DOI:
10.1039/C2RA20362F
(Paper)
RSC Adv., 2012,
2, 5676-5682
Electrochemistry and electrocatalysis of myoglobin on carbon coated Fe3O4 nanospindle modified carbon ionic liquid electrode†
Received
28th February 2012
, Accepted 7th April 2012
First published on 13th April 2012
Abstract
In this paper, carbon coated Fe3O4 nanospindles (C@Fe3O4 NSs) were synthesized by partial reduction of monodispersed hematite nanospindles with carbon coatings and applied to investigate the direct electrochemistry of myoglobin (Mb). A novel modified electrode was prepared by applying carbon coated Fe3O4 nanospindles, Mb, ionic liquid (IL), 1-ethyl-3-methylimidazolium ethylsulfate ([EMIM]EtOSO3) and chitosan (CTS) step by step onto the surface of a carbon ionic liquid electrode (CILE) with another ionic liquid, 1-butyl-3-methylimidazolium tetrafluoroborate ([BMIM]BF4), as the binder. UV-Vis absorption and FT-IR spectra results indicated that Mb retained its native structure in the composite film. The electrochemical behavior of the CTS/IL/Mb/C@Fe3O4/CILE was investigated and a pair of well-defined redox peaks appeared, which indicated that the direct electron transfer of Mb was realized with the underlying electrode. The fabricated CTS/IL/Mb/C@Fe3O4/CILE showed good electrocatalytic activity in the reduction of trichloroacetic acid (TCA) with a linear range from 1.0 to 20.0 mM, which showed potential applications for fabricating novel electrochemical biosensors and bioelectronic devices.
1. Introduction
Direct electrochemistry of redox proteins has attracted considerable attention due to the potential applications of the direct electron transfer mechanism between redox proteins and electrodes in biological systems.1,2 However, direct electrochemistry of most redox proteins on conventional electrodes is a great challenge due to the electroactive center being deeply embedded in the protein structure and the unfavorable orientation of the protein on the electrode surface.3,4 Therefore, various kinds of protein film modified electrodes have been devised to enhance the electron transfer rate of the electroactive center of the proteins,5,6 and the film can provide a suitable microenvironment to maintain the native structure of the protein. Many materials and methods have been used in the protein film fabrication, such as inorganic nanomaterials, biopolymers, polyelectrolytes, solvent casting, self-assembly, electrostatic absorption, covalent binding and sol–gel, etc,7–14 which proved to be an efficient route to the realization of protein electrochemistry.
Recently, ionic liquids (ILs) have aroused great interest in the field of electrochemistry due to their unique physicochemical properties such as preferable chemical stability, high viscosity, good solubility, a relatively wide potential window and high ionic conductivity.15,16 ILs can be used as the supporting electrolyte in the electrochemical process, the modifier on the chemically modified electrodes and the binder in the carbon paste electrode.17–19 Wei and Ivaska have reviewed the applications of ILs in the electrochemical sensor.20 Wang et al. have investigated the electrochemistry and electrocatalysis of heme proteins entrapped in agarose hydrogel films with IL as the electrolyte.21 In our work group, IL was also applied to modify the carbon paste electrode for the investigation of the direct electrochemistry of proteins and the electrochemical detection of double-stranded DNA.22–24 Liu et al. have fabricated a carbon nanotube/IL gel modified microelectrode for bioelectrochemistry.25 Gao et al. have investigated the direct electrochemistry of Hb in a hyaluronic acid and IL composite film.26 Maleki et al. developed an N-octylpyridinium hexafluorophosphate based carbon ionic liquid electrode (CILE) for the detection of some electroactive molecules.27,28
In recent years, nanomaterials have been applied in electrochemical biosensors due to their specific physicochemical properties, such as high surface area, excellent biocompatibility and good thermal and chemical stability.29–32 Nanomaterials with special structures have been used to facilitate electron transfer between the electrode and immobilized proteins.33–35 The electrical properties of nanostructured materials can effectively shorten the electron-tunneling distance and provide an electron-mediating function. Fe3O4 is a versatile semiconductor material which has attracted much attention due to its applications in catalysis,36 molecular adsorption,37 biosensors44–46 and energy storage.38 Up until now, various morphologies of Fe3O4, such as nanocrystals, particles, cubes, porous spheres and nanocomposites, have been successfully synthesized by a variety of methods.39–43 In the field of biosensors, Fe3O4 nanoparticles have successfully served as the matrix for the immobilization of biomolecules. It has also been reported for its specific characteristics, including strong superparamagnetism, biocompatibility, lack of toxicity, low cost and environmental benignity.44–46 Cao et al. have reported an electrochemical biosensor based on the electron transfer of heme proteins immobilized on Fe3O4 nanoparticles.47 Yang et al. have also developed a novel amperometric glucose biosensor by immobilizing glucose oxidase on the surface of Fe3O4 nanoparticles.45 Wang et al. have fabricated a novel tyrosinase biosensor based on a Fe3O4 nanoparticle–chitosan nanocomposite and developed it for the detection of phenolic compounds.48 However, to the best of our knowledge, no paper has reported the use of carbon coated Fe3O4 nanospindles for the direct electrochemistry and electrocatalysis of myoglobin. In this paper, carbon coated Fe3O4 nanospindles were successfully synthesized by introducing carbon coatings onto the surface of iron oxide nanospindles, which was used to fabricate an electrochemical Mb biosensor with CILE as the substrate electrode. An ionic liquid of 1-butyl-3-methylimidazolium tetrafluoroborate ([BMIM]BF4) was further used as the binder for the preparation of CILE, and another ionic liquid, 1-ethyl-3-methylimidazolium ethylsulfate ([EMIM]EtOSO3), was used as the modifier for the fabrication of the composite film. The direct electrochemistry of Mb was achieved on the chitosan (CTS), carbon coated Fe3O4 nanospindles (C@Fe3O4 NSs) and IL composite material modified CILE. The Mb modified electrode showed good electrocatalytic activity in the reduction of trichloroacetic acid (TCA), with a wide linear range and good sensitivity.
2. Experimental
2.1. Reagents and apparatus
1-Ethyl-3-methylimidazolium ethylsulfate ([EMIM]EtOSO3) and 1-butyl-3-methyl-imidazolium tetrafluoroborate ([BMIM]BF4) were purchased from Hangzhou Kemer Chemical Limited Company, China. Horse heart myoglobin (Mb, MW 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800, Sigma), chitosan (CTS, Shanghai Ruji biological technology Co. LTD, China), ferric chloride hexahydrate (FeCl3·6H2O, Tianjin Chemical Reagent Limited Company, China), graphite powder (average particle size 30 μm, Shanghai Colloid Chemical Company, China) and trichloroacetic acid (TCA, Tianjin Kemiou Chemical Limited Company) were used as received. 0.1 mol L−1 phosphate buffer solutions (PBS) with pH 7.0 were used as the supporting electrolyte. All the other chemicals used were of analytical reagent grade and deionized water was used for all aqueous solutions.
800, Sigma), chitosan (CTS, Shanghai Ruji biological technology Co. LTD, China), ferric chloride hexahydrate (FeCl3·6H2O, Tianjin Chemical Reagent Limited Company, China), graphite powder (average particle size 30 μm, Shanghai Colloid Chemical Company, China) and trichloroacetic acid (TCA, Tianjin Kemiou Chemical Limited Company) were used as received. 0.1 mol L−1 phosphate buffer solutions (PBS) with pH 7.0 were used as the supporting electrolyte. All the other chemicals used were of analytical reagent grade and deionized water was used for all aqueous solutions.
All electrochemical measurements were performed on a CHI 440A electrochemical workstation (Shanghai CH Instrument, China) with a conventional three-electrode system composed of a Mb modified electrode as the working electrode, a platinum wire as the auxiliary electrode and a saturated calomel electrode (SCE) as the reference electrode. Ultraviolet-visible (UV-Vis) absorption spectra and Fourier transform infrared (FT-IR) spectra were recorded on a Cary 50 probe spectrophotometer (Varian Company, Australia) and a Tensor 27 FT-IR spectrophotometer (Bruker Company, Germany), respectively. Scanning electron microscopy (SEM) was performed on a JSM-6700F scanning electron microscope (Japan Electron Company, Japan). The X-ray powder diffraction (XRD) spectra were taken on a Japan RigakuD/Maxr-A X-ray diffractometer equipped with graphite monochromatized high-intensity Cu-Kα radiation (λ = 1.54178 Å).
2.2. Synthesis of carbon coated Fe3O4 nanospindles
To prepare the Fe2O3 nanospindles, an 80 mL mix solution containing 0.541 g FeCl3·6H2O and 3.0 mg NaH2PO4·2H2O in deionized water was transferred to a Teflon autoclave with a capacity of 100 mL, and then heated at 100 °C in an electric oven for 48 h. After the autoclave had cooled naturally to room temperature, the monodispersed ferric oxide nanospindles were centrifuged and washed with deionized water and ethanol several times and dried in an electric oven at 60 °C.
The carbon coated Fe3O4 nanospindles were prepared using a hydrothermal approach. In brief, a 150 mg ferric oxide nanospindle was dispersed in 5 mL deionized water by ultrasonication to form a suspension. 500 mg glucose (C6H12O6·H2O) was dissolved in another 15 mL deionized water, and the glucose solution and another 10 mL ethanol were added to the former suspension under gentle stirring. The resulting suspension was transferred to a 40 mL Teflon autoclave, which was then heated at 180 °C in an electric oven for 12 h. The carbon coated iron oxide nanospindles were harvested by centrifugation and washed with deionized water and ethanol several times, then dried at 60 °C in an oven and further annealed in N2 gas at 600 °C for 4 h.
2.3. Preparation of modified electrodes
A CILE was prepared by mixing 0.10 mL [BMIM]BF4, 0.90 mL liquid paraffin and 3.2 g graphite powder together in a mortar for 20 min. A portion of the resulting homogeneous paste was packed firmly into a glass tube (Φ = 4 mm) and a copper wire was inserted through the opposite end to establish an electrical contact. After carefully polishing the surface of the CILE with a piece of weighing paper, the following procedure was performed to fabricate the Mb modified electrode. 10.0 μL of a 2.0 g L−1 C@Fe3O4 NSs suspension solution (dispersed in pH 7.0 PBS) was cast on the surface of the newly prepared CILE and left to dry at room temperature. Then 5.0 μL of a 5.0 mg mL−1 Mb solution was pipetted on the electrode surface and left to allow the water to evaporate gradually. Finally, 5 μL [EMIM]EtOSO3 (6.0% in pH 7.0 PBS) and 7.0 μL CTS (in 1.0% HAc solution) were modified step by step on the surface of the Mb/C@Fe3O4/CILE to form a stable film. The fabricated electrode was denoted as CTS/IL/Mb/C@Fe3O4/CILE and stored at 4 °C when not in use. Other modified electrodes were prepared using a similar procedure for comparison.
2.4. Procedure
Electrochemical measurements were performed in a 10 mL electrochemical cell containing 0.1 mol L−1 PBS. The working buffer solutions were deoxygenated by bubbling highly purified nitrogen thoroughly for 30 min just before the electrochemical experiments and maintained in a nitrogen atmosphere during the experiments. UV-Vis spectroscopic experiments were carried out with a solution mixture of certain concentrations of Mb, CTS, [EMIM]EtOSO3 and C@Fe3O4 NSs with deionized water. The CTS/IL/Mb/C@Fe3O4 film assembled on a glass slide was used for FT-IR measurements.
3. Results and discussion
3.1. Characteristics of carbon coated Fe3O4 nanospindles
The carbon coated iron oxide nanospindles were prepared through brush-fire reduction of hematite spindles with carbon coatings.49 First, the monodispersed ferric oxide nanospindles were synthesized by forced hydrolysis of ferric chloride solutions at certain concentrations with small amounts of phosphate ions at the appropriate temperature. The panoramic morphologies of the as-synthesized ferric oxide nanospindles were recorded by scanning electron microscopy (SEM) with the image results at different magnifications shown in Fig. 1A and B. It is obvious that all particles are uniform spindles alike and have an average particle length of about 450 nm with a diameter of about 100 nm. Then the carbon layers were coated on the outer surface of the ferric oxide nanospindles by pyrolysis of glucose under hydrothermal conditions and carbonation. Fig. 1C shows the SEM micrograph of the carbon coated iron oxide nanospindles. The results indicate that there are uniform and continuous carbon coating layers and no significant change in morphology after the carbon coatings. Energy Dispersive X-Ray Spectroscopy (EDX) measurement results further reveal that the carbon was successfully coated on Fe3O4 (Fig. S1, ESI†). To elucidate the structure of the as-obtained products, X-ray powder diffraction (XRD) experiments were carried out. Fig. 1D shows the typical XRD pattern of carbon coated Fe3O4 nanospindles, which can be readily indexed to cubic Fe3O4 (JCPDS card. 74-748, a = 8.39410 Å). But careful observation can find that there are no obvious XRD peaks corresponding to graphite in the XRD pattern, which indicates that the carbon coatings with enhanced conductivity are not well crystallized.
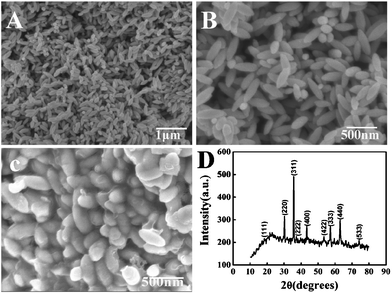 |
| | Fig. 1 (A) and (B) SEM image of Fe2O3 nanospindles at different magnifications; (C) SEM images of carbon coated Fe3O4 nanospindles; (D) XRD pattern of the sample, which is indexed to Fe3O4 (JCPDS no. 74-748). | |
3.2. Spectroscopic results
FT-IR spectroscopy is used to probe into the secondary structure of proteins. The amide I and amide II bands of the protein can exhibit detailed information on the polypeptide chain. The amide I band at 1700–1600 cm−1 results from the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration of the peptide linkage in the backbone of the protein. The amide II band at 1600–1500 cm−1 is caused by the combination of N–H in-plane bending and the C–N stretching vibration of the peptide groups.50 As shown in Fig. 2A, the FT-IR spectra of the amide I and II bands of native Mb (1650 and 1569 cm−1) and those in the composite film (1649 and 1572 cm−1) are located at almost the same position, indicating that the native structure of Mb was not changed in the CTS/IL/C@Fe3O4 film.
O stretching vibration of the peptide linkage in the backbone of the protein. The amide II band at 1600–1500 cm−1 is caused by the combination of N–H in-plane bending and the C–N stretching vibration of the peptide groups.50 As shown in Fig. 2A, the FT-IR spectra of the amide I and II bands of native Mb (1650 and 1569 cm−1) and those in the composite film (1649 and 1572 cm−1) are located at almost the same position, indicating that the native structure of Mb was not changed in the CTS/IL/C@Fe3O4 film.
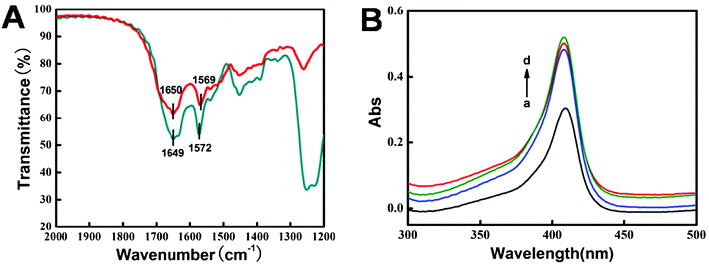 |
| | Fig. 2 (A) FT-IR spectra of Mb (red) and CTS/Mb/IL/C@Fe3O4 film (dark cyan). (B) UV-Vis absorption spectra of (a) Mb, (b) CTS/Mb, (c) CTS/Mb/IL and (d) CTS/Mb/IL/C@Fe3O4 in pH 7.0 PBS. | |
In the UV-Vis absorption spectrum the Soret absorption band from the four iron heme groups of heme proteins may provide information on the conformational integrity of the proteins and the possible denaturation or conformational change of the heme group region.51Fig. 2B shows the UV-Vis spectra of Mb and its mixture with composite materials. The Soret band of Mb molecules in pH 7.0 PBS appears at 408.8 nm (curve a). After Mb was mixed with the CTS, CTS/IL and CTS/IL/C@Fe3O4 (in pH 7.0 PBS), the Soret band appeared at 407.8 nm (curve b–d), indicating that Mb retained its native structure and no significant denaturation had occurred to the protein after being mixed with the composite materials. IL is a biocompatible green solvent with the ability to retain the native structure of the enzyme. CTS is a commonly used biomaterial in electrochemical biosensors with good film-forming ability and biocompatibility, and C@Fe3O4 NSs also possess excellent biocompatibility. So the result also showed that Mb retained its native structure in the composite film.
3.3. Electrochemical characteristics of the modified electrodes
The [Fe(CN)6]3-/4- redox couple is often selected as a probe to evaluate the performance of different electrodes, and the electrochemical response from the Fe3+/Fe2+ redox couple can reflect the conductivity of the electrode interface. Cyclic voltammograms of different modified electrodes are shown in Fig. 3A. A pair of well-defined redox peaks was observed at the bare CILE (curve a) with a peak-to-peak separation (ΔEp) of 0.093 V (vs. SCE), indicating good reversibility of the CILE. On the CTS/Mb/CILE the redox peak current decreased observably (curve b), which indicated that Mb was successfully immobilized on the electrode and the presence of the CTS/Mb composite film hindered the electron transfer rate of [Fe(CN)6]3−/4−. However, on the CTs/Mb/C@Fe3O4/CILE, the electrochemical response was bigger than that on the bare CILE (curve c), which indicated that the presence of C@Fe3O4 NSs could effectively enhance the conductivity of the electrode surface. The C@Fe3O4 NSs used in this experiment are spindle-like with a high specific surface, which can absorb more proteins and provide a three-dimensional structure for protein immobilization. On the CTS/Mb/IL/CILE the redox peak currents also increased (curve d), which could be attributed to the high ionic conductivity of IL. On the CTS/IL/Mb/C@Fe3O4/CILE, the redox peak current increased greatly (curve e) and was bigger than that of the CTS/Mb/C@Fe3O4/CILE and the CTS/IL/Mb/CILE, indicating that the synergistic effect of the C@Fe3O4 NSs and the IL in the composite film enhanced the electron transfer rate of the redox probe.
![(A) Cyclic voltammograms of (a) CILE, (b) CTS/Mb/CILE, (c) CTS/Mb/C@Fe3O4/CILE, (d) CTS/IL/Mb/CILE and (e) CTS/IL/Mb/C@Fe3O4/CILE in a solution mixture of 1.0 mmol L−1 [Fe(CN)6]3-/4- and 0.1 mol L−1 KCl with a scan rate of 100 mV s−1. (B) EIS for (a) CILE, (b) CTS/IL/Mb/CILE, (c) CTS/IL/Mb/Fe2O3/CILE and (d) CTS/IL/Mb/C@Fe3O4/CILE in the presence of 5.0 mmol L−1 [Fe(CN)6] 3−/4− and 0.1 mol L−1 KCl, with the frequencies swept from 105 to 0.1 Hz.](/image/article/2012/RA/c2ra20362f/c2ra20362f-f3.gif) |
| | Fig. 3 (A) Cyclic voltammograms of (a) CILE, (b) CTS/Mb/CILE, (c) CTS/Mb/C@Fe3O4/CILE, (d) CTS/IL/Mb/CILE and (e) CTS/IL/Mb/C@Fe3O4/CILE in a solution mixture of 1.0 mmol L−1 [Fe(CN)6]3-/4- and 0.1 mol L−1 KCl with a scan rate of 100 mV s−1. (B) EIS for (a) CILE, (b) CTS/IL/Mb/CILE, (c) CTS/IL/Mb/Fe2O3/CILE and (d) CTS/IL/Mb/C@Fe3O4/CILE in the presence of 5.0 mmol L−1 [Fe(CN)6] 3−/4− and 0.1 mol L−1 KCl, with the frequencies swept from 105 to 0.1 Hz. | |
Electrochemical impedance spectroscopy (EIS) was performed in a solution mixture of 5.0 mmol L−1 [Fe(CN)6]3−/4− and 0.1 mol L−1 KCl with the frequency range swept from 105 to 0.1 Hz. The Nyquist plots of different modified electrodes are shown in Fig. 3B and the semicircle diameter equalled the electron transfer resistance (Ret). On the CILE the Ret value was obtained as 40.5 Ω (curve a). However, on the CTS/IL/Mb/CILE, the Ret values decreased to 24.1 Ω (curve b), which was attributed to the presence of a highly conductive IL in the composite film. Subsequently, when the C@Fe3O4 NSs were immobilized on the CTS/IL/Mb/CILE, the Ret value decreased dramatically, nearly to zero (curve d), which was much smaller than that of CTS/IL/Mb/Fe2O3/CILE (curve c), indicating its much improved electroactivity. The high electrocatalytic activity is most likely ascribed to good conductivity from the carbon deposited on Fe3O4. The results also indicated that the synergistic effect of C@Fe3O4 NSs and IL in the composite film enhanced the electron transfer rate of the redox probe.
3.4. Direct electrochemistry of CTS/IL/Mb/C@Fe3O4/CILE
Direct electrochemical behaviors of different modified electrodes were carefully investigated in pH 7.0 PBS by cyclic voltammetry and the results are shown in Fig. 4A. No electrochemical responses are observed at bare CILE (curve a), indicating no electroactive substances exist on the electrode surface. On the CTS/Mb/CILE, a pair of small and unsymmetric redox peaks appeared (curve b) and the reduction peak current was greater than the oxidation peak current, which indicated that the direct electron transfer rate between Mb and CILE was very slow. After the addition of C@Fe3O4 NSs in the Mb matrix, the redox peak current (curve c) increased more gradually than that of the CTS/Mb/CILE, with the peak shape having more symmetry, indicating that the presence of C@Fe3O4 NSs can facilitate the electron transfer rate. The C@Fe3O4 NSs used in this experiment display attractive structural, mechanical and electronic properties with increased surface area, which can play a role as an efficient electron-conducting tunnel and facilitate the electron transfer between Mb and the underlying electrode. However, on the CTS/IL/Mb/CILE (curve d) the redox peak current increased greatly, which was attributed to the addition of an IL with higher ionic conductivity. An IL on the surface of a CILE can provide a suitable interface to promote the electron transfer reaction of a protein. Then, IL has good biocompatibility without the denaturation of protein in its matrix. On the CTS/IL/Mb/C@Fe3O4/CILE (curve e), the redox peak currents further increased with the redox peak potentials as −0.168 V (Epa) and −0.298 V (Epc), which indicated that the synergistic augmentation of C@Fe3O4 NSs and IL greatly promoted the electron transfer rate. Then the formal potential (E0'), which is calculated from the midpoint of Epa and Epc, was estimated as −0.233 V (vs. SCE). The results demonstrated that a quasi-reversible electrochemical reaction of electroactive heme Fe(III)–Fe(II) redox couples was realized on the electrode surface, and the composite film on the CILE can greatly promote the efficient direct electron transfer of Mb.
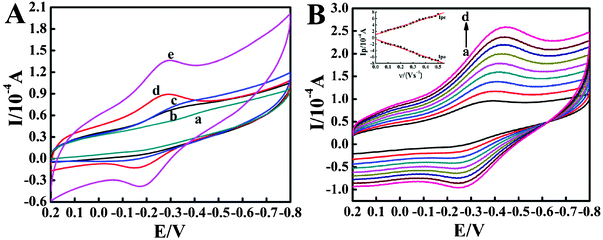 |
| | Fig. 4 (A) Cyclic voltammograms of (a) CILE, (b) CTS/Mb/CILE, (c) CTS/Mb/C@Fe3O4/CILE, (d) CTS/IL/Mb/CILE and (e) CTS/IL/Mb/C@Fe3O4/CILE in pH 7.0 PBS at a scan rate of 100 mV s−1. (B) Cyclic voltammograms of the CTS/IL/Mb/C@Fe3O4/CILE at different scan rates (from a to j: 100, 200, 300, 400, 500, 600, 700, 800, 900, 1000 mV s−1) in pH 7.0 PBS. The inset shows the plot of cathodic and anodic peak current with scan rate (υ). | |
The cyclic voltammetric responses of the CTS/IL/Mb/C@Fe3O4/CILE with different scan rates in pH 7.0 PBS were further recorded, with the results shown in Fig. 4B. It can be seen that a pair of roughly symmetric redox peaks appeared in the scan rate range from 100 to 1000 mV s−1 and the redox peak currents increased gradually with increasing scan rate. The relationships between the redox peak currents and the scan rate were calculated and two linear regression equations were obtained as Ipc (μA) = 124.80 υ (V s−1) + 9.967 (n = 19, γ = 0.997) and Ipa (μA) = −138.01 υ (V s−1) − 4.194 (n = 19, γ = 0.996), respectively. The results indicated that the electrochemical behavior of the immobilized Mb on the modified electrode was a surface-controlled thin-layer process, in which the electroactive Mb Fe(III) in the film was reduced to Mb Fe(II) on the forward cyclic voltammetric scan and then fully reoxidized to Fe(III) on the reversed scan. For the thin layer electrochemical behavior, the total amount of charge (Q) passing through the electrode for the reduction or the oxidation of the electroactive species can be calculated by integration of the cyclic voltammetric peaks of the protein film electrode. Based on the following equation: Q = nAFΓ*, the surface concentration of electroactive Mb in the film was calculated with an average value of 2.237 × 10−9 mol cm−2, while the total amount of Mb on the electrode surface was 9.833 × 10−9 mol cm−2, so 22.7% of the total Mb molecules on the electrode surface took part in the electrochemical reaction. The results demonstrated that the composite film could greatly retain the activity of Mb, and the presence of C@Fe3O4 NSs provides a three-dimensional network for Mb immobilization, then several layers of Mb in the composite film participated in the electron transfer.
With the increase in scan rate, the peak-to-peak separation (ΔEp) also increased gradually, so the electrochemical parameters of the electrode reaction were calculated according to the model of Laviron's equations:52
| | 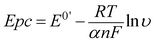 | (1) |
| | 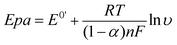 | (2) |
| | 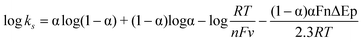 | (3) |
where
α is the electron transfer coefficient,
ks is the apparent heterogeneous
electron transfer rate constant,
R is the gas constant and
T is the absolute temperature. Two straight lines between E
p and ln
υ were obtained with the equations as E
pa(V) = 0.046 ln
υ − 0.398 (n = 19,
γ = 0.994) and E
pc(V) = − 0.062 ln
υ − 0.11 (n = 19,
γ = 0.995), respectively. Then, according to the above equations, the charge transfer coefficient (
α), the number of electrons transferred (
n) and the electrode reaction rate constant (
ks) were calculated as 0.427, 0.966 and 0.610 s
−1, respectively. The value of
ks was much larger than those of the Mb immobilized on other matrices,
13,53,54 indicating that CTS/IL/Mb/C@Fe
3O
4/CILE provided a suitable microenvironment for the increase in the electron transfer rate of Mb.
3.5. Electrocatalytic activity
The electrocatalytic activity of the fabricated CTS/IL/Mb/C@Fe3O4/CILE on the reduction of TCA was further investigated by cyclic voltammetry. Fig. 5 shows the cyclic voltammograms of the Mb modified electrode in 0.1 mol L−1 PBS containing different amounts of TCA. A dramatic increase in the reduction peak current at −0.320 V was observed with the disappearance of the oxidation peak, due to the catalytic reduction of TCA by Mb molecules on the modified electrode. Based on the references,55 the electrocatalytic mechanism is proposed as follows:
| Mb (FeIII) + e− → Mb (FeII) |
| Mb (FeII) + CCl3COOH + H+ → Mb (FeIII) + HCCl2COOH + Cl− |
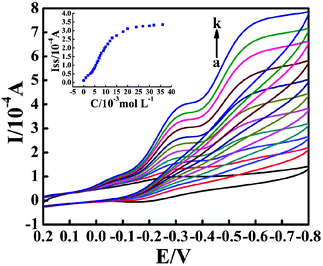 |
| | Fig. 5 Cyclic voltammograms of the CTS/IL/Mb/C@Fe3O4/CILE in pH 7.0 PBS containing different concentrations of TCA (from a to k: 0, 2.0, 3.0, 4.0, 5.0, 6.0, 7.0, 8.0, 10.0, 12.0, 18.0 mmol L−1) with a scan rate of 100 mV s−1. The inset shows the relationship of the catalytic reduction peak current with TCA concentration. | |
The catalytic reductive peak current increased linearly with TCA concentration in the range 1.0 to 20.0 mmol L−1, with the linear regression equation as Iss (μA) = 20.680C (mmol L−1) − 5.764 (n = 18, γ = 0.993), and the detection limit as 0.333 mmol L−1 (3σ). When the TCA concentration was more than 20.0 mmol L−1, the current response turned to level off, which indicated a typical Michaelis–Menten kinetic mechanism. By exploring the slope and intercept of the plot of the reciprocals of the steady-state current versus TCA concentration, the apparent Michaelis–Mentan constant (KMapp) was further calculated as 1.390 mmol L−1, which was smaller than that of some previously reported values.56–58 The smaller value indicates that immobilized Mb molecules on the modified electrode possess higher enzymatic activity due to the higher affinity of Mb molecules to TCA.
3.6. Reproducibility and stability of the biosensor
The stability of CTS/IL/Mb/C@Fe3O4/CILE was evaluated by examining the cyclic voltammetric peak currents after continuous scanning for 50 cycles. Nearly no decrease in the voltammetric response appeared and 98.3% of the initial current response was retained, indicating that the CTS/IL/Mb/C@Fe3O4/CILE was stable in the buffer solution. The Mb modified electrode was stored at 4 °C in a refrigerator when not in use, and 96.8% of the initial current response was retained after 2 weeks of storage. After 1 month of testing, the redox peak current response decreased by about 9.8%. The relatively good stability of the Mb electrode can be attributed to the biocompatibility of the CTS/IL/C@Fe3O4 composite material, which can prevent leakage of the proteins and retain their secondary structure. Six Mb modified electrodes prepared by the same procedure gave a relative standard deviation (RSD) value of 3.4% for the determination of 6.0 mmol L−1 TCA, which indicates that the modified electrode has good repeatability.
4. Conclusion
In this work, a new nanocomposite material containing C@Fe3O4 nanospindles, CTS and ionic liquid [BMIM]EtOSO3 was prepared and further applied to the entrapment of Mb on the surface of CILE using a layer-by-layer casting method, to get a new kind of electrochemical Mb biosensor. UV-Vis and FT-IR spectroscopic results indicated that Mb retained its native structure in the composite film. The resulting CTS/IL/Mb/C@Fe3O4/CILE exhibited good electrochemical behavior with a well-defined quasi-reversible redox couple of Mb FeIII–FeII appearing in pH 7.0 PBS, indicating that a direct electron transfer between Mb with the underlying electrode was realized. Due to the synergistic effects of the different materials, including the high conductivity of IL, the biocompatibility and film forming ability of CTS and the specific characteristics of C@Fe3O4 NSs, the fabricated bioelectrode shows a good catalytic ability in the reduction of TCA, with a lower detection limit and a wide dynamic range. The results reveal the potential application of C@Fe3O4 nanospindles in third generation electrochemical biosensors.
Acknowledgements
We are grateful for the financial support of National Natural Science Foundation of China (No. 50802032; No. 21075071; No. 20973070) and the Key Laboratory of Analytical Chemistry for Biology and Medicine (Wuhan University) of the Ministry of Education (ACBM2010006).
References
- F. A. Armstrong, H. A. O. Hill and N. J. Walton, Acc. Chem. Res., 1988, 21, 407 CrossRef CAS.
- F. W. Scheller, N. Bistolas, S. Q. Liu, M. Janchem, M. Katterle and U. Wollenberger, Adv. Colloid Interface Sci., 2005, 116, 111 CrossRef CAS.
- J. J. Feng, J. J. Xu and H. Y. Chen, J. Electroanal. Chem., 2005, 585, 44 CrossRef CAS.
- X. Y. He and L. Zhu, Electrochem. Commun., 2006, 8, 615 CrossRef CAS.
- Z. H. Dai, H. X. Ju and H. Y. Chen, Electroanalysis, 2005, 17, 862 CrossRef CAS.
- F. Zhao, X. Wu, M. K. Wang, Y. Liu, L. X. Gao and S. J. Dong, Anal. Chem., 2004, 76, 4960 CrossRef CAS.
- H. Sun, H. Y. Ma and N. F. Hu, Bioelectrochem. Bioenerg., 1999, 49, 1 CrossRef CAS.
- Q. L. Wang, G. X. Lu and B. Yang, Langmuir, 2004, 20, 1342 CrossRef CAS.
- L. D. Zhu, C. A. Tian, J. L. Zhai and R. L. Yang, Sens. Actuators, B, 2007, 125, 254 CrossRef.
- H. Y. Liu and N. F. Hu, Anal. Chim. Acta, 2003, 481, 91 CrossRef CAS.
- E. Topoglidis, C. J. Campbell and A. E. G. Cass, Langmuir, 2001, 17, 7899 CrossRef CAS.
- X. J. Liu, Y. X. Huang, L. B. Shang, X. Y. Wang, H. Xiao and G. X. Li, Bioelectrochemistry, 2006, 68, 98 CrossRef CAS.
- C. Y. Liu and J. M. Hu, Biosens. Bioelectron., 2009, 24, 2149 CrossRef CAS.
- X. L. Zhu, I. Yuri, X. Gan, I. Suzuki and G. X. Li, Biosens. Bioelectron., 2007, 22, 1600 CrossRef CAS.
- M. C. Buzzeo, R. G. Evans and R. G. Compton, ChemPhysChem, 2004, 5, 1106 CrossRef CAS.
- M. Galinski, A. Lewandowski and I. Stepniak, Electrochim. Acta, 2006, 51, 5567 CrossRef CAS.
- H. Xu, H. Y. Xiong, Q. X. Zeng, L. Jia, Y. Wang and S. F. Wang, Electrochem. Commun., 2009, 11, 286 CrossRef CAS.
- Z. H. Zhu, L. N. Qu, Q. J. Niu, Y. Zeng, W. Sun and X. T. Huang, Biosens. Bioelectron., 2011, 26, 2119 CrossRef CAS.
- C. X. Guo, Z. S. Lu, Y. Lei and C. M. Li, Electrochem. Commun., 2010, 12, 1237 CrossRef CAS.
- D. Wei and A. Ivaska, Anal. Chim. Acta, 2008, 607, 126 CrossRef CAS.
- S. F. Wang, T. Chen, Z. L. Zhang, D. W. Pang and K. Y. Wong, Electrochem. Commun., 2007, 9, 1709 CrossRef CAS.
- Z. H. Zhu, X. Li, Y. Wang, Y. Zeng, W. Sun and X. T. Huang, Anal. Chim. Acta, 2010, 670, 51 CrossRef CAS.
- Z. H. Zhu, X. Li, Y. Zeng and W. Sun, Biosens. Bioelectron., 2010, 25, 2313 CrossRef CAS.
- Z. H. Zhu, X. Li, Y. Zeng, W. Sun, W. M. Zhu and X. T. Huang, J. Phys. Chem. C, 2011, 115, 12547 CAS.
- Y. Liu, X. Q. Zou and S. J. Dong, Electrochem. Commun., 2006, 8, 1429 CrossRef CAS.
- R. F. Gao, X. D. Shang, G. J. Qiao and J. B. Zheng, Electroanalysis, 2008, 20, 2537 CrossRef CAS.
- N. Maleki, A. Safavi, F. Sedaghati and F. Tajabadi, Anal. Biochem., 2007, 369, 149 CrossRef CAS.
- A. Safavi, N. Maleki, O. Moradlou and F. Tajabadi, Anal. Biochem., 2006, 359, 224 CrossRef CAS.
- M. Arben, Microchim. Acta, 2006, 152, 157 CrossRef.
- P. Martin, S. Samuel, I. Izumi and J. Tang, Sens. Actuators, B, 2007, 123, 1195 CrossRef.
- Y. H. Xiao and C. M. Li, Electroanalysis, 2008, 20, 648 CrossRef CAS.
- G. D. Liu, J. Wang, H. Wu, Y. Y. Lin and Y. H. Lin, Electroanalysis, 2007, 19, 777 CrossRef CAS.
- C. X. Guo, F. P. Hua, C. M. Li and P. K. Shen, Biosens. Bioelectron., 2008, 24, 819 CrossRef CAS.
- S. J. Bao, C. X. Guo and C. M. Li, RSC Adv., 2012, 2, 1014 RSC.
- J. P. Liu, C. X. Guo, C. M. Li, Y. Y. Li, Q. B. Chi, X. T. Huang, L. Liao and T. Yu, Electrochem. Commun., 2009, 11, 202 CrossRef CAS.
- M. Y. Zhu and G. W. Diao, J. Phys. Chem. C, 2011, 115, 18923 CAS.
- Z. H. Ai, K. J. Deng, Q. F. Wan, L. Z. Zhang and S. C. Lee, J. Phys. Chem. C, 2010, 114, 6237 CAS.
- Z. M. Cui, L. Y. Jiang, W. G. Song and Y. G. Guo, Chem. Mater., 2009, 21, 1162 CrossRef CAS.
- G. F. Zou, K. Xiong, C. L. Jiang, H. Li, T. W. Li, J. Du and Y. T. Qian, J. Phys. Chem. B, 2005, 109, 18356 CrossRef CAS.
- Y. F. Zhu, W. R. Zhao, H. R. Chen and J. L. Shi, J. Phys. Chem. C, 2007, 111, 5281 CAS.
- X. A. Li, B. Zhang, C. H. Ju, X. J. Han, Y. C. Du and P. Xu, J. Phys. Chem. C, 2011, 115, 12350 CAS.
- W. W. Wang and J. L. Yao, J. Phys. Chem. C, 2009, 113, 3070 CAS.
- H. Yu, M. Chen, P. M. Rice, S. X. Wang, R. L. White and S. H. Sun, Nano Lett., 2005, 5, 379 CrossRef CAS.
- H. P. Peng, R. P. Liang and J. D. Qiu, Biosens. Bioelectron., 2011, 26, 3005 CrossRef CAS.
- L. Q. Yang, X. L. Ren, F. Q. Tang and L. Zhang, Biosens. Bioelectron., 2009, 25, 889 CrossRef CAS.
- X. C. Tan, J. L. Zhang, S. W. Tan, D. D. Zhao, Z. W. Huang, M. Yan and Z. Y. Huang, Electroanalysis, 2009, 21, 1514 CrossRef CAS.
- D. Cao, P. He and N. Hu, Analyst, 2003, 128, 1268 RSC.
- S. F. Wang, Y. M. Tan, D. M. Zhao and G. D. Liu, Biosens. Bioelectron., 2008, 23, 1781 CrossRef CAS.
- W. M. Zhang, X. L. Wu, J. S. Hu, Y. G. Guo and L. J. Wan, Adv. Funct. Mater., 2008, 18, 3941 CrossRef CAS.
- J. K. Kauppinen, D. J. Moffatt, H. H. Mantsch and D. G. Cameron, Appl. Spectrosc., 1981, 35, 271 CrossRef CAS.
- J. F. Rusling and A. E. F. Nassar, J. Am. Chem. Soc., 1993, 115, 11891 CrossRef CAS.
- E. Laviron, J. Electroanal. Chem., 1979, 101, 19 CrossRef CAS.
- A. B. Moghaddam, M. R. Ganjali, R. Dinarvand, S. Ahadi and A. A. Saboury, Biophys. Chem., 2008, 134, 25 CrossRef CAS.
- X. Q. Li, R. J. Zhao, Y. Wang, X. Y. Sun, W. Sun, C. Z. Zhao and K. Jiao, Electrochim. Acta, 2010, 55, 2173–2178 CrossRef CAS.
- C. H. Fan, Y. Zhuang, G. X. Li and J. Q. Zhu, Electroanalysis, 2000, 12, 1156 CrossRef CAS.
- W. Sun, Y. Wang, X. Q. Li, J. Wu, T. R. Zhan and K. Jiao, Electroanalysis, 2009, 21, 2454 CAS.
- W. Sun, X. Q. Li, Y. Wang, R. J. Zhao and K. Jiao, Electrochim. Acta, 2009, 54, 4141 CrossRef CAS.
- H. Y. Ma, N. F. Hu and J. F. Rusling, Langmuir, 2000, 16, 4969 CrossRef CAS.
Footnotes |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/c2ra20362f/ |
| ‡ Equal contributions to this work. |
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.