Nb2O5-doped MgH2–NaAlH4 (1:1) composite after rehydrogenation. | |
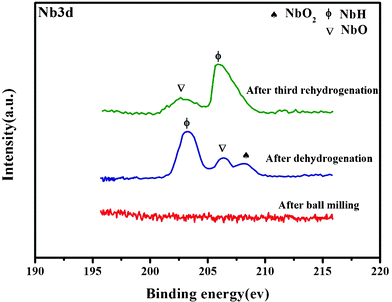
To further analyze the milling and cycling process, the chemical characterization of the Nb-based additive has been also investigated by XPS in the surface and sub surface regions of the milled 1MgH2–1NaAlH4 + 2 mol% Nb2O5 sample before and after hydrogen cycling. Fig. 11 depicts the Nb 3d photoelectron peak region for the as prepared sample as compared to the fully desorbed and fully reabsorbed samples. According to Fig. 11a for the as prepared sample, no Nb catalyst has been detected at the surface. During desorption, the reduced Nb species migrate from the bulk to the surface.56 This is consistent with the results obtained from XRD. It is hard to specify whether the inception of the reduction of Nb2O5 takes place during milling or not. XRD results for the milled sample have not revealed the existence of any reduced Nb species. During the first hydrogen desorption, the reduction of Nb2O5 commences and the originating products disperse in the sample and emerge to the surface. This fact is further strengthened by the XPS spectra from the Mg 2p regions of the doped sample (Fig. 12b), exhibiting the occurrence of MgO after first dehydrogenation. The Na3AlH6 photoelectron peak in Fig. 13(a–d) for the rehydrogenated samples, located at 64.92 eV,57 further proves the reformation of Na3AlH6 during the rehydrogenation process. These observations are in accordance with the results of X-ray diffraction, showing that the enhanced rehydriding capacity of the doped sample is induced by the reformation of Na3AlH6.
The FESEM qualitative and quantitative microstructural investigations have been employed to examine the particle size and morphology of the as-milled and cycled samples. Fig. 14a and b indicates that the pure MgH2 and NaAlH4 particles are irregularly shaped with a mean particle size of more than 30 μm. Fig. 14c,d brings out the microstructural features of the as-milled undoped 1MgH2–1NaAlH4 and 2MgH2–1NaAlH4 powders. Remarkably, the stoichiometry has exhibited an evident influence on the particle size distribution reached after the milling process. This noticeable difference may be ascribed to the higher brittleness of the MgH2 powders which, indeed, improves the efficiency of the ball milling procedure. The FESEM analysis carried out on the cross-section of 1MgH2–1NaAlH4 + 2 mol% Nb2O5 composite has revealed the substantial refinement induced by the Nb2O5 nanoparticles (Fig. 14e). The size of most of the particles is less than 5 μm for the sample with Nb2O5 dopant, indicating the significant diminution of particle size compared to that of undoped unary as well as binary samples. It is vividly discernible in the inset of Fig. 14e that the oxide particles are embedded heterogeneously in the composite matrix, resulting in the substantial surface modifications exhibiting many deformed and disordered surface regions. Moreover, the hardness of Nb2O5 is much higher than that of MgH2 and NaAlH4. Therefore, the fraction of embedded Nb2O5 nanoparticles will enhance the hardness and brittleness of hydride particles that will ultimately shift the balance between fracturing and agglomeration to smaller particle sizes. Moreover, Nb2O5 is well known for its good lubricant and dispersive properties that prevent the agglomeration and cold welding of hydride particles and thereby facilitates the refinement of matrix particles during milling. Fig. 14f brings out the microstructural features of the doped materials after third rehydrogenation. The small pores are observed at the matrix surface are imputable to the repeated volume shrinkage and expansion during the sorption cycles. Fig. 15(a–b) is the surface EDS spectra collected at the positions circled in Fig. 14e,f. The variation in the Nb peak positions following the cycling (Fig. 15b) further corroborates that Nb2O5 has been reduced during cycling.
 |
| | Fig. 14 Field emission scanning electron microscopy (FESEM) images of (a) as-received MgH2, (b) as-received NaAlH4, (c) undoped MgH2–NaAlH4 (1:1) composite, (d) undoped MgH2–NaAlH4 (2:1) composite, (e) Nb2O5-doped MgH2–NaAlH4 (1:1) composite after ball milling, and (f) Nb2O5-doped MgH2–NaAlH4 (1:1) composite after third rehydrogenation. | |
The above experimental results suggest that all the stoichiometries follow the same initial low-temperature reaction pathway (decomposition of NaAlH4). Nevertheless, the differences in the respective pathways begin to emerge at elevated temperatures (the formation of NaMgH3), depending upon the proportion of the potential reactant (MgH2). The improvement in dehydrogenation kinetics of binary composite is believed to be due to the interaction between MgH2 and alanate that changes the thermodynamics of the reactions by either lowering the enthalpy of the dehydrogenation reaction or leads the balance of the reaction towards one direction. Moreover, the smaller powder agglomerates are obtained for the 2:1 composites (Fig. 14), which accounts for their enhanced dehydrogenation kinetics compared to that of other samples. The results of the present investigation also reveal that additive, i.e. Nb2O5 nanopowders can lead to the generation of surface defects by inducing a substantial refinement of powder particles during the high-energy milling, which are expected to further ameliorate the kinetics as well as the hydrogen sorption capability. It is documented in our previous work9,10,43 that the hydrogen desorption/absorption is closely associated with the surface defects and the refinement, and the hydrogen sorption capability increases with the increase in the defects in the nanostructures. In addition, the lubricant, dispersive, and hardness properties of Nb2O5 can facilitate a further reduction of hydride particles by preventing the agglomeration of matrix particles during milling, which has to be considered also an important factor for enhancing the reaction kinetics. Nonetheless, other factors, such as the nature and local electronic structure of the added oxide as well as its reduction during heating also have to be taken into account. Since the Nb2O5 is reduced during the heating, the possible candidates for catalysis remain in the non-stoichiometric magnesium–niobium oxide or even other possible niobium phases with lower oxidation states formed during cycling. Similarly, in previous studies by ourselves and others,10,42 it is suggested that the partially reduced Nb species with a wide range of valence might play a major role as a catalyst. Thereby, the finely dispersed oxygen-deficient Nb species may contribute to kinetic improvement by facilitating the diffusion of hydrogen through the diffusion barriers both in dehydrogenation and rehydrogenation processes.
Conclusions
In conclusion, we have examined the hydrogen storage properties and reaction pathways of three distinct stoichiometries within the MgH2–NaAlH4 binary composite system both undoped and doped with Nb2O5 nanopowders. The premilled reactant stoichiometry of the MgH2/NaAlH4 system has a profound impact on the reaction kinetics and the sorption properties because of the reactant availability. It has also been demonstrated that the dehydrogenation kinetics of MgH2 and NaAlH4 can be improved by combining them with each other. Apart from the existence of the mutual interactions between two hydrides, the crystallite size of the undoped binary composites is also found to be smaller than that of the pristine components, which explains for the superior hydrogen performance of the mixed samples. Significant improvements in the dehydrogenation/rehydrogenation properties of the MgH2–NaAlH4 system have been achieved by adding a small amount of Nb2O5 nanoparticles. The onset as well as the peak temperatures of hydrogen desorption shift to lower temperatures. The kinetics of hydrogen desorption with Nb2O5 additions are found to be 2–6 times as fast as the undoped MgH2 and MgH2–NaAlH4 samples. The rehydrogenation properties of the MgH2–NaAlH4–Nb2O5 composite are also improved significantly as compared to the MgH2–NaAlH4 composite. The reversible capacity of 4.4 wt% is acquired for the MgH2–NaAlH4–Nb2O5 composite, which is larger than that of MgH2–NaAlH4 (3.3 wt%). Moreover, this catalytically enhanced rehydrogenation capacity persists well during three de/rehydrogenation cycles. XRD, XPS, and FESEM-EDS analyses suggest that hydrogen cycling has induced the reduction of Nb2O5, which leads to the formation of oxygen-deficient reduced niobium oxide species. Accordingly, it is believed that the fine dispersion of these oxygen-deficient niobium oxide nanoparticles facilitates the dehydrogenation process by serving as the active sites for nucleation and growth of the dehydrogenated product associated with the shortening of the diffusion paths among the reaction ions and thus reducing the kinetic barriers and rendering the amelioration of dehydrogenation kinetics. Besides, the nanocrystalline reduced niobium oxide phases present in the mixture can also provide the active catalytic sites for the hydrogen dissociation and surface adsorption, which improves the kinetics of the rehydrogenation as well. Moreover, the lubricant, dispersive, and hardness properties of Nb2O5 increase the surface defects and grain boundaries by a large reduction in the particle size, creating a larger surface area for hydrogen to interact, thereby decreasing the temperature for decomposition.
Acknowledgements
This work is financially supported by the University of Science and Technology Beijing (USTB). The authors also thank the Higher Education Commission (HEC) of Pakistan for the financial support to Rafi-ud-din.
References
-
R. A. Varin, T. Czujko and Z. S. Wronski, Nanomaterials for Solid State Hydrogen Storage; Springer Science + Business Media, New York, 2009 Search PubMed.
- J. Yang, A. Sudik, C. Wolverton and D. J. Siegel, Chem. Soc. Rev., 2010, 39, 656–675 RSC.
- A. W. C. Berg and C. O. Arean, Chem. Commun., 2008, 668–681 RSC.
- S.-I. Orimo, Y. Nakamori, J. R. Eliseo, A. Züttel and C. M. Jensen, Chem. Rev., 2007, 107, 4111–4132 CrossRef CAS.
- T. Umegaki, J. M. Yan, X. B. Zhang, H. Shioyama, N. Kuriyama and Q. Xu, Int. J. Hydrogen Energy, 2009, 34, 2303 CrossRef CAS.
- L. Jun, J. C. Young, Z. F. Zhigang, Y. S. Hong and R. J. Ewa, AM. CHEM. SOC., 2009, 131, 15843–15852 CrossRef.
- J. F. Mao, Z. Wu, T. J. Chen, B. C. Weng, N. X. Xu and T. S. Huang, J Phys Chem C, 2007, 111, 12495–12498 CAS.
- M. David, B. J. Daniel, S. Toyoto, N. Dag, K. Daisuke, S. Tetsuo, K. Naoyuki and Y. Hitoshi, J. Mater. Chem., 2009, 19, 8150–8161 RSC.
- Rafi-ud-din, L. Zhang, L. Ping and Q. Xuanhui, J. Alloys Compds., 2010, 508, 119–129 CrossRef CAS.
- Rafi-ud-din, Q. Xuanhui, L. Ping, L. Zhang and M. Ahmad, J Phys Chem C, 2011, 115, 13088–13099 CAS.
- L. Li, F. Qiu, Y. Wang, Y. Wang, G. Liu, C. Yan and C. An, J. Mater. Chem., 2012, 22, 3127–3132 RSC.
- J. Chen, N. Kuriyama, Q. Xu, H. T. Takeshita and T. Sakai, J. Phys. Chem. B, 2001, 105, 11214–11220 CrossRef CAS.
- F. Schuth, B. Bogdanovic and M. Felderhoff, Chem. Commun., 2008, 668–681 Search PubMed.
- J. F. Mao, X. B. Yu, Z. P. Guo, C. K. Poh, H. K. Liu, Z. Wu and J. Ni, J. Phys. Chem. C, 2009, 113, 10813–10818 CAS.
- P. Chen, Z. T. Xiong, L. F. Yang, G. T. Wu and W. F. Luo, J Phys Chem B, 2006, 110, 14221–14225 CrossRef CAS.
- H. Y. Leng, T. Ichikawa, S. Hino, N. Hanada, S. Isobe and H. Fujii, J Power Sources, 2006, 156, 166–170 CrossRef CAS.
- M. Au, A. R. Jurgensen, W. A. Spencer, D. L. Anton, F. E. Pinkerton and S. J. Hwang, J Phys Chem C, 2008, 112, 18661–18671 CAS.
- X. B. Yu, G. L. Xia, Z. P. Guo and H. K. Liu, J Mater Res, 2009, 24, 2720–2727 CrossRef CAS.
- Y. H. Guo, X. B. Yu, L. Gao, G. L. Xia, Z. P. Guo and H. K. Liu, Energy Environ. Sci., 2010, 3, 465–470 CAS.
- X. D. Kang, Z. Z. Fang, L. Y. Kong, H. Cheng, X. D. Yao, G. Lu and P. Wang, Adv. Mater., 2008, 20, 2756–2759 CrossRef CAS.
- J. Zhao, J. Shi, X. Zhang, F. Cheng, J. Liang and Z. Tao, Adv. Mater, 2010, 22, 394–397 CrossRef CAS.
- B. Peng, J. Liang, Z. Tao and J. Chen, J. Mater. Chem., 2009, 19, 2877–288 RSC.
- F. Zeppelin, H. Reule and M. Hirscher, J. Alloys Compds., 2002, 330, 723–726 CrossRef.
- C. X. Shang, M. Bououdina1, Y. Song and Z. X. Guo, Int. J. Hydrogen Energy, 2004, 29, 73–80 CrossRef CAS.
- J. F. Mao, Z. Guo, X. Yu, H. Liu, Z. Wu and J. Ni, Int. J. Hydrogen Energy, 2010, 35, 4569–4575 CrossRef CAS.
- L. Xie, Y. Liu, X. Zhang, J. Qu, Y. Wang and X. Li, J. Alloys Compds., 2009, 482, 388–392 CrossRef CAS.
- M. Polanski and J. Bystrzycki, J. Alloys Compds., 2009, 486, 697–701 CrossRef CAS.
- D. L. Croston, D. M. Grant and G. S. Walker, J. Alloys Compds., 2010, 492, 251–258 CrossRef CAS.
- O. Friedrichs, T. Klassen, J. C. Sanchez-Lopez, R. Bormann and A. Fernandez, J. scripta mat., 2006, 54, 1293–1297 CrossRef CAS.
- L. P. Ma, P. Wang and H. M. Cheng, Int. J. Hydrogen Energy, 2010, 35, 3046–3050 CrossRef CAS.
- N. Recham, V. V. Bhat, M. Kandavel, L. Aymard, J. M. Tarascon and A. Rougier, J. Alloys Compds., 2008, 464, 377–382 CrossRef CAS.
- J. Lu and Z. G. Z. Fang, J. Phys. Chem., 2005, 109, 20830–20834 CrossRef CAS.
- Z. T. Xiong, G. T. Wu, J. J. Hu and P. Chen, J. Power Sources, 2006, 159, 167–170 CrossRef CAS.
- P. Wang, L. P. Ma, Z. Z. Fang, X. D. Kang and P. Wang, Energy Environ. Sci., 2009, 2, 120–123 CAS.
- G. S. Walker, D. M. Grant, T. C. Price, X. Yu and V. Legrand, J. Power Sources, 2009, 194, 1128–1134 CrossRef CAS.
- D. Pottmaier, C. Pistidda, E. Groppo, S. Bordiga and G. Spoto, Int. J. Hydrogen Energy, 2011, 36, 7891–7896 CrossRef CAS.
- J. F. Mao, Z. Guo, H. Leng, Z. Wu, Y. Guo, X. Yu and H. Liu, J. Phys. Chem. C, 2010, 114, 11643–11649 CAS.
- J. F. Mao, Z. Guo, X. Yu, M. Ismail and H. Liu, Int. J. Hydrogen Energy, 2011, 36, 5369–5374 CrossRef CAS.
- Y. Zhang, T. Qi-Feng, L. Shu-Sheng and S. Li-Xian, J. Power Sources, 2008, 185, 1514–1518 CrossRef CAS.
- M. Ismail, Y. Zhao, X. B. Yu and S. X. Dou, RSC Advances, 2011, 1, 408–415 RSC.
- M. Ismail, Y. Zhao, X. B. Yu, J. F. Mao and S. X. Dou, Int. J. Hydrogen Energy, 2011, 36, 9045–9050 CrossRef CAS.
- O. Friedrichs, F. Aguey-Zinsou, J. R. Ares Fernandez, J. C. Sanchez-Lopez, A. Justo, T. Klassen, R. Bormann and A. Fernandez, Acta mat., 2006, 54, 105–110 CrossRef CAS.
- A. Mashkoor, Rafi-ud-Din, P. Caofeng and Z. Jing, J. Phys. Chem. C, 2010, 114, 2560 Search PubMed.
- C. Milanese, S. Garroni, A. Girella, G. Mulas, V. Berbenni, G. Bruni, S. Surinach, M. D. Baro and A. Marini, J. Phys. Chem. C, 2011, 115, 3151–3162 CAS.
- H. E. Kissinger, Anal. Chem., 1958, 29, 1702 CrossRef.
- S. Zheng, Y. Li, F. Fang, G. Zhou, X. Yu, G. Chen, D. Sun, L. Ouyang and M. Zhu, J. Mater. Res., 2010, 25, 2047–2053 CrossRef CAS.
- X. Fan, X. Xiao, L. Chen, S. Li and Q. Wang, J. Alloys Compds., 2011, 509, 386–389 Search PubMed.
- G. Sandrock, K. Gross and G. Thomas, J. Alloys Compd., 2002, 339, 299–308 CrossRef CAS.
- D. Pukazhselvan, M. Hudson, A. Sinha and O. Srivastava, Energy, 2010, 35, 5037–5042 CrossRef CAS.
- O. Kircher and M. Fichtner, J. Alloys Compd., 2005, 404, 339–342 CrossRef.
- X. Fan, X. Xiao, L. Chen, S. Li, H. Ge and Q. Wang, J. Phys. Chem. C, 2011, 115, 2537–2543 CAS.
- X. Fan, X. Xiao, L. Chen, L. Han, S. Li, H. Ge and Q.Wang, Int. J. Hydrogen Energy, 2011, 36, 10861–10869 CrossRef CAS.
- Y. Luo, P. Wang, L. Ma and H. Cheng, J. Alloys Compd., 2008, 453, 138–142 CrossRef CAS.
- M. Fan, S. Liu, Y. Zhang, J. Zhang, L. Sun and F. Xu, Energy, 2010, 35, 3417–3421 CrossRef CAS.
- N. Hanada, T. Ichikawa, S. Hino and H. Fujii, J. Alloys Compd., 2006, 420, 46–49 CrossRef CAS.
-
http://srdata.nist.gov/xpx/
.
- C. Wan, X. Ju, Y. Qi, S. Wang, X. Liu and L. Jiang, J. Alloys Compd., 2009, 486, 436–441 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.