Hydrogen bond directed self-assembly of cyclic dipeptide derivatives: gelation and ordered hierarchical architectures†
Shivaprasad
Manchineella
and
T.
Govindaraju
*
Bioorganic Chemistry Laboratory, New Chemistry Unit, Jawaharlal Nehru Centre for Advanced Scientific Research, Jakkur, Bangalore 560064, India. E-mail: tgraju@jncasr.ac.in; Fax: +918022082627
First published on 1st May 2012
Abstract
Cyclo(Gly-L-Lys) ε-amino derivatives (1 and 2) were synthesised and exploited as low molecular weight (LMW) gelators and ordered hierarchical structures. Cyclic dipeptide (CDP) derivative 1 forms organogels in hexane, carbon tetrachloride, toluene and chloroform at minimal gelation concentrations (MGC) 5.5 wt%, 4.4 wt%, 2.6 wt% and 3.2 wt% respectively. Organogel (toluene) and hydrogel were obtained using CDP 2 at a MGC of 3.2 wt% and 2.5 wt% respectively. Interestingly, CDP 2 exhibits excimer emission in chloroform and self-assembles into ordered microporous hierarchical structures. The insight into supramolecular self-assembly of CDPs into nanofibres and ordered hierarchical structures was achieved by microscopy analysis. Presence of a network of strong intermolecular hydrogen bonding and aromatic π–π interactions was validated through concentration and temperature dependent NMR studies.
In recent years, the scientific community has shown a strong interest in the development of low molecular weight (LMW) organo- and hydrogelators as a class of soft materials.1 The LMW gelators can be prepared with ease starting from easily accessible starting materials. Unlike in the case of high molecular weight polymeric gelators wherein covalent bonding and large association constants are required for gelation, the LMW gels are formed by noncovalent interaction-driven supramolecular self-assembly of designed small molecules. Noncovalent interactions such as hydrogen bonding, aromatic π–π interactions and van der Waals forces are mainly responsible for the self-assembly of small molecules.2 The self-assembled fibrillar structures then entangle the solvent molecules owing to high surface tension and capillary action.3 Temperature, light, enzymes, pH and shear force can be used as external stimuli for triggering the process of gelation.4–7 The LMW gels formed through supramolecular self-assembly are reversible, the property which makes them most interesting soft materials. The applications of LMW gels are vast and includes drug delivery, cell culture, templates for the synthesis of inorganic nanomaterials and in the synthesis and stabilisation of nanoparticles.8–10 Among LMW gelators, small peptide based gels are promising owing to their structural similarity with various beneficial biological materials as well as those implicated in diseased states such as Alzheimer's, Parkinson's, type II diabetes and prion diseases.11–13
The well-ordered hierarchical architectures are of great importance in the field of nanoscience owing to their numerous applications. The top-down approach, the classic way of preparing such architectures, limits the synthesis of ordered architectures in the case of organic molecules owing to their instability. A supramolecular self-assembly-based bottom-up approach for the synthesis of ordered microporous hierarchical architectures is increasingly attracting the interest of researchers owing to its ease of preparation and solution phase processing in comparison to expensive and tedious procedures involved in top-down approaches. These ordered microporous structures will find potential applications as catalysts, microreactors and tissue engineering scaffolds.14,15 Despite the advantage of bottom-up approaches over top-down approaches there are only few reports available for the synthesis of regular hierarchical structures using small molecules and peptides in the literature.16
Amongst the peptide-based LMW gelators, dipeptide and cyclic dipeptide (CDP) derived molecules presents a class of efficient gelators for organic solvents, organic fluids and ionic liquids.17,18 In particular, CDPs also familiar as diketopiperazines (DKP) are the smallest possible cyclic form of peptides and are known to form intermolecular hydrogen bonded molecular ladders and layers.19–21 CDPs are quite often observed in nature with a specific biological activity and in vitro CDPs are being exploited as antibiotics and asymmetric catalysts.22,23 The outstanding structural stability and resistance towards the proteolytic enzymes makes them attractive materials for the preparation of soft materials. We have recently reported two designer cyclic dipeptides and their molecular self-assembly into robust two dimensional nano- and meso-sheets and one dimensional fibre-bundles with structural hierarchies similar to that of graphene and natural fibres respectiely.21 Similar to any LMW gelators, noncovalent interactions are the main driving force behind the gelation using CDPs, which are further strengthened by their unique structural features. The rigid structure and strong intermolecular interactions through hydrogen bonding among other noncovalent interactions enable CDPs to form ordered morphologies.21
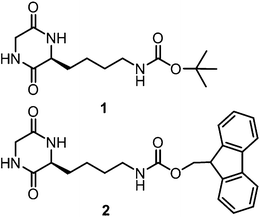
Herein we report the synthesis and systematic gelation study of 1 and 2. CDPs 1 and 2 form gels through their self-assembled nanostructures and 2 also forms ordered microporous hierarchical structures in chloroform. The synthesis of CDPs 1 and 2 was accomplished through a sequence of reactions (Scheme S1 and S2, ESI†). A sequential heating and cooling procedure was used as a trigger for the formation of organogels of CDPs. Specifically, gelation was achieved by heating a specified concentration of CDP solution in a closed vial at around 45–50 °C and cooling the obtained clear solution to room temperature. The ability of CDPs 1 and 2 to form organogels was eventually determined by inverting the vials upside down. The absence of any solvent flow in the vials confirmed the formation of the organogel (Fig. 1). Table 1 shows the minimum gelation concentrations (MGCs) of CDPs in different solvents which were obtained through the inversion tube method (Fig. 1a–e). The organogels of 1 in chloroform and other solvents were found to be stable for several weeks while that of 2 for a few weeks. Next, the organogel of 1 with hexane was facilitated over a large area petri dish. The formed gel could be lifted using a spatula without any deformation or loss of structural integrity of the soft material (Fig. 1f). In addition, we have further demonstrated the physical entrapment of guest molecules using an organogel of 1 in chloroform. CDP 1 containing 0.1 wt% of rhodamine B and curcumin was found to undergo gelation in chloroform physically entrapping these dye and drug molecules as indicated by their visible and fluorescent colours (Fig. 1g–j). A weighed amount of 2 (50 mg mL−1) dissolved in DMSO and water was added to achieve hydrogelation at MGC (Fig. 1e). Thus the obtained hydrogel was found to be relatively less stable. The molecularly dissolved sample of 2 (10 μM) in chloroform showed an emission band (Emax) at 310 nm and a weak emission band at 460 nm when excited at 260 nm (Fig. 2). The fluorescence emission band at 310 nm red-shifted (15 nm) and the intensity decreased gradually with a large enhancement at 460 nm as a function of concentration (10–100 μm). The emission peak at 460 nm is the characteristic excimer emission of the Fmoc-group along with the J-type aggregation which is an indication of strong intermolecular interaction.24 CDP 1 did not form hydrogels under the studied experimental conditions. The reversibility (gel–sol) of the obtained gels can be achieved either by increasing the temperature of the gels above 45 °C or by sonication for 15 min. The LMW gelators enable their molecular self-assembly into gels either by formation of nanofibres, nanoribbons or nanosheets. Insight into the morphology of self-assembled CDP organogelators in different solvents was obtained by field emission scanning electron microscopy (FESEM) analysis.
 | ||
| Fig. 1 Photographs of organogels. (a) 1 in carbontetrachloride (4.4 wt%), (b) 1 in toluene (2.6 wt%), (c) 1 in chloroform (3.2 wt%), (d) 2 in toluene (3.2 wt%), (e) 2 in water (2.5 wt%), (f) 1 in hexane (5.5 wt%), formed over a large area petri dish and was mechanically lifted using a spatula, (g) and (h) rhodamine B and curcumin entraped organogel of 1 in choloroform under visible light and (i) and (j) corresponding fluorescent organogels under UV light. | ||
 | ||
| Fig. 2 Emission spectra of 2 in chloroform with increasing concentration (10, 20, 30, 50, 60, 70, 80, 90 and 100 μM). (Full spectra are not shown to avoid the overlapped first overtone band of 310 nm at around 521 nm.) | ||
The xerogel of 1 in chloroform revealed the presence of an entangled network of nanofibres having a diameter of a few nanometres and extending to >100 μm in length (Fig. 3a). The xerogel morphology of 1 in carbontetrachloride, hexane and toluene was found to be nanoribbons extending to >100 μm length scale (Fig. 3b–d). The nanoribbons were very closely entangled and in the case of toluene, the formation of bundled structures was observed (Fig. 3d; inset). The xerogel morphology of 2 in toluene showed the presence of tightly packed nanofibres (Fig. 3e). FESEM analysis of 2 (100 μM) showed uniform distribution of microporous hierarchical architectures (Fig. 3f). The ordered microporous hierarchical structures observed in FESEM analysis were of 1–2 μm in size.
 | ||
| Fig. 3 FESEM images of xerogels of 1 in chloroform (a), carbontetrachloride (b), hexane (c), toluene, inset showing bundled nanoribbon structures (d), 2 in toluene (e), and ordered microporous hierarchical arrays of 2 in chloroform (f). | ||
The crucial role of hydrogen bonding in the formation of network of supramolecular nanostructures leading to the formation of organogels was understood by concentration and temperature dependent NMR spectroscopy.18,251H NMR spectra of 1 recorded at 25 °C with varying concentrations (3.5 to 56.0 mM) in dimethyl sulfoxide-d6 showed linear downfield shifts of amide and carbamate protons (Fig. S2, ESI†). Such downfield shifts of protons with an increase in concentration is clear evidence of the presence of intermolecular hydrogen bonding between CDP units. CDP 1 forms a molecular ladder through amide-protons (NH1 and NH2) and amide-carbonyls via pair of intermolecular hydrogen bonding to two of its neighbours. Similarly, carbamate-proton (NH3) and carbamate-carbonyl were involved in the formation of another linear chain of single intermolecular hydrogen bonding. 1H NMR spectra of 1 (56.0 mM) recorded as a function of temperature (25 to 50 °C) showed an overall upfield shift of 0.12 ppm for amide-protons (Fig. 4a). Such an upfield shift of amide-protons was attributed to the weakening of intermolecular hydrogen bonding at higher temperatures. Thus downfield and upfield shifts of amide- and carbamate-protons, with an increase in concentration and temperature respectively, strongly support the presence of intermolecular hydrogen bonding and its role in the self-assembly of 1 to form supramolecular nanostructures. The concentration dependent 1H NMR spectra of 2 (2.9–24.5 mM) were recorded at 25 °C. Splitting and downfield shifts of amide- and aromatic-protons (Fmoc) of CDP 2 were observed (Fig. 4b). The downfield shift of amide-protons (0.03 ppm and 0.01 ppm) as a function of concentration, was attributed to the presence of strong intermolecular hydrogen bonding, similar to that observed for CDP 1. The splitting of Fmoc-protons, as indicated in Fig. 4b, was attributed to the presence of strong aromatic π–π interactions. Interestingly, a doublet (2H, 7.66–7.69 ppm) appeared for two aromatic protons of Fmoc-functionality as depicted in Fig. 4b split into two separate doublets (H) for each protons along with the doublet corresponding to one of the protons being downfield shifted from 7.67–7.69 to 7.83–7.85 ppm (H) at a 24.5 mM concentration of 2 (Fig. 4b(v)). Further, we observed the downfield shift and merging of the previously unaffected doublet with that of the already downfield shifted aromatic proton doublet after aging the NMR sample (Fig. 4b(vi)). This is an indication of time dependent strengthening and stabilization of aromatic interactions. Furthermore, merging of the unaffected proton with an already downfield shifted proton after aging the sample clearly indicates the initial J-type aggregation followed by transformation towards excimer-like arrangement as indicated by the excimer emission of 2 (Fig. 2). The temperature dependent 1H NMR spectra recorded for 2 (24.5 mM) from 25–60 °C showed an upfield shift of CDP core amide-protons and carbamate-proton and two of them merge with other upfield aromatic proton signals at temperatures >40 °C (Fig. 4c). These concentration and temperature dependent NMR spectroscopic studies discussed above prove the presence of strong intermolecular hydrogen bonding and aromatic π–π interactions and play a major role in the molecular self-assembly of 1 and 2. We propose a model for the self-assembly of 1 and subsequent formation of nanofibers as shown in Fig. 5. The hydrophobic t-butoxycarbonyl (Boc) group of CDP 1 is presumed to be the driving force for the organization of [N–H–O] hydrogen bonded molecular chains into fibrillar structures. These self-assembled nanofibrillar structures then entangle the solvent molecules owing to high surface tension and capillary action to form nanofibrous network as found in the organogels.3
 | ||
| Fig. 4 1H NMR studies of 1 and 2. Temperature dependent NMR spectra of 1 in DMSO-d6 (a). Concentration (b) and temperature (c) dependent NMR spectra of 2 in DMSO-d6. I: 25 °C, II: 30 °C, III: 40 °C, IV: 50 °C, V: 60 °C and i: 2.9 mM, ii: 4.9 mM, iii: 7.6 mM, iv: 12.0 mM, v: 24.5 mM of 2. vi: 24.5 mM of 2 after one day aging. | ||
 | ||
| Fig. 5 Proposed model for the hydrogen bond directed self-assembly of 1 into nanofibres which subsequently form organogels. | ||
In summary, we have demonstrated intermolecular hydrogen bond directed self-assembly of cyclic dipeptide derivatives 1 and 2 into nanofibres, nanoribbons and their subsequent gelation. Concentration and temperature dependent NMR studies clearly demonstrate the involvement of strong intermolecular hydrogen bonding and aromatic π–π interactions in the molecular self-assembly of 1 and 2. Aromatic and hydrogen bond interactions render the self-assembly of 2 into an ordered microporous hierarchical architecture, which corresponds to excimer emission of 2 in solution. We have also demonstrated that organogels can be used to physically entrap dye and drug molecules such as rhodamine B and curcumin. The gels reported herein can be used as entrapping agents, drug delivery systems and thermoresponsive soft materials.
Acknowledgements
We thank Prof. C. N. R. Rao, FRS for constant support, JNCASR, Department of Biotechnology (DBT), India, Innovative Young Biotechnologist Award (IYBA) to T.G. for financial support, and CSIR, New Delhi, for awarding JRF to SM.References
- (a) P. Terech, R. G. Weiss, Molecular gels: materials with self-assembled fibrillar networks, Springer, Dordrecht, 2006 Search PubMed; (b) W. H. Binder and O. W. Smrzka, Angew. Chem., Int. Ed., 2006, 45, 7324 CrossRef CAS; (c) D. J. Adams and P. D. Topham, Soft Matter, 2010, 6, 3707 RSC; (d) J. W. Steed, Chem. Commun., 2011, 47, 1379 RSC.
- E. L. Johnson, D. J. Adams and P. J. Cameron, J. Mater. Chem., 2011, 21, 2024 RSC.
- Z. Xie, A. Zhang, L. Ye and Z. Feng, Soft Matter, 2009, 5, 1474 RSC.
- (a) S. Yagai, T. Nakajima, K. Kishikiva, S. Kohmoto, T. Karatsu and A. Kitamura, J. Am. Chem. Soc., 2005, 127, 11134 CrossRef CAS; (b) Y. Huang, Z. Qiu, Y. Xu, J. Shi, H. Lin and Y. Zhang, Org. Biomol. Chem., 2011, 9, 2149 RSC.
- (a) A. Aggeli, M. Bell, L. M. Carrick, C. W. G. Fishwic, R. Harding, P. J. Mawar, S. E. Radford, A. E. Stron and N. Boden, J. Am. Chem. Soc., 2003, 125, 9619 CrossRef CAS; (b) D. Khatua, R. Maiti and J. Dey, Chem. Commun., 2006, 4903 RSC.
- (a) J. Brinksma, B. L. Feringa, R. M. Kellogg, R. Vreeker and J. V. Esch, Langmuir, 2000, 16, 9249 CrossRef CAS; (b) V. Percec, M. Peterca, M. E. Yurchenko, J. G. Rudick and P. A. Heiney, Chem.–Eur. J., 2008, 14, 909 CrossRef CAS.
- (a) S. Toledano, R. J. Williams, V. Jayawarna and R. V. Uljin, J. Am. Chem. Soc., 2006, 128, 1070 CrossRef CAS; (b) Z. Yang, P.-L. Ho, G. Liang, K. H. Chow, Q. Wang, Y. Cao, Z. Guo and B. Xu, J. Am. Chem. Soc., 2007, 129, 266 CrossRef CAS.
- (a) R. V. Uljin and A. M. Smith, Chem. Soc. Rev., 2008, 37, 664 RSC; (b) A. Vintiloiu and J. C. Leroux, J. Controlled Release, 2008, 125, 179 CrossRef CAS.
- (a) K.-H. Park, K. Na and H.-M. Chung, Biotechnol. Lett., 2005, 27, 227 CrossRef CAS; (b) M. W. Tibbitt and K. S. Anseth, Biotechnol. Bioeng., 2009, 103, 655 CrossRef CAS; (c) S. Denath, S. Anshupriya, S. Dutta and P. K. Das, Chem.–Eur. J., 2008, 14, 6870 CrossRef; (d) K. Tanmoy, S. Denath, D. Das, S. Anshupriya and P. K. Das, Langmuir, 2009, 25, 8639 CrossRef.
- (a) M. Kimura, S. Kobayashi, T. Kuroda, K. Hanabusa and H. Shirai, Adv. Mater., 2004, 16, 335 CrossRef CAS; (b) S. Ray, A. K. Das and A. banergee, Chem. Commun., 2006, 2816 RSC.
- (a) X. Li, Y. Kuang, H.-C. Lin, Y. Gao, J. Shi and B. Xu, Angew. Chem., Int. Ed., 2011, 50, 9365 CrossRef CAS; (b) B. Adhikari, J. Nanda and A. Banergee, Chem.–Eur. J., 2011, 17, 11488 CrossRef CAS.
- (a) L. Kreplak, J. Doucet, P. Dumas and F. Briki, Biophys. J., 2004, 87, 640 CrossRef CAS; (b) S. Keten, Z. Xu, B. Ihle and M. J. Buehler, Nat. Mater., 2010, 9, 359 CrossRef CAS.
- C. M. Dobson, Nature, 2003, 426, 884 CrossRef CAS.
- (a) S. J. Haswell and V. Skelton, TrAC, Trends Anal. Chem., 2000, 19, 389 CrossRef CAS; (b) S. S. Babu, S. Mahesh, K. K. Kartha and A. Ajayaghosh, Chem.–Asian J., 2009, 4, 824 CrossRef CAS.
- V. P. Shastri, I. Martin and R. Langer, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 1970 CrossRef CAS.
- (a) R. J. H. Hafkamp, M.C. Feiters and R. J. M. Nolte, Angew. Chem., Int. Ed. Engl., 1994, 33, 986 CrossRef; (b) W.-Y. Yang, J. H. Ahn, Y.-S. Yoo, N. K. Oh and M. Lee, Nat. Mater., 2005, 4, 399 CrossRef CAS.
- (a) K. Hanabusa, Y. Matsumoto, T. Miki, T. Koyama and H. Shirai, J. Chem. Soc., Chem. Commun., 1994, 1401 RSC; (b) K. Hanabusa, M. Matsumoto, M. Kimura, A. Kakehi and H. Shirai, J. Colloid Interface Sci., 2000, 224, 231 CrossRef CAS.
- Z. Xie, A. Zhang, L. Ye and Z.-G Feng, Soft Matter, 2009, 5, 1474 RSC.
- (a) E. Benedetti, P. Corradini and C. Pedone, J. Phys. Chem., 1969, 73, 2891 CrossRef CAS; (b) K. B. Joshi and S. Verma, Tetrahedron Lett., 2008, 49, 4231 CrossRef CAS.
- J. C. M.-Donald and G. M. Whitesides, Chem. Rev., 1994, 94, 2383 CrossRef.
- (a) T. Govindaraju, M. Pandeeswar, G. Jaipuria and H. S. Atreya, Supramol. Chem., 2011, 23, 487–492 CrossRef CAS; (b) T. Govindaraju, Supramol. Chem., 2011, 23, 759 CrossRef CAS.
- (a) Y. Sasaki, Y. Akutsu, M. Matsui, K. Suzuki, S. Sakurada, T. Sato and K. Kisara, Chem. Pharm. Bull., 1982, 30, 4435 CrossRef CAS; (b) C. Prasad, Peptides, 1995, 16, 151 CrossRef CAS; (c) F. R. Lucietto, P.J. Milne, G. Kilian, C. L. Frost and M. V. D. Venter, Peptides, 2006, 27, 2706 CrossRef CAS.
- (a) K. Tanaka, A. Mori and S. Inoue, J. Org. Chem., 1990, 55, 181 CrossRef CAS; (b) H. J. Kim and W. R. Jackson, Tetrahedron: Asymmetry, 1992, 3, 1421 CrossRef CAS; (c) M. Durini, F. A. Sahr, M. Kuhn, M. Civeria, C. Gennari and U. Piarulli, Eur. J. Org. Chem., 2011, 5599 CrossRef CAS.
- A. M. Smith, R. J. Williams, C. Tang, P. Coppo, R. F. Collins, M. L. Turner, A. Saiani and R. V. Uljin, Adv. Mater., 2008, 20, 37 CrossRef CAS.
- G. T. R. Palmore, T.-J. M. Luo, M. T. M.- Wieser, E. A. Picciotto and C. M. R. Paz, Chem. Mater., 1999, 11, 3315 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: Synthetic details, FESEM, NMR and mass data. See DOI: 10.1039/c2ra20342a/ |
| This journal is © The Royal Society of Chemistry 2012 |