Synthesis and characterization of peroxotungsten(VI) complexes bound to water soluble macromolecules and their interaction with acid and alkaline phosphatases
Siva Prasad
Das
,
Seshadri Reddy
Ankireddy
,
Jeena Jyoti
Boruah
and
Nashreen S.
Islam
*
Department of Chemical Sciences, Tezpur University, Tezpur 784028, Assam, India. E-mail: nsi@tezu.ernet.in; nashreen.islam@rediffmail.com; Fax: +91-3712-267006; Tel: +91-3712-267007; +91-9435380222
First published on 1st June 2012
Abstract
A set of peroxotungsten(VI) complexes in a macroligand environment has been prepared and characterized by elemental analysis (CHN and energy dispersive X-ray spectroscopy), spectral studies (UV–vis, IR, 13C NMR) and thermal gravimetric analysis (TGA), as well as SEM studies. The compounds were obtained by anchoring of peroxotungsten species to the pendant functional groups of water soluble polymers, such as poly(vinyl sulfonate) (PVS), poly(acrylate) (PA), poly(methylacrylate) (PMA) and poly(acrylamide) (PAm). The stability of the compounds in solutions of pH values ranging between 1.2 to 8.0 has been ascertained. The polymeric compounds, as well as a pair of previously reported monomeric and dinuclear pW complexes, were screened for their effect on two different membrane bound phosphatases viz., wheat thylakoid membrane acid phosphatase (ACP) and rabbit intestine alkaline phosphatase (ALP). Each of the tested complexes behaved as active inhibitors of the enzymatic function of the model enzymes. The two classes of enzymes exhibited significantly different sensitivity towards the inhibitors. The IC50 and Ki values were more than 50 orders of magnitude lower for ACP than for ALP, which shows the greater affinity of the complexes for the enzyme binding site of ACP compared to ALP. The kinetic data enabled us to group the complexes into two classes on the basis of their mechanistic preferences. The group comprised of polymeric pW complexes behave as classic non-competitive inhibitors of ACP and ALP, while the free heteroligand pW compounds show mixed inhibition, combining competitive and non-competitive pathways.
Introduction
The current interest in tungsten compounds has, to a great extent, been fuelled by the fascinating finding that tungstates and peroxotungstates (pW), like vanadate and peroxovanadates (pV), are capable of mimicking the bioeffects of insulin in rat adipocytes.1 The normoglycemic properties of tungstates have been extensively investigated.2 Moreover, compounds of tungsten have been reported to exhibit antiviral3 and anti-obesity activity.4 It has been demonstrated by Claret et al. that the anti-obesity effects of tungstate are expressed without any adverse effects, such as gastrointestinal discomfort, which is the main undesirable side effect of vanadate.4 These exciting observations highlighted this group as promising new therapeutic agents.2bThe importance of enzyme inhibition as a mode of action for inorganic drugs has been increasingly recognized in recent years.5 It has been suggested that the strong inhibitory action of tungstate on glucose-6-phosphatase hydrolysis may be an important part of its insulin-like effect.2c Foster and co-workers reported that tungstate acts as one of the most potent competitive inhibitors of multifunctional glucose-6-phosphatase hydrolysis known.2c A definite correlation was also found between the abilities of vanadate and pV, as well as tungstate and pW, to inhibit protein phosphatases and their in vivo insulin mimetic activities,6 although the exact mechanism of action is yet to be unravelled. Peroxovanadates have proved to be more potent agents than vanadates in different in vitro studies.1a,7 However, most of the synthetic pV compounds suffer from the disadvantages of being hydrolytically unstable or toxic,2b,6b,8 limiting their utility as therapeutic agents. It is therefore intriguing to note that, despite the knowledge that peroxotungstates display superior bio-relevant properties, such as a higher hydrolytic stability,1a bioavailability and low toxicity compared to pV,2b the direct in vitro effect of discreet pW compounds on different enzymes, including phosphohydrolases, remained unexplored. However, we have previously reported our observations on the phosphatase inhibitory activity exhibited by heteroligand dinuclear and mononuclear pW compounds.9 The inhibitor potency of these compounds was found to be significantly higher than that of tungstate or bare peroxotungstate species formed in solution.9b,c We have also recently studied the effect of a series of polymer-anchored pV and pMo compounds on the activity of mammalian ALP.10 Interestingly, these compounds are the first known examples of peroxo-metal derivatives anchored to water soluble polymers.10,11 The most notable finding of our study was that the polymer-anchored and neat peroxo compounds induced their inhibitory effects on ALP through distinct pathways. Significantly, the immobilization of the pV and pMo species enhanced their stability, as well as their capacity to resist degradation under the effect of the enzyme catalase.10
There is a scarcity of information pertaining to well defined peroxometallates anchored to polymer matrices in spite of the importance and potential application of metal-incorporated macromolecules in diverse areas ranging from catalysis12 to medicine.13 Awareness regarding the utility of water soluble polymers as supports in chemistry and biology has been increasing in recent years.14 The binding of active drug molecules, including low molecular weight metal complexes, to soluble macromolecular carriers is of importance since such systems can be expected to overcome limitations, such as toxic side effects, by improving the distribution of the drugs in the body and prolonging their activity.13b,15 Polymers usually lack some of the inconvenient properties of monomeric species, such as lability, volatility, toxicity and odour.12a Anchoring of peroxometallates to water soluble polymers has been recently recognized as a promising new field.15a
As a direct sequel to our work on peroxometal-incorporated water soluble polymers, in the present study we endeavored to form stable and well-defined pW compounds in macroligand environments with an objective to explore their interaction with various enzymes, including phosphatases, vis-à-vis the free monomeric species as well as dimeric pW compounds. Two types of membrane-associated enzymes, viz., wheat thylakoid membrane ACP and rabbit intestine ALP, were chosen for our investigation because, in addition to being excellent models to investigate the toxic metal inhibitory effect in membrane proteins, these enzymes play key roles in a variety of biological phenomena.16 Thylakoid membrane phosphatase is unique to photosynthesis.16c,17 Non-specific alkaline phosphatases (ALP), one of the ubiquitous groups of phosphohydrolases, are used extensively in immunoassays18 and are involved in the development of several diseases. To date, there has been no previous study that has reported the in vitro effect of pW compounds on acid phosphatases.
Experimental
Materials
The chemicals used were all reagent grade products. The sources of the chemicals are given below: sodium tungstate, glycylglycine, (E. Merck, Mumbai, India), cysteine (CDH, New Delhi, India), hydrogen peroxide (30%) (Ranbaxy, New Delhi, India), poly(sodium acrylate) (Mw = 2100) (Fluka), poly(sodium methacrylate) (Mw = 4000), poly(acrylamide) (Mw = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), poly(sodium vinyl sulfonate) (Mw = 4000), acid phosphatase from wheat thylakoid membrane (ACP), alkaline phosphatase from rabbit intestine (ALP), p-nitrophenyl phosphate (p-NPP), tungstic acid (Sigma–Aldrich Chemical Co., Milwaukee, USA), sodium thiosulphate, potassium hydrogen phosphate, potassium dihydrogen phosphate, glacial acetic acid, sodium acetate and MgCl2 (SD Fine Chemicals, Mumbai, India). Na2[W2O3(O2)4(cystine)]·4H2O (DWC) and [WO(O2)2(glycylglycine)]·3H2O (MWG) were prepared by the method described in our earlier papers.9b,c
000), poly(sodium vinyl sulfonate) (Mw = 4000), acid phosphatase from wheat thylakoid membrane (ACP), alkaline phosphatase from rabbit intestine (ALP), p-nitrophenyl phosphate (p-NPP), tungstic acid (Sigma–Aldrich Chemical Co., Milwaukee, USA), sodium thiosulphate, potassium hydrogen phosphate, potassium dihydrogen phosphate, glacial acetic acid, sodium acetate and MgCl2 (SD Fine Chemicals, Mumbai, India). Na2[W2O3(O2)4(cystine)]·4H2O (DWC) and [WO(O2)2(glycylglycine)]·3H2O (MWG) were prepared by the method described in our earlier papers.9b,c
Synthesis of [WO(O2)2(carboxylate)]–PA (PAW) [PA = poly(sodium acrylate)], [WO(O2)2(carboxylate)]–PMA (PMAW) [PMA = poly(sodium methacrylate)], [WO(O2)2(amide)]–PAm (PAmW) [PAm = poly(acrylamide)], and [WO(O2)2(sulfonate)]–PVS (PVSW) [PVS = poly(sodium vinyl sulfonate)]
In a typical reaction, H2WO4 (1.0 g, 4.0 mmol for PAW and PMAW; 5.28 g, 21.12 mmol for PAmW; 2.88 g, 11.53 mmol for PVSW) was dissolved in a minimum volume of 30% H2O2 (10.0 mL, 88.2 mmol for PAW and PMAW; 52.8 mL, 465.69 mmol for PAmW; 28.8 mL, 254.11 mmol for PVSW) in a 250 mL beaker with constant stirring at room temperature (ca. 30 °C). The pH of the clear solution obtained was recorded to be 1.21. Concentrated sodium hydroxide (ca. 8 M) was then added to the above solution dropwise with constant stirring to raise the pH of the reaction medium to 5.0. Keeping the temperature of the reaction mixture below 4 °C in an ice bath, 1.5 g of the respective polymer was added to it. The reaction mixture was kept for 24 h under continuous stirring at a temperature below 4 °C to provide sufficient contact time for the interaction of the reactants. On adding pre-cooled acetone (ca. 50 mL) to this mixture under vigorous stirring, a white pasty mass separated out. After being allowed to stand for about 30 min, the supernatant liquid was decanted and the residue was treated repeatedly with acetone with scratching. The microcrystalline product was separated by centrifugation, washed with cold acetone and dried in vacuo over concentrated sulfuric acid. The compounds were subsequently dried by heating to 70 °C under a nitrogen atmosphere.Elemental analysis
The compounds were analyzed for C, H and N using an elemental analyzer (Perkin—Elmer 2400 series II). The tungsten content was estimated gravimetrically19 as BaWO4. The peroxide contents for the compounds were determined by adding a weighed amount of the compound to a cold solution of 1.5% boric acid (w/v) in 0.7 M sulfuric acid (100 mL) and titration with a standard cerium(IV) solution.20Physical and spectroscopic measurements
The IR spectra were recorded with samples as KBr pellets in a Nicolet model 410 FT-IR spectrophotometer. The spectra were recorded at ambient temperature by making pressed pellets of the compounds. Spectroscopic determinations of the initial rate of ACP and ALP catalyzed hydrolysis of p-NPP were carried out in a Cary model Bio 100 spectrophotometer, equipped with a Peltier-controlled constant temperature cell. The absorbance values were denoted as A405, for example, at the wavelength indicated. The SEM characterization was carried out using a JEOL JSM-6390LV scanning electron micrograph with an attached energy-dispersive X-ray detector. Scanning was done within the 10–20 μM range and images were taken at a magnification of 15–20 kV. Data were obtained using INCA software. The standardization of the data is an integral part of the SEM–EDX instrument employed. The 13C-NMR spectra were recorded on a JEOL JNM-ECS400 spectrometer at a carbon frequency of 100.5 MHz, 131072 X-resolution points, 8000 scans, 1.04 s of acquisition time and 2.0 s relaxation delay with the 1H decoupling method in D2O. Thermogravimetric analysis was done in Perkin–Elmer STA 6000 system at a heating rate of 10 °C min−1 under an atmosphere of nitrogen using an aluminium pan. Prior to TGA analysis, the samples were dried by heating under a nitrogen atmosphere at 70 °C.
Stability of the complexes toward decomposition in solution
The stability of the compounds in distilled water, at their natural pH, was studied by determining the peroxide content in aliquots drawn from the respective compound solutions containing PAW (0.122 mg mL−1), PMAW (0.160 mg mL−1), PAmW (0.156 mg mL−1) or PVSW (0.085 mg mL−1) at varying time intervals by the method already described above. The initial peroxide concentration in each of the test solutions was maintained at 0.4 mM. As a measure of the stability of the compounds in solution, changes in the absorbance of their electronic spectral band at ca. 230–250 nm at ambient temperature were recorded in 30 min time intervals for a period of 12 h. The stability of the compounds in solution at pH 1.2 and 2.1 (50 mM KCl/HCl buffer), 3.1 (50 mM citrate buffer) and 4.4, 7.0 or 8.0 (50 mM phosphate buffer) was measured similarly.Measurement of acid phosphatase activity
Phosphatase activity was assayed spectrophotometrically using p-NPP as a substrate. In the standard assay, the reaction mixture contained acetate buffer (0.1 M, pH = 4.6), acid phosphatase (18.38 μg protein mL−1) and the compound solution (where the concentration varies between 0.1–80 μM). The substrate, p-NPP, was added to the reaction solution, which was pre-incubated for 5 min. Subsequently, the reaction was stopped after incubation for 30 min at 30 °C, by addition of 0.9 mL of NaOH solution (0.5 M) and the absorbance was read at 405 nm in order to measure the p-NP produced. The molar extinction coefficient of the p-nitrophenolate ion at 405 nm in alkaline medium is 18000 M−1 cm−1.21 The activity in the absence of an inhibitor was considered to be a control (100%). For the polymer-bound compounds, concentrations were determined on the basis of the actual peroxometal loading (mmol g−1). All the assays were performed in triplicate. The IC50 values were graphically determined as the half-maximal inhibitory concentration of the inhibitor species giving 50% inhibition. The data in the figures are presented as the means ± SE from three separate experiments.
Measurement of the alkaline phosphatase activity
Phosphatase activity was assayed spectrophotometrically using p-NPP as a substrate. The continuous production of p-nitrophenol (p-NP) was determined at 30 °C by measuring the absorbance at 405 nm in a reaction mixture containing ALP from rabbit intestine (3.3 μg protein mL−1) and p-NPP (2 mM) in an incubation buffer (25 mM glycine + 2 mM MgCl2, pH 10.0). The initial reaction rates were obtained by starting the reaction via the addition of ALP to the reaction solution, which was pre-incubated for 5 min. The initial reaction rate of p-NPP hydrolysis in the absence of the inhibitors, V0, was determined and used as a control. The effects of pW and bare ligands were assessed by adding different concentrations of each species to the ALP assay. For the polymer-bound compounds, concentrations were determined on the basis of the actual peroxometal loading (mmol g−1). The IC50 values were graphically determined as the half-maximal inhibitory concentration of the inhibitor species giving 50% inhibition. All the assays were performed in triplicate. The data in the figures are presented as the means ± SE from three separate experiments.Determination of the kinetic parameters
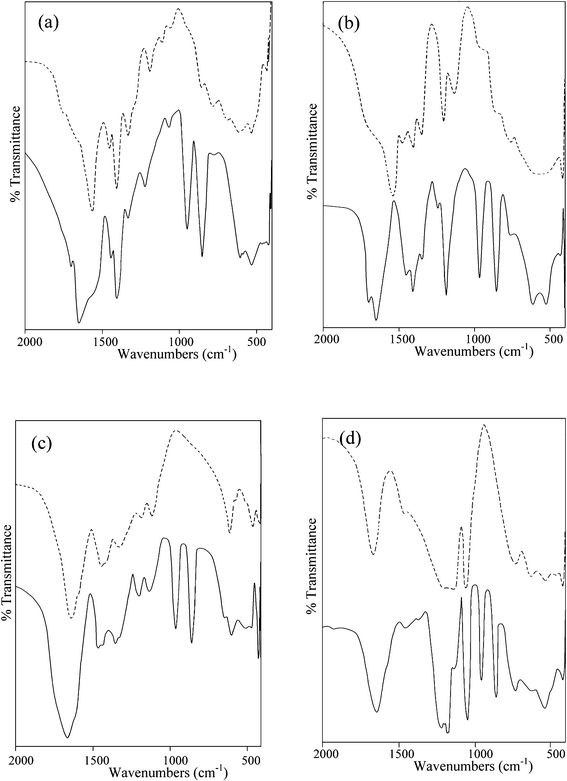
The enzyme kinetic studies were carried out using Cary 100 Bio Enzyme Kinetics software. The kinetic parameters, Vmax and Km, were determined using a Lineweaver–Burk plot following a rearrangement of the Michaelis–Menten equation: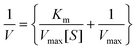 | (1) |
The Vmax parameter is the maximum velocity and Km is the Michaelis constant, its value being equivalent to the substrate concentration at which the velocity is equal to half of Vmax. Vmax and Km can be obtained from the intercept and slope, respectively. In the present case, the expression for the rate of the reaction is given by
 | (2) |
Results and discussion
Synthesis and characterization
The synthesis of the polymer-bound tungsten complexes were achieved by employing a methodology based on the reaction of H2WO4, 30% H2O2 and the respective polymer in aqueous medium at near neutral pH. The acrylate, acrylamide and sulfonate-based polymers were chosen as supports for the present work mainly due to their chemical stability, easy availability and, most importantly, their appropriate functional groups for the facile attachment of metal complexes.22 In a solution of tungsten and excess H2O2, the formation of monomeric or dimeric diperoxotungsten species is favoured at pH ≥ 5.23 Also, the mode and extent of co-ordination offered by water soluble polymers used as support in the present study are known to be pH-sensitive.24 For the synthesis of the title compounds, a pH of ca. 5 was found to be optimal for the formation of diperoxotungsten species and their anchoring to the pendant functional groups of the polymers. Maintenance of required time and temperature at ≤ 4 °C and limiting the water content to that contributed by 30% H2O2 and alkali hydroxide solutions were the other essential components of the procedure. The compounds were obtained by solvent -induced precipitation, which is an effective way of isolating soluble polymeric compounds.14 The compounds remain stable in the solid state for several weeks stored dry in closed containers at < 30 °C.The elemental analysis data for each of the polymeric compounds, PAW, PMAW, PAmW and PVSW, indicated the presence of two peroxide groups per tungsten centre. The metal loading on the compounds, based on elemental analysis and confirmed by EDX analysis, are presented in Table 1. The metal:ligand ratio obtained for the compound PVSW was 1:1, indicating near 100% anchoring of the ligand sites. The ratio was observed to be 1:4 for PAW and PMAW, and rather low in case of the compound PAmW (1:7).
| Compound | % Found from elemental analysis (% obtained from EDX spectra) | Tungsten loadinga (mmol g−1 of polymer) | |||||
|---|---|---|---|---|---|---|---|
| C | H | N | Na | W | O22− | ||
a
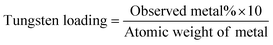 . .
|
|||||||
| PAW | 21.86 | 2.66 | — | — | 30.01 | 10.23 | 1.63 |
| (22.61) | — | — | (12.47) | (30.06) | — | ||
| PMAW | 23.18 | 3.16 | — | — | 23.00 | 7.92 | 1.25 |
| (23.50) | — | — | (15.40) | (23.14) | — | ||
| PAmW | 33.61 | 4.10 | 13.61 | — | 24.68 | 8.24 | 1.28 |
| (34.28) | — | (14.23) | — | (23.44) | — | ||
| PVSW | 5.51 | 0.89 | — | 43.52 | 15.00 | 2.36 | |
| (5.89) | — | — | (6.44) | (43.96) | — | ||
SEM and energy dispersive X-ray (EDX) analysis
Scanning electron microscopy showed the morphological changes occurring on the surfaces of the polymers after loading of the peroxotungstates to the polymer matrices. From the micrographs, it was evident that metal ions are distributed across the surface of the polymers. The surfaces of the pW-incorporated polymers exhibited considerable roughening in contrast to the smooth and flat surfaces of the pristine polymers, as was observed in case of pV- and pMo-anchored polymers (Fig. 1).10 This indicated that the metal ions are distributed across the surface of the polymers. Data obtained on the composition of the compounds from the energy dispersive X-ray spectroscopy, which provides in situ chemical analysis of the bulk, were consistent with the elemental analysis values (Table 1). EDX analysis was carried out focusing multiple regions over the surface of the polymer. The data presented in Table 1 is the average of the data from these regions. | ||
| Fig. 1 (A) Scanning electron micrographs of (a) PA, (b) PAW, (c) PMA and (d) PMAW. EDX spectra of (e) PAW and (f) PMAW. (B) Scanning electron micrographs of (a) PAm, (b) PAmW, (c) PVS and (d) PVSW. EDX spectra of (e) PAmW and (f) PVSW. | ||
IR and electronic spectral studies
The electronic spectrum of each of the title compounds recorded in aqueous solution exhibited a weak intensity broad band at 230–250 nm, which was attributable to the peroxo-to-metal (LMCT) transition. The band was observed in the region characteristic of a diperoxotungstate species.23a,25The IR spectra of the compounds displayed typical features indicating the presence of a side-on bound peroxotungsten moiety (Fig. 2). Apart from the strong absorption at ca. 960 cm−1, which is attributable to the ν(W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) mode of the terminal oxo group, the ν(O–O), νasym(W–O2) and νsym(W–O2) modes were observed at ca. 870, ca. 610 and ca. 530 cm−1, respectively.23b,c Reliable empirical assignments could be derived for the characteristic IR bands observed for the title compounds by comparison of the IR spectra of the anchored complexes to the spectra of pure polymers and the available literature data on metal compounds with co-ordination environments comprised of the ligands relevant to the present study.
O) mode of the terminal oxo group, the ν(O–O), νasym(W–O2) and νsym(W–O2) modes were observed at ca. 870, ca. 610 and ca. 530 cm−1, respectively.23b,c Reliable empirical assignments could be derived for the characteristic IR bands observed for the title compounds by comparison of the IR spectra of the anchored complexes to the spectra of pure polymers and the available literature data on metal compounds with co-ordination environments comprised of the ligands relevant to the present study.
 | ||
| Fig. 2 FTIR spectra of (a) PA (dashed line) and PAW (solid line), (b) PMA (dashed line) and PMAW (solid line), (c) PAm (dashed line) and PAmW (solid line) and (d) PVS (dashed line) and PVSW (solid line). | ||
The spectra of free polymers PA and PMA and the corresponding metal-anchored compounds, PAW and PMAW, exhibited typical bands between 1710 and 1540 cm−1, due to νasym(COO), and 1415 and 1406 cm−1, due to the νsym(COO) mode. It has been well established that the (Δν = νasym − νsym) relationship with the carboxylato co-ordination, derived from a thorough investigation of carboxylato complexes having known crystal structures,26 also holds for polycarboxylates,27 as well as for polyacrylates.28 A close analogy was observed in the spectral patterns exhibited by PAW and PMAW. In the spectrum of pure polyacrylate, νasym(COO) and νsym(COO) modes are observed at 1565 and 1409 cm−1, respectively (Δν = 156 cm−1); whereas, in case of neat PMA, the same peaks appeared at 1540 and 1415 cm−1, respectively (Δν = 125 cm−1). After incorporation of the pW species into these matrices the spectra of both the compounds showed a distinct shift of the νasym(COO) band to a higher frequency in the region of 1650 to 1660 cm−1, along with some broadening. The resulting Δν (240 cm−1 for PAW and 252 cm−1 for PMAW) were much greater relative to free PA and PMA and gave a clear indication of the presence of unidentate coordinated carboxylate groups in the compounds. The broadening of the band in the spectra of the compounds and a shoulder identified at 1570 cm−1 in the spectrum of PAW are likely to be due to the presence of uncoordinated carboxylates in the compound. In PAW and PMAW, the presence of free –COOH groups was evident from the additional band appearing in the vicinity of ca. 1705 cm−1.
In the spectrum of pristine poly(acrylamide), the ν(CO) appeared as an intense band at 1643 cm−1. The amide groups of poly(acrylamide) have two potential alternative metal binding sites, viz. an amide nitrogen or the carbonyl oxygen.29 Co-ordination through the lone pair of nitrogen is known to cause an increase in the ν(CO) (amide-I) band frequency; whereas bonding via the carbonyl oxygen shifts the carbonyl absorption to a lower value.29a–c In the spectrum of PAmW with complexed amide groups, in addition to the band at 1643 cm−1, a new characteristic band appeared in the carbonyl region at 1659 cm−1. This later band is attributable to a shift of the amide-I absorption to a higher frequency resulting from the co-ordination of the W(VI) ion with the N (amide) atom. N–H stretching could not be assigned with certainty as it occurred in the O–H frequency region. The ν(C–N) was identified as a medium intensity band at 1447 cm−1.
In the spectrum of neat poly(vinyl sulfonate), the bands corresponding to the S–O stretching of the pendant sulfonate group occur at 1210 and 1127 cm−1.29a The band at ca. 1040 cm−1 is due to the symmetric stretching vibration of the sulfonate anion.29f The spectrum of PVSW shows a distinct splitting pattern displaying bands at 1220 and 1188 cm−1 in addition to the antisymmetric vibration of S–O at 1128 cm−1, which we attribute to a complexed sulfonate group.29a,f,30
Metal oxygen vibrations in the spectra of each of pW compound were identified as weak bands observed in the far IR region between 500 and 400 cm−1. The presence of lattice water in the title complexes was evident from the appearance of strong and broad ν(OH) absorptions displayed at 3500–3400 cm−1.
TGA–DTG analysis
The TG–DTG plots for the title compounds and the corresponding thermogravimetric analysis data presented in Fig. 3 and Table 2 show that the polymer-incorporated pW compounds gradually undergo multistage decomposition upon heating up to a temperature of 750 °C. The first stage of the decomposition occurs in the temperature range of ca. 74–101 °C, which corresponds to the liberation of lattice water from the complexes. In the second decomposition stage, in the temperature range of 112–210 °C, the compounds lose their co-ordinated peroxo groups completely. The absence of peroxide in the decomposition product isolated at this stage was confirmed from the IR spectral analysis. The loss of peroxide is followed by a three stage decomposition occurring in the temperature range of 324–551 °C for PAW and 301–543 °C for PMAW, which may be ascribed to decarboxylation involving free, as well as coordinated, carboxylate functionalities accompanied by rupturing of the polymers as has been observed previously.10 Further evidence in support of the decarboxylation of polymers was provided by the IR spectra recorded after heating the compounds separately to the final decomposition temperature, which showed the disappearance of the peaks originating from the carboxylate stretching in the spectra of the original compounds.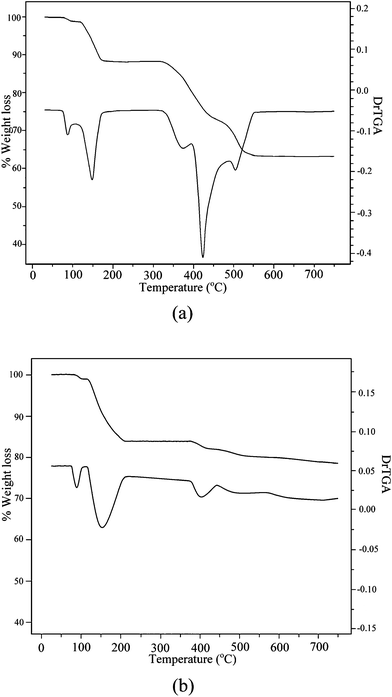 | ||
| Fig. 3 TG–DTG plot of (a) PAW and (b) PVSW. | ||
| Compound | Temperature range (°C) | Observed weight loss (%) | Final residue (%) |
|---|---|---|---|
| PAW | 76–96 | 1.10 | |
| 119–172 | 10.46 | 63.53 | |
| 324–551 | 24.91 | ||
| PMAW | 74–101 | 0.97 | |
| 112–187 | 8.05 | 65.86 | |
| 301–543 | 25.12 | ||
| PAmW | 76–101 | 1.01 | |
| 115–190 | 8.21 | 43.33 | |
| 200–330 | 14.24 | ||
| 330–451 | 33.21 | ||
| PVSW | 76–101 | 1.02 | |
| 116–210 | 15.24 | 78.55 | |
| 378–440 | 1.88 | ||
| 440–570 | 3.31 |
PAmW, with poly(acrylamide) as a macroligand and after the loss of coordinated peroxo groups, undergoes two weight loss processes in the temperature range of ca. 200–451 °C, attributable to the breakdown of the polymer ligand. On the basis of the thermal decomposition patterns reported for some poly(acrylamides),31 the first stage of the decomposition in the temperature range of 200–330 °C is ascribed to the release of water, ammonia and small amounts of carbon dioxide from the side-chain amide groups with the polymer chains remaining intact.31 In the second stage of decomposition (330–451 °C), main chain breakdown occurs accompanied by a majority of weight loss (33.21%). In the case of PVSW, a two stage decomposition was observed to occur after the loss of peroxide. By analogy with the available data pertaining to the thermal degradation of poly(vinyl sulfonate) sodium salts, these decompositions, in the range of 378–440 °C and the last one commencing at 440 °C and ending at 570 °C, are attributed to the loss of the sulfonate group and a rupturing of the polymer accompanied by the evolution of ethylene, water, SO2 and CS2.32
The final residue remaining after complete loss of the components, viz. lattice water, the coordinated peroxide and the polymeric functional, from all the complexes shows the presence of oxotungstate species. Thermogravimetric analysis data of the compounds provides further evidence in support of their composition and assigned formula. It is notable that the decomposition patterns exhibited by the pW complexes closely resemble those exhibited by the corresponding pV- or pMo-containing analogues.10
Based on the above data, structures of the type shown in Fig. 4 have been proposed for the polymer-anchored pW complexes.
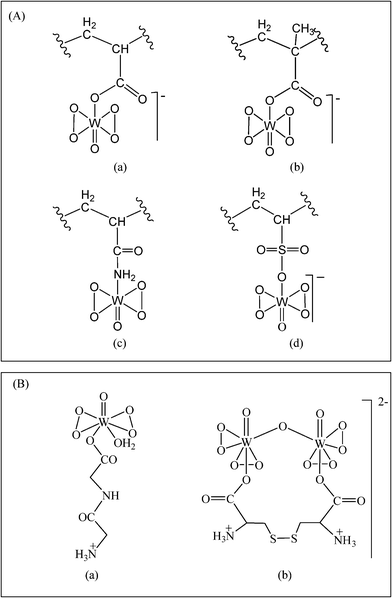 | ||
Fig. 4 Peroxotungsten compounds under investigation in the current study. (A) Proposed structures of polymer-anchored compounds, (a) PAW, (b) PMAW, (c) PAmW and (d) PVSW. “ ” represents the polymer chain. (B) Structures of (a) MWG9b and (b) DWC.9c ” represents the polymer chain. (B) Structures of (a) MWG9b and (b) DWC.9c | ||
13C NMR studies
The study of co-ordination induced 13C NMR chemical shifts has been recognized as an effective tool to understand the nature of co-ordination of ancillary ligands in peroxo metal compounds.23c,33 The 13C NMR spectra of the pure polymers, PA and PMA, display resonances due to the carboxylate carbon atoms centered at 184 and 187 ppm, respectively (Table 3), in addition to the characteristic signals corresponding to the chain carbon atoms.34 The assignments of the major peaks were made on the basis of available literature data.23c,33,34 Two closely spaced peaks observed in this region are likely to be due to the presence of free carboxylate, as well as the –COOH groups of the polymers in solution. The spectrum of the pW-anchored polymeric compounds, PAW and PMAW, provided evidence for the presence of complexed, as well as free, carboxylate groups by displaying a new peak at the lower field of ca. 215 ppm, in addition to the characteristic carboxylate resonance corresponding to the free carboxylate groups as observed in the pure polymers (Table 3). A strong metal–ligand interaction was indicated by the substantial downfield shift, Δδ(δcomplex − δfree carboxylate) = 30.88 ppm in the case of PA-bound compounds and 28.58 ppm in the metal-anchored PMA compound relative to the free carboxylate peak of the pristine polymer.| Compound | Chemical shift (ppm) | ||||
|---|---|---|---|---|---|
| Carboxylate/amide carbon | CH2 | CH | CH3 | ||
| Free | Complexed | ||||
| PA | 184.50 | 36.10 | 45.52 | ||
| PAW | 185.15 | 216.03 | 36.72 | 46.12 | |
| PMA | 187.41 | 17.36 | 56.54 | ||
| PMAW | 186.64 | 215.22 | 17.12 | 55.93 | |
| PAm | 179.48 | 34.88 | 41.66 | ||
| PAmW | 179.51 | 200.26 | 34.89 | 41.66 | |
| PVS | 30.97 | 54.51 | |||
| PVSW | 30.92 | 54.54 | |||
Detailed information are available in the literature on the 13C NMR spectral analysis of poly(acrylamide) under varying pH conditions.35 The spectrum of pW-incorporated PAmW displayed, in addition to the resonance at 179.51 ppm corresponding to the free amide groups as observed in the pure poly(acrylamide) (Table 3), a new peak at 200.26 ppm. This resonance is attributable to the carbon atom of the tungsten co-ordinated amide group. In the spectrum of PVSW, no significant change was noted in the positions of the peaks corresponding to the CH and CH2 groups of the polymer chain compared to the pure polymer, poly(vinyl sulfonate). This may not be unusual considering that W atoms are bound to the polymer through the sulfonate groups and hence are well separated from the chain carbon atoms of the polymer support.
The resonance, due to the complexed carboxylate or amide occurring as a singlet in the spectra of the polymer-anchored compounds (PAW, PMAW and PAmW) provided evidence for a single carbon environment, which obviously results from a single mode of the metal–ligand co-ordination and is in agreement with the proposed structure.
Stability of the compounds in solution
One of the most important criteria required to be met by water soluble metal complexes to be useful as therapeutic or bio-relevant agent is the hydrolytic stability.36 We have therefore considered it imperative to ascertain the stability of the compounds in solution not only at pH ca. 5, the natural pH attained by the solution on dissolving the compounds, but also under varying pH conditions ranging from 1.2 to 8.0. The stability of the compounds with respect to the loss of peroxide in solution have been examined by estimating their peroxide content and absorbance in the 230–250 nm region of the electronic spectra at specified time intervals. The studies revealed that the peroxide content and position and intensity of their electronic spectral bands remained unaltered even after a period of over 12 h. Moreover, the 13C NMR spectra of the compounds recorded after 24 h of solution preparation showed no change in the spectral patterns, suggesting that metal co-ordination to the amide or carboxylate groups remained unchanged in the compounds in solution. Thus, all the gathered evidence clearly attests to the stability of the compounds in solution. It is pertinent to mention that Shisheva et al.36 observed earlier that orally administered pV was ineffective in inducing normoglycemia in STZ-rats possibly because it could not survive the strong acidity of the stomach. There is a continued search for peroxo derivatives easily formed and stable to degradation, suitable for clinical applications.Inhibition of acid and alkaline phosphatases by the peroxotungsten compounds
Using the established enzyme assay system and p-NPP as a substrate, the effect of the polymer-anchored compounds on the activities of membrane-associated proteins, viz., wheat thylakoid membrane ACP as well as rabbit intestine ALP, was examined vis-á-vis the effect induced separately by the previously reported free mononuclear or dinuclear pW compounds, MWG and DWC, as well as the tungstate anion. The data presented in Fig. 5 and 6 indicate that the pW compounds, irrespective of being polymer-bound or free, were able to markedly inhibit the activity of each of the model enzymes in a dose-dependent manner. The inhibitory potential of the inhibitor species was quantified by determining the half-maximal inhibitory concentration (IC50), which represents a 50% suppression of the original enzyme activity, for each of the compounds (Table 4). The IC50 values for the polymeric compounds are reported in terms of their actual peroxometal loading. It is apparent from the data that enzymes have different sensitivity to the inhibitor complexes. It is notable that the ACP activity was efficiently inhibited by the pW compounds at doses lower than 1 μM.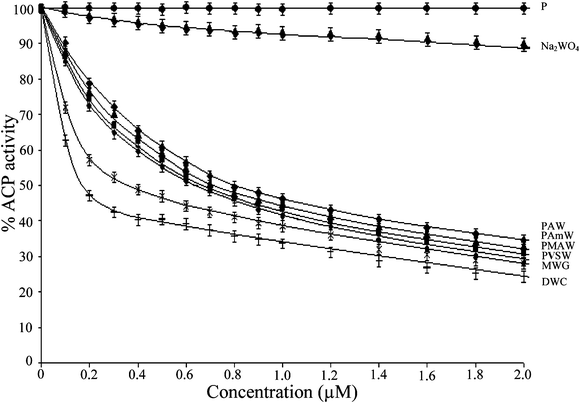 | ||
| Fig. 5 The effect of PAW, PMAW, PAmW, PVSW, MWG, DWC, Na2WO4 and the free polymers (P) on the activity of ACP. The ACP catalyzed rates of hydrolysis of p-NPP at pH 4.6 were determined at 30 °C by measuring A405 in a reaction mixture containing ACP (18.38 μg mL−1) and p-NPP (2 mM) in acetate buffer (0.1 M, pH = 4.6) in the absence or presence of stated concentrations of the inhibitors. The effects of the additions are represented as the percent values (rounded to integers) of the control (Δp-NPP = 3.13 μM min−1). The data are presented as the means ± SE from three separate experiments. For polymeric compounds, the concentrations are based on the peroxometal loading. | ||
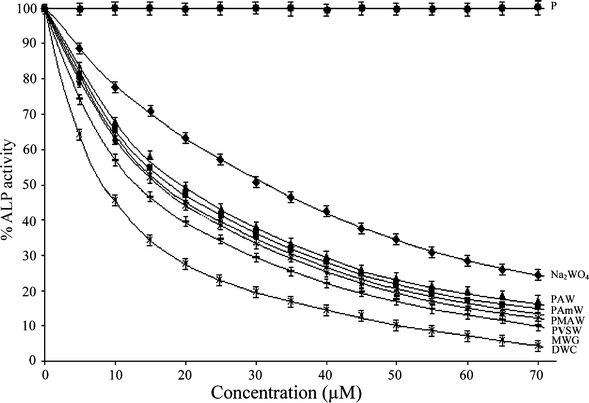 | ||
| Fig. 6 The effect of PAW, PMAW, PAmW, PVSW, MWG, DWC, Na2WO4 and the free polymers (P) on the activity of ALP from rabbit intestine. The ALP catalyzed rates of hydrolysis of p-NPP at pH 10.0 were determined at 30 °C by measuring A405 in a reaction mixture containing ALP (3.3 μg mL−1) and p-NPP (2 mM) in incubation buffer (25 mM glycine + 2 mM MgCl2, pH 10.0) in the absence or presence of stated concentrations of the inhibitors. The data are presented as the means ± SE from three separate experiments. For polymeric compounds, the concentrations are based on the peroxometal loading. | ||
| Enzyme | Compound | IC50 (μM) | K i (μM) | K ii (μM) | K ii/Ki | Types of inhibition |
|---|---|---|---|---|---|---|
| a Note: the ACP catalyzed rates of hydrolysis of p-NPP at pH 4.6 were determined at 30 °C by measuring A405 in a reaction mixture containing ACP (18.38 μg mL−1) and p-NPP (2 mM) in acetate buffer (0.1 M, pH = 4.6) in the presence of stated concentrations of the inhibitors. The ALP catalyzed rates of hydrolysis of p-NPP at pH 10.0 were determined at 30 °C by measuring A405 in a reaction mixture containing ALP (3.3 μg mL−1) and p-NPP (2 mM) in incubation buffer (25 mM glycine + 2 mM MgCl2, pH 10.0) in the presence of stated concentrations of the inhibitors. The Vmax and Km in absence of inhibitor were found to be 7.9 μM min−1 and 2.85 mM, respectively. For polymeric compounds concentrations are on the basis of the peroxometal loading. | ||||||
| ACPa | PAW | 0.81 | 0.83 | 0.82 | 0.98 | Non-competitive |
| PMAW | 0.67 | 0.72 | 0.70 | 0.97 | Non-competitive | |
| PAmW | 0.71 | 0.78 | 0.77 | 0.99 | Non-competitive | |
| PVSW | 0.64 | 0.67 | 0.61 | 0.92 | Non-competitive | |
| MWG | 0.36 | 0.20 | 0.56 | 2.80 | Mixed inhibition | |
| DWC | 0.17 | 0.10 | 0.34 | 3.40 | Mixed inhibition | |
| Na2WO4 | 15.23 | 10.45 | 10.10 | 0.96 | Non-competitive | |
| Free polymer | — | — | — | — | — | |
| ALPa | PAW | 19.33 | 17.54 | 17.13 | 0.97 | Non-competitive |
| PMAW | 16.12 | 15.55 | 15.08 | 0.96 | Non-competitive | |
| PAmW | 17.97 | 16.23 | 16.01 | 0.98 | Non-competitive | |
| PVSW | 15.95 | 14.95 | 14.43 | 0.96 | Non-competitive | |
| MWG 9a | 14.20 | 14.70 | 48.20 | 3.20 | Mixed inhibition | |
| DWC 9c | 8.20 | 6.46 | 19.10 | 2.95 | Mixed inhibition | |
| Na2WO4 9c | 31.68 | 17.15 | — | — | Competitive | |
| Free polymer | — | — | — | — | — | |
In order to evaluate the mode of inhibitory action of the complexes on the activity of the model enzymes used, the kinetic parameters, Km and Vmax were determined in the absence, as well as in presence, of peroxo metal compounds using Lineweaver–Burk (L–B) double reciprocal plots. Enzyme kinetic investigation is a major tool in enabling us to distinguish between the inhibition mechanisms of enzyme-catalyzed reactions. Kinetic measurements for the acid phosphatase-catalysed hydrolysis of p-NPP at several different substrate concentrations in the presence of each of the inhibitors yielded straight lines with a point of intersection in the second quadrant (Fig. 7 and 8). As demonstrated by the L–B plots (Fig. 7 and 8), an increase in concentration of each of the polymeric complexes led to a substantial decrease in Vmax, although Km remained unaffected suggesting a non-competitive type of inhibition by these complexes. On the other hand, the free monomeric complex, MWG, as well as dinuclear complex, DWG, exerted mixed inhibitory effects combining competitive and non-competitive modes. Tungstate also exhibited non-competitive type inhibition of ACP activity under these conditions.
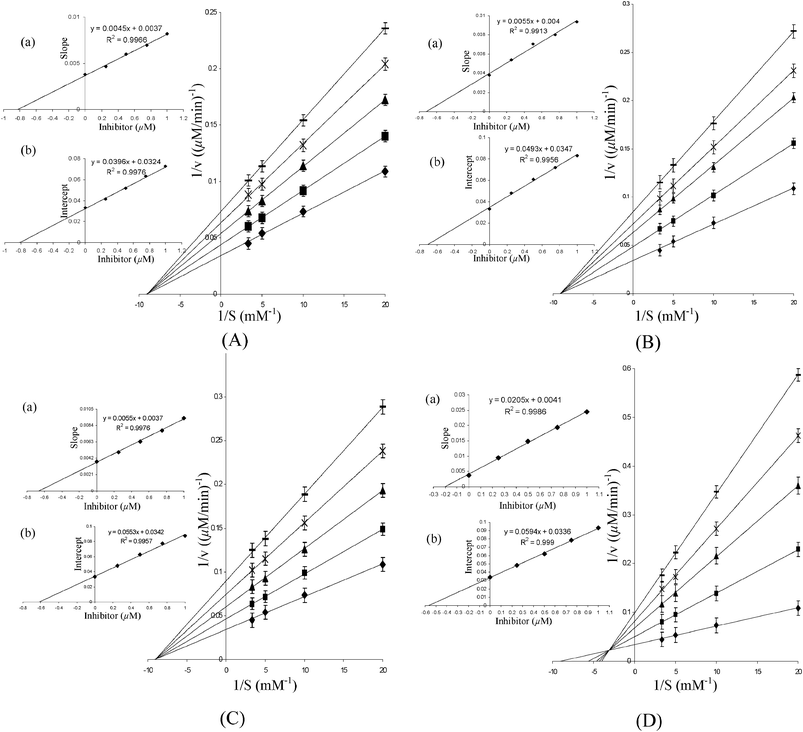 | ||
| Fig. 7 Lineweaver–Burk plots for the inhibition of ACP activity in the absence and presence of (A) PAW, (B) PMAW, (C) PVSW and (D) MWG. The inset represents the secondary plot of the initial kinetic data of the Lineweaver plot. The reaction mixture contained acetate buffer (0.1 M, pH 4.6) and p-NPP (50–300 μM). The reaction was started by adding ACP (18.38 μg mL−1) to the reaction solution, which was pre-incubated for 5 min and the rate of hydrolysis in the presence of ◆ 0 μM; ■ 0.25 μM; ▲ 0.50 μM; × 0.75 μM; – 1.00 μM inhibitors were obtained. The values are expressed as the mean ± SE from three separate experiments. Inset: (a) the slopes were plotted against inhibitor concentrations and Ki values were obtained from the x-intercepts of these re-plots. (b) The vertical intercepts were plotted against the inhibitor concentration and Kii values were obtained from the x-intercepts of these re-plots. For polymeric compounds, the concentrations are on the basis of the peroxometal loading. | ||
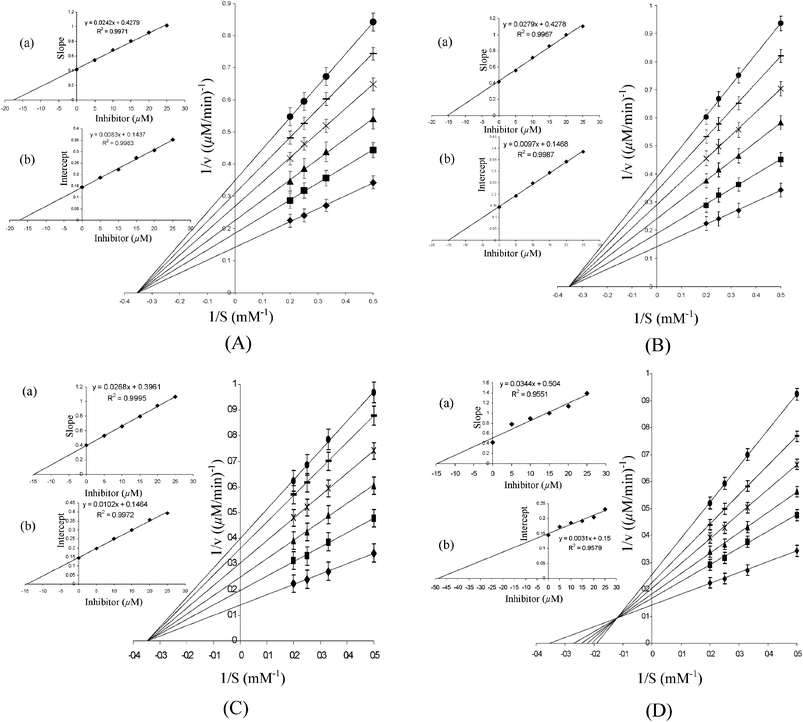 | ||
| Fig. 8 Lineweaver–Burk plots for the inhibition of ALP activity in the absence and presence of (A) PAW, (B) PMAW, (C) PVSW and (D) MWG. The inset represents secondary plot of the initial kinetic data of the Lineweaver plot. The reaction mixture contained glycine buffer (25 mM glycine + 2 mM MgCl2, pH 10.0) and p-NPP (2–5 mM). The reaction was started by adding ALP (3.3 μg mL−1) to the reaction solution, which was pre-incubated for 5 min and the rate of hydrolysis in the presence of ◆ 0 μM; ■ 5 μM; ▲ 10 μM; × 15 μM; – 20 μM; ● 25 μM inhibitors were obtained. The values are expressed as the mean ± SE from three separate experiments. Inset: (a) the slopes were plotted against the inhibitor concentrations and Ki values were obtained from the x-intercepts of these re-plots. (b) The vertical intercepts were plotted against the inhibitor concentration and Kii values were obtained from the x-intercepts of these re-plots. For polymeric compounds, the concentrations are on the basis of the peroxometal loading. | ||
It was important to assess the affinity of the enzyme for the inhibitor by determining the inhibitor constants. All compounds exhibited affinity for the acid phosphatase in a close order of magnitude as observed from the Ki and Kii values (Table 4). The inhibitor constant, Ki, for the competitive part of inhibition was determined from the secondary plot of the slope of the primary plot (1/V versus 1/[S]) against the inhibitor concentration with intercept on the inhibitor axis being −Ki (Fig. 7 and 8). The value of Kii, the inhibitor constant for non-competitive inhibition, was obtained from a linear secondary plot of 1/Vmax against the inhibitor concentration of each inhibitor, with the intercept on the inhibitor axis being equivalent to −Kii (Fig. 7 and 8). For each of the polymer-anchored pW complexes, the value of Ki was found to be equal to Kii, which is typical of a non-competitive inhibitor. For free peroxotungstates, DWC or MWG, Kii > Ki as is the case with a mixed type of inhibitor with the major mode of inhibition being the competitive type. The inhibitors could thus be arranged in the following order of potency: DWC > MWG > PVSW > PMAW > PAmW > PAW > Na2WO4. Importantly, although the effect of the individual ligand on the enzyme activity is practically negligible under the assay conditions used, our results show that there is a marked influence of the co-ligand environment on the inhibitory potency of the intact metal complexes.
The mode of inhibitory action of the polymeric pW compounds on rabbit intestinal alkaline phosphatase was similar to that found for the wheat thylakoid membrane acid phosphatase. However, significant differences were noted in the IC50 values and inhibitor constants for the two classes of enzyme. The values for ALP were observed to be more than 50 orders of magnitude higher than for ACP, which clearly indicated a greater affinity of the complexes for the enzyme binding site of ACP compared to ALP. Table 4 presents the kinetic data for the inhibition of the ALP-catalysed hydrolysis of p-NPP by the pW complexes and Na2WO4. From the Lineweaver–Burk plots in Fig. 7 and 8, it has been confirmed that polymeric pW compounds are classic non-competitive inhibitors of ALP function. Dinuclear DWC is noted to induce mixed inhibition on the enzyme, as has been observed earlier for MWG.9a Interestingly, our previous findings have also shown polymer-bound pV and pMo to be non-competitive inhibitors of ALP.10 The kinetic data for the ALP inhibition by the pW compounds indicated the following sequence: DWC > MWG > PVSW > PMAW > PAmW > PAW > Na2WO4.
Acid phosphatases from human prostate, wheat germ and other sources have been isolated as phosphohistidyl enzymes containing a histidine-rich active site.37 The mammalian acid phosphatase enzyme possesses dinuclear iron active sites16a,b and highly conserved amino acid sequences.38 Alkaline phosphatase, on the other hand, is a zinc metalloenzyme with a broad substrate specificity, which catalyzes the hydrolysis of organic phosphate monoesters possibly via an enzyme–phosphate intermediate. The maximum activity of the enzyme is observed at pH ≥ 8. Phosphotransferase activity and protein phosphatase activity are some of the other probable functions assigned to the enzyme. Phosphatases are, in general, inhibited by oxyanions, such as vanadate,6,37 molybdate and tungstates.39 The competitive inhibition exhibited by these ions on ALP has been attributed to the formation of penta or hexa co-ordinated structures, which are described as structural analogues of phosphate.39,40 Gresser and co-workers observed metal oxyanion inhibition of mammalian acid and alkaline phosphatases to be strictly competitive.39a In agreement with previous reports, we have previously found that vanadate, molybdate and tungstate inhibit the activity of mammalian ALP in a competitive manner.5,41 While no exception was noted to this observation among the existing literature for ALP inhibition, in the case of ACP, the type of inhibition induced by these anions was found to be different to that produced by phosphate in certain cases.16c,38a,39a For instance, molybdate and tungstate were observed to be non-competitive inhibitors of bovine spleen purple acid phosphatase.35 Interestingly, Wang and co-workers reported molybdate and vanadate to be uncompetitive and non-competitive inhibitors, respectively, with regards to the activity of wheat thylakoid membrane ACP.16c In the present study, we observed tungstate to exert non-competitive inhibition on ACP. The information available thus shows that different oxometallates may reveal different mechanistic preferences in these classes of enzymes, in spite of sharing common structural features.
Among the peroxometallates, the inhibitory effect of peroxo derivatives of vanadium on phosphohydrolases have been extensively investigated.6a,b,42 Very little information is available pertaining to similar studies carried out on pMo or pW systems.1a,9,10 Earlier studies suggested that the inhibitory ability of different vanadium derivatives, including peroxovanadates, to modulate ACP or ALP activity depend on several factors, such as the oxidation state of the metal, the co-ordination geometry, the stability of the compounds under physiological conditions and the nature of the phosphoproteins.5,6b The majority of the synthetic pV compounds tested were competitive inhibitors of the enzyme, although there are examples of diperoxovanadate compounds showing mixed inhibition of Green-crab ALP, with Ki and Kii values in the millimolar range.42a,b
The trend emerging out of our present investigation reveals that the compounds tested can be grouped into two classes on the basis of their effect on ACP and ALP hydrolysis. The first class comprising free monomeric or dimeric peroxometallates exhibits mixed inhibition due to the presence of competitive and non-competitive binding sites, whereas the group of polymer-bound metal peroxo compounds are classical non-competitive inhibitors of the phosphoproteins. A competitive inhibitor typically has close structural similarities to the normal substrate of the enzyme. A non-competitive inhibitor usually binds reversibly at a site other than the active site and causes a change in the overall three-dimensional shape of the enzyme that leads to a decrease in the catalytic activity. Since the inhibitor binds at a different site to the substrate, the enzyme may bind to the inhibitor and the substrate or both the inhibitor and the substrate together. In the case of the macromolecular metal complexes, factors, such as the structural analogy with the transition state or the phosphate mimicry, are unlikely to be the cause responsible for the non-competitive mode of ALP or ACP inhibition exhibited mainly because of their large size. It is relevant to recall that the non-competitive inhibition induced by molybdate and tungstate on bovine spleen purple acid phosphatase was earlier ascribed to the larger size of these ions causing them to bind at a site different from the active site.38a Moreover, it has been suggested that highly oxidative compounds, such as molybdate and tungstate, probably interact with the Fe2+ of the ACP enzyme and oxidize it to the ferric form, causing the inactivation of the enzyme in a non-competitive manner.38a Significantly, the compounds reported herein, irrespective of being neat or polymer-anchored, were found to be active oxidants of bromide43 and sulfide44, as well as GSH.9b,c It may therefore be reasonable to expect that the observed enzyme inhibition by the pW compounds originates from their interaction with oxidation-prone essential groups, such as –SH.42d,45 The presence of such groups in the active centre of the enzymes, or in the stabilization of the quaternary structure, are essential for enzyme activity.16c It was observed earlier that peroxovanadate effectively inhibits tyrosine phosphatase by oxidizing the critical cysteine residue in the catalytic domain of the enzyme.6a,42e,46 Based on our findings and taking into account the reports documenting the importance of the redox properties of peroxo compounds in the inhibition of protein phosphatases, we have previously proposed that the oxidant activity of the tested peroxo compounds of V, Mo and W is one of the possible factors responsible for making the compounds effective inhibitors of ALP.6a,b,9,10 However, we refrain from drawing any conclusion on the exact mechanism of inhibition in view of the fact that the mode of action of such compounds with complex biomolecules are obviously not simple and many experiments will be needed to unravel the actual mechanism. Although enzyme model systems may not be very effective predictors of in vivo activities, the phosphatase inhibition studies can nevertheless be expected to provide an academic basis for further investigation of the functions of the enzymes in several other reaction courses.
Conclusions
The present work afforded a set of water soluble, hydrolytically stable and well-defined peroxo derivatives of tungsten supported on macromolecular matrices. The findings of the detailed study of their inhibition kinetics confirmed that the title compounds, as well as the previously reported neat pW complexes, serve as potent inhibitors of two different types of phosphohydrolases, viz., ALP and ACP. Interestingly, the two classes of complex examined, i.e. anchored and free monomeric or dinuclear peroxotungstates, exhibit distinct mechanistic preferences for phosphatase inhibitory activity. The neat complexes induced a mixed type of inhibition of the function of both model enzymes, whereas the macro-complexes served as classic non-competitive inhibitors. The pW complexes, irrespective of being supported or free, displayed more than 50 fold greater affinity for ACP than for ALP. Taken together, from the information generated from the present study and the results of our previous investigation on polymer-anchored pV and pMo compounds,10 it is hoped that the supported peroxometallates may serve as excellent selective probes for the non-competitive site of inhibition of the model systems investigated. Moreover, in view of their easy method of preparation and stability in acidic and higher pH solutions, the compounds may be expected to emerge as suitable candidates for in vivo studies. It is pertinent to mention that polymer-bound pV has recently been investigated for its vasomodulatory effect on rat aorta.47 Some of these compounds were observed to elicit the dual responses of vasoconstriction and relaxation.Acknowledgements
Financial support from the Department of Biotechnology, New Delhi, India is gratefully acknowledged. We are grateful to Prof. T. Ramasarma, INSA Hon. Scientist, Indian Institute of Science (IISc), Bangalore, India and Prof. G. Ramakrishna, Laboratory of Cancer Biology, Centre for DNA Fingerprinting and Diagnostics, Hyderabad, India for valuable discussion. We are thankful to the University Grants Commission (UGC), New Delhi, India for providing Senior Research Fellowship (RGNSRF) to SPD.References
- (a) J. Li, G. Elberg, D. Gefel and Y. Shechter, Biochemistry, 1995, 34, 6218–6225 CrossRef CAS; (b) A. Maniatakou, S. Karaliota, M. Mavri, C. Raptopoulou, A. Terzis and A. Karaliota, J. Inorg. Biochem., 2009, 103, 859–868 CrossRef CAS; (c) K. Nomiya, H. Torii, T. Hasegawa, Y. Nemoto, K. Nomura, K. Hashino, M. Uchida, Y. Kato, K. Shimizu and M. Oda, J. Inorg. Biochem., 2001, 86, 657–667 CrossRef CAS.
- (a) A. Barbera, J. E. Rodriguez-Gil and J. J. Guinovart, J. Biol. Chem., 1994, 269, 20047–20053 CAS; (b) M. Jelikic-Stankov, S. Uskokovic-Markovic, I. Holclajtner-Antunovic, M. Todorovic and P. Djurdjevic, J. Trace Elem. Med. Biol., 2007, 21, 8–16 CrossRef CAS; (c) J. D. Foster, S. E. Young, T. D. Brandt and R. C. Nordlie, Arch. Biochem. Biophys., 1998, 354, 125–132 CrossRef CAS.
- J. Rhule, C. Hill, D. Judd and R. Schinazi, Chem. Rev., 1998, 98, 327–357 CrossRef CAS.
- M. Claret, H. Corominola, I. Canals, J. Saura, S. Barcelo-Batllori, J. J. Guinovart and R. Gomis, Endocrinology, 2005, 146, 4362–4369 CrossRef CAS.
- A. Y. Louie and T. J. Meade, Chem. Rev., 1999, 99, 2711–2734 CrossRef CAS.
- (a) D. C. Crans, J. J. Smee, E. Gaidamauskas and L. Yang, Chem. Rev., 2004, 104, 849–902 CrossRef CAS; (b) D. C. Crans, in Vanadium Compounds Chemistry, Biochemistry, and Therapeutic Application, ed. A. S. Tracy and D. C. Crans, Oxford University Press, New York, 1998, pp. 82–103 Search PubMed; (c) D. Rehder, M. Bashipoor, S. Jantzen, H. Schmidt, M. Farahbakhsh and H. Nekola, in Vanadium Compounds, Chemistry, Biochemistry, and Therapeutic Applications, ed. A. S. Tracy and D. C. Crans, Oxford University Press, New York, 1998, pp. 60–71 Search PubMed.
- G. Fantus, S. Kadota, G. Deragon, B. Foster and B. I. Posner, Biochemistry, 1989, 28, 8864–8871 CrossRef.
- (a) K. H. Thompson, J. H. McNeill and C. Orvig, Chem. Rev., 1999, 99, 2561–2572 CrossRef CAS; (b) C. Djordjevic, N. Vuletic, M. L. Renslo, B. C. Puryear and R. Alimard, Mol. Cell. Biochem., 1995, 153, 25–29 CrossRef CAS; (c) S. Sarmah, D. Kalita, P. Hazarika, R. Borah and N. S. Islam, Polyhedron, 2004, 23, 1097–1107 CrossRef CAS; (d) S. Sarmah, P. Hazarika, N. S. Islam, A. V. S. Rao and T. Ramasarma, Mol. Cell. Biochem., 2002, 236, 95–105 CrossRef CAS.
- (a) D. Kalita, S. P. Das and N. S. Islam, Biol. Trace Elem. Res., 2009, 128, 200–219 CrossRef CAS; (b) P. Hazarika, D. Kalita and N. S. Islam, J. Enzyme Inhib. Med. Chem., 2008, 23, 504–513 CrossRef CAS; (c) P. Hazarika, D. Kalita, S. Sarmah and N. S. Islam, Mol. Cell. Biochem., 2006, 284, 39–47 CrossRef CAS.
- J. J. Boruah, D. Kalita, S. P. Das, S. Paul and N. S. Islam, Inorg. Chem., 2011, 50, 8046–8062 CAS.
- D. Kalita, S. Sarmah, S. P. Das, D. Baishya, A. Patowary, S. Baruah and N. S. Islam, React. Funct. Polym., 2008, 68, 876–890 CrossRef CAS.
- (a) A. Skorobogaty and T. D. Smith, Coord. Chem. Rev., 1984, 53, 55–226 CrossRef CAS; (b) A. D. Pomogalilo, Catalysis by Polymer Immobilized Metal Complexes, Gordon and Breach Scientific Publishers, Amsterdam, 1998 Search PubMed; (c) D. C. Sherrington, in Supported Reagents and Catalyst in Chemistry, ed. B. K. Hondett, A. P. Keybett, J. H. Clark and K. Smith, Royal Society of Chemistry, Cambridge, 1998, pp. 220 Search PubMed; (d) A. Choplin and F. Quignard, Coord. Chem. Rev., 1998, 180, 1679–1702 CrossRef.
- (a) R. Breslow, S. Belvedere, L. Gershell and D. Leung, Pure Appl. Chem., 2000, 72, 333–342 CrossRef CAS; (b) B. Schechter, R. Arnon and M. Wilchek, React. Polym., 1995, 25, 167–175 CrossRef CAS.
- (a) D. E. Bergbreiter, Chem. Rev., 2002, 102, 3345–3384 CrossRef CAS; (b) T. J. Dickerson, N. N. Reed and K. D. Janda, Chem. Rev., 2002, 102, 3325–3344 CrossRef CAS.
- (a) V. Conte and B. Floris, Dalton Trans., 2011, 40, 1419–1436 RSC; (b) J. Jagur-Grodzinski, React. Funct. Polym., 1999, 39, 99–138 CrossRef CAS; (c) D. Avichezer, B. Schechter and R. Arnon, React. Funct. Polym., 1998, 36, 59–69 CrossRef CAS; (d) Y. Ohya, T. Masunaga, T. Baba and T. Ouchi, J. Macromol. Sci., Part A: Pure Appl. Chem., 1996, 33, 1005–1016 CrossRef.
- (a) W. N. Lipscomb and N. Strater, Chem. Rev., 1996, 96, 2375–2433 CrossRef CAS; (b) D. E. Wilcox, Chem. Rev., 1996, 96, 2435–2458 CrossRef CAS; (c) M-J. Fei, J-S. Chen and X-Y. Wang, J. Integr. Plant Biol., 2006, 48, 294–299 CrossRef CAS.
- (a) J. Bennett, Nature, 1977, 269, 344–346 CrossRef CAS; (b) A. Regasamy, R. Selvam and A. Gnanam, Arch. Biochem. Biophys., 1981, 209, 230–236 CrossRef; (c) J. F. Allen, Biochim. Biophys. Acta, Bioenerg., 1992, 1098, 275–335 CrossRef CAS; (d) J. Y. Ye and Y. J. Wang, Acta. Biochem. Biophys., 1997, 29, 40–45 CAS.
- (a) M. P. Whyte, Endocr. Rev., 1994, 15, 439–461 CAS; (b) K. M. Holtz and E. R. Kantrowitz, FEBS Lett., 1999, 462, 7–11 CrossRef CAS; (c) H. N. Fennley, The Enzymes, 3rd ed. ed. P. D. BouerAcademic Press, New York, 1971, 4, pp. 417–447 Search PubMed; (d) M. Li, W. Ding, B. Baruah, D. C. Crans and R. Wang, J. Inorg. Biochem., 2008, 102, 1846–1853 CrossRef CAS; (e) Jr. G. R. Beck, E. C. Sullivan, E. Moran and B. Zerler, J. Cell. Biochem., 1998, 68, 269–280 CrossRef; (f) A. I. Vovk, V. I. Kalchenko, S. A. Cherenok, V. P. Kukhar, O.V. Muzychka and M.O. Lozynsky, Org. Biomol. Chem., 2004, 2, 3162–3166 RSC.
- G. H. Jeffery, J. Basset, J. Mendham and R. C. Denny, Vogel's textbook of quantitative inorganic analysis including elementary instrumental analysis,London, Longman Group Ltd, 4th edn, 1978, pp. 486–487 Search PubMed.
- M. K. Chaudhuri, S. K. Ghosh and N. S. Islam, Inorg. Chem., 1985, 24, 2706–2707 CrossRef CAS.
- C. V. Ferreira, E. M. Taga and H. Aoyama, Plant Sci., 1999, 147, 49–54 CrossRef CAS.
- (a) Z. S. Nurkeeva, V. V. Khutoryanskiy, G. A. Mun, M. V. Sherbakova, A. T. Ivaschenko and N. A. Aitkhozhina, Eur. J. Pharm. Biopharm., 2004, 57, 245–249 CrossRef CAS; (b) M. J. Fonseca, A. Cabanes, M. A. Alsina and F. Reig, Int. J. Pharm., 1996, 133, 265–268 CrossRef CAS; (c) E. Turos, J. Y. Shim, Y. Wang, K. Greenhalgh, G. S. K. Reddy, S. Dickey and D. V. Lim, Bioorg. Med. Chem. Lett., 2007, 17, 53–56 CrossRef CAS; (d) S. Garg, K. Vermani, A. Garg, R. A. Anderson, W. B. Rencher and L. J. D. Zaneveld, Pharm. Res., 2005, 22, 584–595 CrossRef CAS.
- (a) M. H. Dickman and M. T. Pope, Chem. Rev., 1994, 94, 569–584 CrossRef CAS; (b) N. J. Campbell, A. C. Dengal, C. J. Edwards and W. P. Griffith, J. Chem. Soc., Dalton Trans., 1989, 1203–1207 RSC; (c) A. C. Dengel, W. P. Griffith, R. D. Powell and A. C. Skapski, J. Chem. Soc., Dalton Trans., 1987, 991–995 RSC.
- (a) B. L. Rivas and I. Moreno-Villoslada, J. Appl. Polym. Sci., 1998, 70, 219–225 CrossRef CAS; (b) B. L. Rivas and I. Moreno-Villoslada, J. Phys. Chem. B, 1998, 102, 6994–6999 CrossRef CAS; (c) B. L. Rivas and I. Moreno-Villoslada, Chem. Lett., 2000, 29, 166–167 CrossRef.
- B. F. Sels, D. E. De Vos, M. Buntinx and P. A. Jacobs, J. Catal., 2003, 216, 288–297 CrossRef CAS.
- (a) K. Nakamoto, Infrared and Raman Spectra of Inorganic and Co-ordination Compounds, Part B,Wiley and Sons, New York, 5th edn, 1997, pp. 60 Search PubMed; (b) G. B. Deacon and R. J. Phillips, Coord. Chem. Rev., 1980, 33, 227–250 CrossRef CAS.
- (a) C. Djordjevic, M. Lee and E. Sinn, Inorg. Chem., 1989, 28, 719–723 CrossRef CAS; (b) P. Schwendt, P. Svancarek, L. Kuchta and J. Marek, Polyhedron, 1998, 17, 2161–2166 CrossRef CAS.
- (a) H. Li and C. P. Tripp, Langmuir, 2004, 20, 10526–10533 CrossRef CAS; (b) F. Jones, J. B. Farrow and W. Van-Bronswijk, Langmuir, 1998, 14, 6512–6517 CrossRef CAS.
- (a) Y. Feng, A. Schmidt and R. A. Weiss, Macromolecules, 1996, 29, 3909–3917 CrossRef CAS; (b) R. B. Penland, S. Mizushima, C. Curran and J. V. Quagliano, J. Am. Chem. Soc., 1957, 79, 1575–1578 CrossRef CAS; (c) W. E. Bull, S. K. Madan and J. E. Willis, Inorg. Chem., 1963, 2, 303–306 CrossRef CAS; (d) R. Murugan, S. Mohan and A. Bigotto, J. Korean Phys. Soc., 1998, 31, 505–512 Search PubMed; (e) R. M. Silverstein, G. C. Bassler and T. C. Morrill, Spectrometric identification of Organic Compounds, 5th ed.John Wiley and Sons, New York, 1991, pp. 122 Search PubMed; (f) R. M. Silverstein, G. C. Bassler and T. C. Morrill, Spectrometric identification of Organic Compounds,John Wiley and Sons, New York, 5th edn, 1991, pp. 129 Search PubMed.
- (a) B. L. Rivas, G. V. Seguel and K. E. Geckeler, J. Appl. Polym. Sci., 2002, 85, 2546–2551 CrossRef CAS; (b) Z. M. Sun, J. G. Mao, Y. Q. Sun, H. Y. Zeng and A. Clearfield, Inorg. Chem., 2004, 43, 336–341 CrossRef CAS.
- (a) J. D. Van Dyke and K. L. Kasperski, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 1807–1823 CrossRef CAS.
- D. D. Jiang, Q. Yao, M. A. McKinney and C. A. Wilkie, Polym. Degrad. Stab., 1999, 63, 423–434 CrossRef CAS.
- (a) D. Bayot, B. Tinant and M. Devillers, Inorg. Chim. Acta, 2004, 357, 809–816 CrossRef CAS; (b) L. L. G. Justino, M. L. Ramos, M. M. Caldeira and V. M. S. Gil, Inorg. Chim. Acta, 2003, 356, 179–186 CrossRef CAS; (c) S. E. Jacobson, R. Tang and F. Mares, Inorg. Chem., 1978, 17, 3055–3063 CrossRef CAS; (d) L. Pettersson, I. Andersson and A. Gorzsas, Coord. Chem. Rev., 2003, 237, 77–87 CrossRef CAS; (e) V. Conte, F. D. Furia and S. Moro, J. Mol. Catal., 1994, 94, 323–333 CrossRef CAS.
- (a) A. Bodor, I. Banyai and I. Toth, Coord. Chem. Rev., 2002, 228, 175–186 CrossRef CAS; (b) Z. H. Zhou, Y. F. Deng, Z. X. Cao, R. H. Zhang and Y. L. Chow, Inorg. Chem., 2005, 44, 6912–6914 CrossRef CAS; (c) L. L. G. Justino, M. L. Ramos, M. M. Caldeira and V. M. S. Gil, Inorg. Chim. Acta, 2000, 311, 119–125 CrossRef CAS; (d) M. Matzapetakis, C. P. Raptopoulou, A. Terzis, A. Lakatos, T. Kiss and A. Salifoglou, Inorg. Chem., 1999, 38, 618–619 CrossRef CAS.
- F. O. Garces, K. Sivadasan, P. Somasundaran and N. J. Turro, Macromolecules, 1994, 27, 272–278 CrossRef CAS.
- A. Shisheva, O. Ikonomov and Y. Shechter, Endocrinology, 1994, 134, 507–510 CrossRef CAS.
- Y. Shechter, I. Goldwaser, M. Mironchik, M. Fridkin and D. Gefel, Coord. Chem. Rev., 2003, 237, 3–11 CrossRef CAS.
- (a) J. B. Vincent, M. W. Crowder and B. A. Averill, Biochemistry, 1991, 30, 3025–3034 CrossRef CAS; (b) J. Shabanowitz, N.-Z. Zhu, T. Zirino, B. A. Averill, S. T. Daurat-Larroque, J. G. Shewale, R. M. Roberts and K. Brew, Biochem. Biophys. Res. Commun., 1987, 144, 1154–1160 CrossRef; (c) C. M. Ketcham, R. M. Roberts, R. C. M. Simmen and H. S. Nick, J. Biol. Chem., 1989, 264, 557–563 CAS; (d) D. K. Lord, N. C. P. Cross, M. A. Bevilacgua, S. H. Rider, P. A. Gorman, A. V. Groves, D. W. Moss, D. Sheer and T. M. Cox, Eur. J. Biochem., 1990, 189, 287–293 CrossRef CAS.
- (a) P. J. Stankiewicz and M. J. Gresser, Biochemistry, 1988, 27, 206–212 CrossRef CAS; (b) G. Soman, Y. C. Chang and D. J. Graves, Biochemistry, 1983, 22, 4994–5000 CrossRef CAS.
- (a) Y. S. Heo, J. M. Ryu, S. M. Park, J. H. Park, H. C. Lee, K. Y. Hwang and J. Kim, Exp. Mol. Med., 2002, 34, 211–223 CAS; (b) R. L. Van-Etten, P. P. Waymack and D. M. Rehkop, J. Am. Chem. Soc., 1974, 96, 6782–6785 CrossRef CAS.
- (a) V. Ravindranath, in Methods in enzymology, ed. L. Packer, Academic, New York, 1994, 233, pp. 613 Search PubMed; (b) V. Lopez, T. Stevens and R. N. Lindquist, Arch. Biochem. Biophys., 1976, 175, 31–38 CrossRef CAS.
- (a) X. W. Zhou, Q. X. Chen, Z. Chen, Z. Q. He and H. M. Zhou, Biochemistry, 2000, 65, 1424–1428 CAS; (b) X. W. Zhou, Z. L. Zhuang and Q. X. Chen, J. Protein Chem., 1999, 18, 735–740 CrossRef CAS; (c) C. M. Vescina, V. C. Salice, A. M. Cortizo and S. B. Etcheverry, Biol. Trace Elem. Res., 1996, 53, 185–191 CrossRef CAS; (d) G. Huyer, S. Liu, J. Kelly, J. Moffat, P. Payette, B. Kennedy, G. Tsaprailis, M. J. Gresser and C. Ramachandran, J. Biol. Chem., 1997, 272, 843–851 CrossRef CAS; (e) A. S. Tracey, G. R. Willsky and E. S. Takeuchi, Vanadium: Chemistry, Biochemistry, Pharmacology and Practical Application, CRC Press and Taylor & Francis Group, Boca Raton, 2007 Search PubMed; (f) B. I. Posner, R. Faure, J. W. Burgess, A. P. Bevan, D. Lachance, G. Zhang-Sun, I. G. Fantus, J. B. Ng, D. A. Hall, B. Soo Lum and A. Shaver, J. Biol. Chem., 1994, 269, 4596–4604 CAS; (g) A. P. Bevan, P. G. Drake, J. F. Yale, A. Shaver and B. I. Posner, Mol. Cell. Biochem., 1995, 153, 49–58 CrossRef CAS.
- P. Hazarika, D. Kalita, S. Sarmah, R. Borah and N. S. Islam, Polyhedron, 2006, 25, 3501–3508 CrossRef CAS.
- S. P. Das, J. J. Boruah, H. Chetry and N. S. Islam, Tetrahedron Lett., 2012, 53, 1163–1168 CrossRef CAS.
- C. M. Jonsson, L. C. Paraiba and H. Aoyama, Ecotoxicology, 2009, 18, 610–619 CrossRef CAS.
- G. E. Meister and A. Butler, Inorg. Chem., 1994, 33, 3269–3275 CrossRef CAS.
- V. Khanna, M. Jain, M. K. Barthwal, D. Kalita, J. J. Boruah, S. P. Das, N. S. Islam, T. Ramasarma and M. Dikshit, Pharmacol. Res., 2011, 64, 274–282 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |