A covalently linked graphene-oligo(phenylenevinylene) adduct: self-organization and photo-physical properties†
H. S. S. Ramakrishna
Matte
ab,
Ankit
Jain
a and
Subi J.
George
*a
aNew Chemistry Unit, Jawaharlal Nehru Centre for Advanced Scientific Research, Jakkur P. O., Bangalore, 560 064, India. E-mail: george@jncasr.ac.in
bChemistry and Physics of Materials Unit, Jawaharlal Nehru Centre for Advanced Scientific Research, Jakkur P. O., Bangalore, 560 064, India
First published on 9th May 2012
Abstract
We report a simple method of covalent functionalization of graphene with an organic donor such as oligo(phenylenevinylene) (OPV). We present morphological proof for the unprecedented self-organization of these hybrids into higher order assemblies, with a periodic spacing of ∼2.3 nm, intercalated with OPV molecules. After systematic characterization of the resulting hybrid material, we provide unequivocal proof for possible electronic communication between the donor OPV molecules and the graphene sheets. The extent of functionalization and degree of fluorescence quenching were determined by hydrolyzing the EG-OPV hybrid. We also discuss the formation of head-to-tail cyclo-addition dimer of OPV molecules possibly due to the topochemical constraint set by the assembled EG-OPV.
Introduction
Graphene has generated a great sensation in the last few-years because of its fascinating properties with potential applications.1–4 Molecular functionalization of graphene, resulting in its solubilization and assembly, plays a crucial role in exploring applications in nano-electronic devices and other areas. Hybrid materials of graphene have the advantage of multifunctionality comprising properties of both graphene and the functionalizing agent.5–8 There has been some recent effort to carry out covalent and non-covalent functionalization of graphene with aromatic molecules, which helps to modify its electronic properties, exfoliate sheets and stabilize individual graphene-molecule hybrids.8–17 Interaction of graphene with electron donor and acceptor molecules has been exploited to control its electronic properties through ground-state molecular charge-transfer.18 Excited-state processes involving graphene and chromophoric electron donors such as oligo(phenylenevinylene) (OPV)10,15,19–21 and porphyrin and the concomitant photo-induced electron transfer process from the chromophore to graphene, which could be of value in the design of photovoltaics, have also been reported.10–13 We consider it is desirable to carry out the covalent functionalization of graphene with electron donor molecules using reaction protocols similar to those established in the case of carbon nanotubes,22–27 and examine the structural and spectroscopic aspects of the resulting hybrid materials. In this article, we report on the structure, reactivity as well as photo-physical properties of a novel hybrid obtained by covalently integrating electron-rich oligo(phenylenevinylene) amine (OPV-amine) with graphene. The graphene sample that we used (Exfoliated graphene, EG) contains 5 layers on average and was prepared by thermal exfoliation of GO. The OPV molecule, apart from serving as a useful probe, also helps in the exfoliation of graphene layers, rendering the resulting hybrid solution-processable. Interestingly, we find that non-covalent π–π stacking interactions of the phenylenevinylene conjugated backbones results in the unprecedented organization of graphene sheets into bi- or few-layer hybrids sandwiched by OPV molecules.Results and discussion
The three dodecyl chains at the gallic wedge of the OPV-amine molecule were excepted to render the resulting material soluble in organic solvents. The OPV-graphene adduct (EG-OPV), in which the components are linked by amide bonds, was prepared by the reaction between the OPV-amine and acid chloride functionalized EG (EG-COCl), in dimethylformamide (Scheme 1).28EG-OPV, so obtained, was purified by washing several times with chloroform, followed by centrifugation, in order to ensure the complete removal of physically absorbed OPV ligands, taking advantage of the higher solubility of OPV-amine in CHCl3.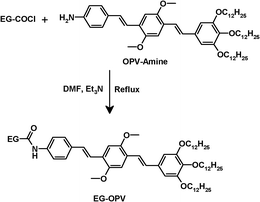 | ||
| Scheme 1 Synthetic route for the EG-OPV hybrid. | ||
Fig. 1a shows the IR spectra of EG-acid and EG-OPV which provide evidence for functionalization of graphene through amide bonds. The IR spectrum of EG-acid shows a band at 1632 cm−1, due to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching frequency of the carboxyl groups, which shifts to a higher frequency in the corresponding acyl chloride derivative. The spectrum of EG-OPV, shows the characteristic amide I and II bands at 1572 and 1639 cm−1, respectively. These observations clearly suggest the formation of the amide bond between OPV-amine and graphene. In Fig. 1b, we compare the Raman spectra of EG-acid and the EG-OPV hybrid. The Raman spectrum of EG-acid shows three bands namely D-, G- and 2D bands at 1342, 1585 and 2672 cm−1 respectively.3,4 In the case of EG-OPV, the G-band shifts to 1572 cm−1 while the 2D band appears at 2647 cm−1.29 It is well studied and understood from the literature that there will be significant changes in the Raman spectra of graphene when it interacts with either donor or acceptor molecules. Raman G-bands soften on interaction with donor molecules where as it stiffens in the case of acceptor molecules. From the above results it is clear that there is a softening in the G and 2D bands because the molecule attached is OPV which is a good donor. Softening of the G-band can be explained in terms of electron–phonon coupling in graphene which causes Kohn anomalies in the phonon dispersion which can give rise to phonon softening.29 Interestingly, the 2D Raman band becomes sharp accompanied by a large increase in the I2D/IG intensity ratio. This is clearly indicative of the decrease in the number of layers due to exfoliation. It is indeed well known that the 2D band becomes prominent in single-layer graphene.3,4
O stretching frequency of the carboxyl groups, which shifts to a higher frequency in the corresponding acyl chloride derivative. The spectrum of EG-OPV, shows the characteristic amide I and II bands at 1572 and 1639 cm−1, respectively. These observations clearly suggest the formation of the amide bond between OPV-amine and graphene. In Fig. 1b, we compare the Raman spectra of EG-acid and the EG-OPV hybrid. The Raman spectrum of EG-acid shows three bands namely D-, G- and 2D bands at 1342, 1585 and 2672 cm−1 respectively.3,4 In the case of EG-OPV, the G-band shifts to 1572 cm−1 while the 2D band appears at 2647 cm−1.29 It is well studied and understood from the literature that there will be significant changes in the Raman spectra of graphene when it interacts with either donor or acceptor molecules. Raman G-bands soften on interaction with donor molecules where as it stiffens in the case of acceptor molecules. From the above results it is clear that there is a softening in the G and 2D bands because the molecule attached is OPV which is a good donor. Softening of the G-band can be explained in terms of electron–phonon coupling in graphene which causes Kohn anomalies in the phonon dispersion which can give rise to phonon softening.29 Interestingly, the 2D Raman band becomes sharp accompanied by a large increase in the I2D/IG intensity ratio. This is clearly indicative of the decrease in the number of layers due to exfoliation. It is indeed well known that the 2D band becomes prominent in single-layer graphene.3,4
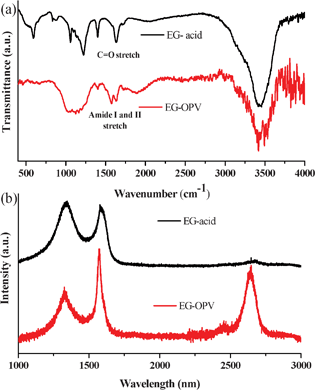 | ||
| Fig. 1 (a) FT-IR and (b) Raman spectra of EG-acid and EG-OPV hybrid. | ||
Detailed microscopic studies of EG-OPV generated from DMSO solution show the characteristic morphology of graphene. TEM images reveal the presence of mostly bi-and multi-layered sheets of the hybrids along with the stacked graphene sheets, with an interlayer spacing varying from 2–2.4 nm which is ∼7 times higher than the usual layer separation in few-layer graphene (Fig. 2a). AFM studies on the EG-OPV hybrid showed the formation of graphene sheets having lateral dimensions extended up to micrometers (Fig. 2b). The height profile shown in Fig. 2c corresponds to ∼2.64 nm, which may be due to a single layer of EG-OPV hybrid. Further analysis of the EG-OPV hybrid at different places also revealed a step wise increase in the height profile as can be seen in Fig. 2d. It is noteworthy that the step nearly corresponds to the length of the OPV rigid backbone.
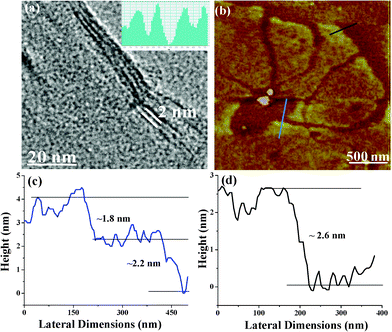 | ||
| Fig. 2 (a) TEM, (b) AFM image of EG-OPV hybrid, height profiles of black and blue lines are shown in (c) and (d) respectively. Additional figures are presented in the supporting information (Fig. S1†). | ||
AFM, TEM and Raman results suggest intercalation of the organic molecules between the graphene layers, by orienting perpendicular to the plane of the graphene as can be seen in the schematic (inset, Fig. 3b). Interestingly, a higher order organization of EG-OPV hybrid to multilayered sheets with ∼2.4 nm spacing could also be visualized in the TEM images (Fig. 3b). Organization of these hybrids could be driven by the π–π-stacking interactions between the interdigitated OPV-molecules, from neighboring sheets.30 This argument firmly supports from the observation of topochemical photodimerization of head-tail oriented OPV ligands of the two adjacent graphene sheets (Fig. 3a, vide infra).
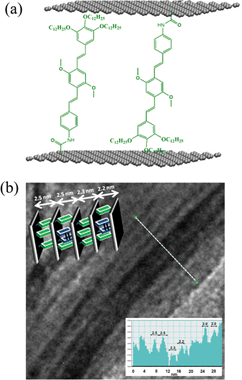 | ||
| Fig. 3 (a) Proposed molecular organization of OPV chromophores in the hybrid and (b) TEM of the layered graphene structures (insets of (b) shows the interlayer distances across the white line and the graphic representation of the arrangement.) | ||
In order to probe the interactions between OPV donor molecules and graphene in the hybrid, we have performed steady-state and time resolved photo-luminescence studies of the hybrids. The absorption spectrum of EG-OPV shows clear signatures of both EG and OPV absorptions. However, no new band attributable to a ground state charge transfer interaction is seen (Fig. S2†). Photoluminescence spectrum of the EG-OPV hybrid in DMSO shows very weak emission (see inset Fig. 4a) with maxima at 460 and 490 nm, when excited at 410 nm (Fig. 4a). These emission spectral features are characteristic of isolated OPV molecules, considering that red-shifted emissions (∼500–550 nm) of these molecules are found in the presence of intermolecular interactions.30 This suggests spatially dispersed attachment of OPV chromophores to the graphene, with a perpendicular orientation of its molecular axis (free hanging) rather than a face-on orientation on the graphene plane, which is also supported by the microscopic analysis. The relative intensities of the emission bands are, however, different compared to the standard amide substituted OPV molecules (OPV-B) possibly due to the covalent functionalization.23 The weak photoluminescence of the chromophore-graphene hybrids is attributed to fluorescence quenching of the molecules by graphene.10,31 In order to quantify the quenching, we have carried out in situ hydrolysis of the EG-OPV hybrid in the cuvette using KOH, resulting in the turn-on OPV fluorescence. We have quantified the quenching to be 78% in the EG-OPV hybrid in DMSO (Fig. 4b). This experiment allowed us to determine the number of OPV molecules attached to the graphene per unit weight. After comparing from a known concentration of OPV-amine we elucidated that 0.28 μM of OPV is functionalized per mg of graphene sample (Fig. S3†). This corresponds to about 5.1 × 1014 molecules of OPV being bonded to 1 mg of graphene (∼0.846 nmol of OPV per milligram of graphene).
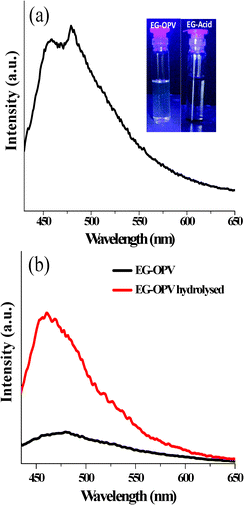 | ||
| Fig. 4 (a) Fluorescence spectrum of EG-OPV in DMSO, λexc = 410 nm (inset depicts the photographs of EG-acid and EG-OPV under UV illumination) and (b) the quenching quantification experiment of EG-OPV. | ||
Such efficient quenching of OPV fluorescence indicates that there is a strong interaction between the excited state of OPV and graphene and could only be due to an excited state phenomenon since we do not observe any evidence for ground state charge-transfer from the absorption spectrum (Fig. S2†). Fig. 5 shows the fluorescence decay profiles of the EG-OPV hybrid and the control sample OPV-B (OPV-amide), in DMSO (λexc = 410 nm) monitored at 500 nm. It can be clearly seen that EG-OPV decays in a multi exponential manner when compared to the mono exponential decay trace of nascent OPV (τ = 1.7 ns). Fluorescence decay measurements on the hybrid could be fitted to a bi-exponential decay with lifetimes of 0.6 and 1.6 ns. The decay of EG-OPV hybrid is clearly indicated by a short component and a long component. This multi-exponential decay, apart from being characteristic of hybrid carbon based materials, suggests electronic communication between OPV and graphene. Fluorescence quenching of the excited OPV can arise from photo-induced electron transfer or energy transfer.10,13,32 Similar photoluminescence quenching has been observed in non-covalent hybrids of graphene with OPVs, where an electron transfer mechanism has been shown to occur.10
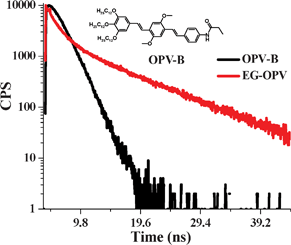 | ||
| Fig. 5 Time-resolved fluorescence decay profiles of the EG-OPV hybrid and OPV-B excited at 410 nm and emission collected at 500 nm. Inset shows the molecular structure of OPV-B. | ||
1H-NMR and mass spectral analysis of the OPV obtained after the hydrolysis of EG-OPV showed the presence of dimeric species. Signatures of the cyclobutane methylene could be seen in the 1H-NMR spectrum, indicating the likely occurrence of [2+2] cycloaddition of OPV (Fig. S4 and S5†).33 The mass spectrum also showed peaks due to the dimeric species. The cycloaddition process was further evident from the detailed photophysical characterization of the hybrid. When EG-OPV solution was excited at 360 nm, presence of a lesser conjugated species was evident by the blue emission, with maxima around 410 nm and 425 nm (Fig. 6a). This emission is characteristic of less conjugated stilbene derivatives, which would be formed due to cycloaddition reaction.33 Excitation spectra collected at this blue-emission (λmon = 412 nm) showed a maximum at 360 nm corresponding to the absorption of stilbene derivatives, which further confirmed its origin (Fig. 6b). The dimerization reaction can result from the head-to-tail organization of the OPV molecules in the hybrid (as shown in Fig. 3a), which brings the electron-rich (next to trialkoxy benzene) and electron-deficient (next to amide bonds) double bonds close to each other. Additionally, though we do not have a direct proof for a head to tail dimeric species, topological considerations suggest that this is the only possible conformation. The formation of the cyclo-adduct also confirms the π–π interaction between the OPV molecules, which helps the organization of graphene-OPV hybrid sheets into multi-layers. This observation allows us to predict the possibility of using unprecedented aggregation tendency of graphene for bringing photo-labile groups together to procure hybrids with unique properties.
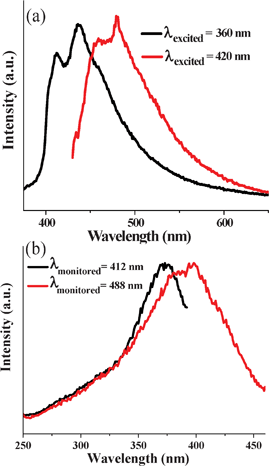 | ||
| Fig. 6 Normalized (a) emission and (b) excitation spectra of EG-OPV hybrids. | ||
Conclusions
In conclusion, we have covalently functionalized graphene with OPV. The conjugation results in exfoliation and electronic communication between the two components. In addition the intermolecular interactions and the perpendicular orientation of the OPV chromophores resulted in the self-organization of graphene in periodic intervals. Furthermore, the photochemical dimerization of the OPVs occurs probably due to topological confinement of these molecules as the graphene conjugates come close, resulting in interdigitation.Experimental section
Graphene oxide (GO) was synthesized by employing the literature procedure and was subjected to thermal exfoliation in a furnace which was preheated to 1050 °C under argon flow to obtain few-layer graphene (EG).34 The graphene sample contained on an average 5 layers, as determined by transmission electron microscopy (TEM) and atomic force microscopy (AFM). Acid functionalized graphene (EG-Acid) was obtained by taking conc. nitric acid (2 ml), conc. sulfuric acid (2 ml) and water (16 ml) and adding the mixture to 50 mg of EG. The mixture was heated in a microwave oven for 10 min followed by overnight incubation in conventional oven at 100 °C. The product obtained was washed with distilled water and centrifuged repeatedly to remove traces of acid. This product was refluxed with excess SOCl2 for 12 h to convert the acid groups in EG-Acid to more reactive acid chloride. The unreacted SOCl2 was removed under vacuum.14 OPV-amine was synthesized according to the reported procedure.35–37EG-OPV hybrid was prepared by the reaction of OPV-amine with EG-COCl in dry N,N-dimethylformamide (DMF) and triethyl amine under reflux conditions for 24 h.28Characterization
Infrared spectra were recorded using a Bruker IFS 66 v/S spectrometer. Raman spectra of samples were recorded with a 633 nm Ar laser using JobinYvon Lab Ram HR spectrometer. Transmission electron microscope (TEM) images were obtained with a JEOL JEM 3010, operating with an accelerating voltage of 300 kV. AFM measurements were carried on Vecco digital instruments, di Innova. Samples for AFM measurements were prepared by spin coating the EG-OPV in chloroform on a silicon substrate. Electronic absorption spectra were recorded on Perkin Elmer Lambda 900 UV-Vis-NIR Spectrometer. UV-Vis spectra were recorded in 1 cm path length quartz cuvette. Photoluminescence (PL) spectra were recorded with a Perkin-Elmer model LS55 luminescence spectrometer. Fluorescence decay was recorded in a time-correlated single-photon-counting spectrometer of Horiba-JobinYvon. 1H NMR was recorded on a Bruker AVANCE-400 MHz spectrometer at room temperature.Acknowledgements
We thank Prof. C. N. R. Rao (FRS) for the scientific discussion and suggestions throughout this work. The authors thank Prof. P. Ramamurthy, National centre for Ultra fast process (NCUFP) Chennai, for providing the facilities for lifetime studies. We are grateful to DST and JNCASR for financial support.References
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS.
- A. K. Geim, Science, 2009, 324, 1530–1534 CrossRef CAS.
- C. N. R. Rao, A. K. Sood, K. S. Subrahmanyam and A. Govindaraj, Angew. Chem., Int. Ed., 2009, 48, 7752–7777 CrossRef CAS.
- S. K. Pati, T. Enoki and C. N. R. Rao ed.Graphene and its fascinating properties, World Scientific, Singapore, 2011 Search PubMed.
- Z. Z. Sun, D. K. James and J. M. Tour, J. Phys. Chem. Lett., 2011, 2, 2425–2432 CrossRef CAS.
- Y. F. Xu, Z. B. Liu, X. L. Zhang, Y. Wang, J. G. Tian, Y. Huang, Y. F. Ma, X. Y. Zhang and Y. S. Chen, Adv. Mater., 2009, 21, 1275 CrossRef CAS.
- Z. B. Liu, Y. F. Xu, X. Y. Zhang, X. L. Zhang, Y. S. Chen and J. G. Tian, J. Phys. Chem. B, 2009, 113, 9681–9686 CrossRef CAS.
- L.-H. Liu and M. Yan, J. Mater. Chem., 2011, 21, 3273 RSC.
- A. Ghosh, K. V. Rao, S. J. George and C. N. R. Rao, Chem.–Eur. J., 2010, 16, 2700–2704 CrossRef CAS.
- H. S. S. Ramakrishna Matte, K. S. Subrahmanyam, K. V. Rao, S. J. George and C. N. R. Rao, Chem. Phys. Lett., 2011, 506, 260–264 CrossRef.
- A. Ghosh, K. V. Rao, R. Voggu and S. J. George, Chem. Phys. Lett., 2010, 488, 198–201 CrossRef CAS.
- N. V. Kozhemyakina, J. M. Englert, G. Yang, E. Spiecker, C. D. Schmidt, F. Hauke and A. Hirsch, Adv. Mater., 2010, 22, 5483–5487 CrossRef CAS.
- A. Wojcik and P. V. Kamat, ACS Nano, 2010, 4, 6697–6706 CrossRef CAS.
- K. S. Subrahmanyam, A. Ghosh, A. Gomathi, A. Govindaraj and C. N. R. Rao, Nanosci. Nanotechnol. Lett., 2009, 1, 28–31 CrossRef CAS.
- S. K. Samanta, K. S. Subrahmanyam, S. Bhattacharya and C. N. R. Rao, Chem.–Eur. J., 2012, 18, 2890–2901 CrossRef CAS.
- Z. X. Zhang, H. L. Huang, X. M. Yang and L. Zang, J. Phys. Chem. Lett., 2011, 2, 2897–2905 CrossRef CAS.
- H. Yang, C. Shan, F. Li, D. Han, Q. Zhang and L. Niu, Chem. Commun., 2009, 3880–3882 RSC.
- C. N. R. Rao and R. Voggu, Mater. Today, 2010, 13, 34–40 CrossRef CAS.
- S. Srinivasan, S. S. Babu, V. K. Praveen and A. Ajayaghosh, Angew. Chem., Int. Ed., 2008, 47, 5746–5749 CrossRef CAS.
- S. K. Samanta, A. Pal, S. Bhattacharya and C. N. R. Rao, J. Mater. Chem., 2010, 20, 6881 RSC.
- S. Srinivasan, V. K. Praveen, R. Philip and A. Ajayaghosh, Angew. Chem. Int. Ed., 2008, 47, 5750–5754 CAS.
- W. R. Collins, W. Lewandowski, E. Schmois, J. Walish and T. M. Swager, Angew. Chem., 2011, 123, 9010–9014 CrossRef.
- J. M. Englert, C. Dotzer, G. Yang, M. Schmid, C. Papp, J. M. Gottfried, H.-P. Steinraick, E. Spiecker, F. Hauke and A. Hirsch, Nat. Chem., 2011, 3, 279–286 CrossRef CAS.
- S. Niyogi, E. Bekyarova, J. Hong, S. Khizroev, C. Berger, W. de Heer and R. C. Haddon, J. Phys. Chem. Lett., 2011, 2, 2487–2498 CrossRef CAS.
- J. Geng and H.-T. Jung, J. Phys. Chem. C, 2010, 114, 8227–8234 CAS.
- Y. Xu, Z. Liu, X. Zhang, Y. Wang, J. Tian, Y. Huang, Y. Ma, X. Zhang and Y. Chen, Adv. Mater., 2009, 21, 1275–1279 CrossRef CAS.
- C. N. R. Rao and A. Govindaraj, Nanotubes and Nanowires, The Royal Society of Chemistry, London, 2011 Search PubMed.
- U. Maitra, A. Jain, S. J. George and C. N. R. Rao, Nanoscale, 2011, 3, 3192–3197 RSC.
- B. Das, R. Voggu, C. S. Rout and C. N. R. Rao, Chem. Commun., 2008, 5155–5157 RSC.
- A. Ajayaghosh and S. J. George, J. Am. Chem. Soc., 2001, 123, 5148–5149 CrossRef CAS.
- C.-H. Lu, H.-H. Yang, C.-L. Zhu, X. Chen and G.-N. Chen, Angew. Chem., Int. Ed., 2009, 48, 4785–4787 CrossRef CAS.
- R. S. Swathi and K. L. Sebastian, J. Chem. Phys., 2009, 130, 086101–086103 CrossRef CAS.
- S. J. George, T. F. A. de Greef, R. Bovee, J. L. J. van Dongen, A. P. H. J. Schenning and E. W. Meijer, Chem.–Asian J., 2009, 4, 910–917 CrossRef CAS.
- K. S. Subrahmanyam, S. R. C. Vivekchand, A. Govindaraj and C. N. R. Rao, J. Mater. Chem., 2008, 18, 1517–1523 RSC.
- A. Ajayaghosh and V. K. Praveen, Acc. Chem. Res., 2007, 40, 644–656 CrossRef CAS.
- F. J. M. Hoeben, M. Wolffs, J. Zhang, S. De Feyter, P. Leclere, A. Schenning and E. W. Meijer, J. Am. Chem. Soc., 2007, 129, 9819–9828 CrossRef CAS.
- D. G. Rodriguez and A. P. H. J. Schenning, Chem. Mater., 2011, 23, 310–325 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: TEM, ESI-Mass Analysis, NMR and additional photophysical studies. See DOI: 10.1039/c2ra20455j |
| This journal is © The Royal Society of Chemistry 2012 |