Anchored palladium nanoparticles onto single walled carbon nanotubes: Efficient recyclable catalyst for N-containing heterocycles†
Subhankar
Santra
a,
Priyadarshi
Ranjan
a,
Parthasarathi
Bera
b,
Prasenjit
Ghosh
c and
Swadhin K.
Mandal
*a
aS. Santra, P. Ranjan, Dr S. K. Mandal, Department of Chemical Sciences, Indian Institute of Science Education and Research-Kolkata, Mohanpur-741252, India. E-mail: swadhin.mandal@iiserkol.ac.in; Tel: 91-9903676563; Fax: (+)91-33-25873020
bDr P. Bera, Surface Engineering Division, CSIR-National Aerospace Laboratories, Bangalore-560017, India
cDr P. Ghosh, Department of Chemistry and Physics, Indian Institute of Science Education and Research–Pune, Pune-411021, India
First published on 13th June 2012
Abstract
A convenient process has been developed to decorate palladium nanoparticles onto carboxylic acid functionalized single walled carbon nanotubes (SWNTs) via the thermolysis of palladium acetate. This single walled carbon nanotube–palladium nanoparticles (SWNT–PdNPs) composite has been investigated through a number of instrumental techniques such as transmission electron microscopy (TEM), scanning electron microscopy (SEM), atomic force microscopy (AFM), UV-vis-NIR spectroscopy, resonance Raman spectroscopy and X-ray photoelectron spectroscopy (XPS), which disclose that the palladium nanoparticles are attached by carboxylic acid groups onto the surface of SWNTs. The SWNT–PdNPs composite has been tested as an efficient heterogeneous nanocatalytic system for copper free acyl Sonogashira reaction. A library of ynones was synthesized in high yields under mild reaction condition. The catalyst was recovered and recycled successfully up to seven times. This simple protocol was further broadened in the synthesis of trimethylsilyl-ynones (TMS-ynones) and explored in the one-pot multicomponent synthesis of 2,4-disubstituted pyrimidines.
Introduction
Tiny sized metal nanoparticles have appeared as the new face of catalysis in recent years1 towards various organic transformations2 as well as in electronics, information technology, sensor development and biomedical sciences due to their potential tunability in terms of size, shape, and activity.3 The high surface to volume ratio of palladium nanoparticles and their versatility to act as catalysts for a variety of organic transformations have made them one of the most admired one in modern organic synthesis, especially in the field of nanocatalysis. In this perspective, dispersed palladium nanocatalysts in solution tend to lose their catalytic activity because of palladium metal aggregation and precipitation.4 In addition, they experience the inherent problem of catalyst separation from the reaction mixture for reprocessing. The heterogenization of the homogeneous palladium catalyst is a smart key to conquer such troubles. This can be accomplished by employing metal nanoparticles on a solid support. Many different materials have already been employed to support metal nanoparticles such as mesoporous silica,5 zeolites,6 activated carbons,7 metal oxides,8 ionic liquids,9 microporous polymers,10 dendrimers11etc., while chemically functionalized single walled carbon nanotubes (SWNTs) appear as potential candidates for the stabilization of metal nanoparticles owing to their large surface area and presence of organic functional groups. Functional groups such as carboxyl moieties attached to SWNTs may be helpful in gripping the metal nanoparticles to its surface. The high electrical conductivity with a unique combination of mechanical, thermal, optical, optoelectronic and electronic properties have made carbon nanotubes a special material.12 It is well documented that pristine SWNTs mostly exist as bundles having strong intermolecular cohesive forces (0.5 eV nm−1) and thus exhibit inert surfaces and poor dispersion in various solvents resulting in a futile decoration of metal nanoparticles on their surface.13 Therefore, chemical functionalization of carbon nanotubes for specific application has been a major growth area.14 The presence of defects at the tube surface, mainly acid groups through oxidation, brings benefits such as introducing anchor points for a metal nanoparticle's attachment as the carboxylic acid functionality has already displayed an extensive range of metal coordination chemistry as a versatile ligand.15 Thus, these carboxylic acid groups chemically attached to SWNTs may behave as nucleation centers for anchoring metal nanoparticles. Numerous efforts have been devoted towards the fabrication of SWNT-based metal nanocomposites including vapour deposition,16 impregnation,17 plasma treatment,18 supercritical fluid method,19 polymer wrapping,20 self-assembly,21 electrochemical decoration22 and thermal decomposition of metal salts.23 Among all these methods, thermal decomposition or pyrolysis of metal salts remains as the most straightforward and easy handling method to anchor metal nanoparticles on the surface of carbon nanotubes.24 The metal nanoparticle–nanotube heterogeneous architectures have been employed in fuel cells,25 electrocatalytic reactions,26 different gas sensors27 and only a few reports include catalytic applications in Heck, Suzuki, Stille, and Sonogashira coupling reactions.28 However, the catalytic application of nanotube–metal nanoparticle composites in acyl Sonogashira coupling reactions has been missing till to date. In the present study, a simple synthetic process was adopted to anchor palladium nanoparticles (PdNPs) onto the surface of carboxylic acid functionalized SWNTs following our recent approach to synthesize heterogeneous PdNPs anchored in a polymer matrix.29 We tested the SWNT–PdNPs as a catalyst in acyl Sonogashira reaction under copper free condition to synthesize a library of ynones. The “ynones” are multipurpose isolable intermediates in the synthesis of pharmaceutically prominent and biologically active N-heterocyclic compounds, such as pyrroles,30 pyrazoles,31 isoxazoles,32 pyrimidines,33 quinolines,34 and tetrahydro-β-carbolines.35 Synthetic methods for the preparation of ynones utilize well defined palladium catalysts for coupling of terminal alkynes with an acid chloride (acyl Sonogashira reaction)29,36 or with organic halides in the presence of carbon monoxide (carbonylative Sonogashira reaction).37 A recent literature survey unveils that most of these studies exploited the use of copper as a co-catalyst, which in turn makes the separation of the products more tedious, generating alkyne homocoupling bi-products. Nevertheless, the acyl Sonogashira reaction remains the more straightforward process for the generation of ynones avoiding poisonous carbon monoxide gas, and it can also be extended to design sequential reactions in a one-pot fashion leading to pharmaceutically important heterocycles.29 Thus we embellish the carboxylic acid functionalized SWNT's surface with palladium nanoparticles to quench the thirst in developing copper free recyclable palladium catalysts for acyl Sonogashira coupling with mild reaction conditions yielding a library of ynones with excellent yield. Additionally, SWNT-PdNPs composite displays promising catalytic efficiency for trimethylsilylacetylene (analogous of terminal alkyne) furnishing derivatives of trimethylsilyl-ynones (TMS-ynones) which are further utilized in the synthesis of 2,4-disubstituted pyrimidines in high yields through multicomponent and sequential one-pot processes (Scheme 1).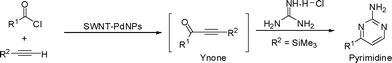 | ||
| Scheme 1 One-pot synthesis of 2,4-disubstituted pyrimidines catalyzed by SWNT-PdNPs using acyl Sonogashira reaction protocol under copper free condition. | ||
Results and discussion
As a part of our ongoing interest in developing palladium nanocatalysts for different organic transformations,29,38 herein, we utilized carboxylic acid functionalized SWNTs as templates for anchoring palladium nanoparticles via thermolysis of palladium acetate under inert atmosphere avoiding the use of any external hazardous reducing agents (Scheme 2). | ||
| Scheme 2 A schematic representation for the synthesis of SWNT–PdNPs considering a small part of the nanotube–nanoparticle architectures. | ||
In the current strategy, SWNT–PdNPs nanocomposite can be accomplished after mixing carboxylic acid functionalized SWNTs and palladium acetate in dry DMF followed by one hour sonication and thermal treatment at 95 °C for four hours. The as-synthesized SWNT–PdNPs were characterized by transmission electron microscopy (TEM), energy dispersive X-ray spectrum (EDX), scanning electron microscopy (SEM), atomic force microscopy (AFM), ICP-AES, X-ray photoelectron spectroscopy (XPS), UV-vis-NIR spectroscopy, and resonance Raman spectroscopy.
TEM images, recorded on a carbon–copper grid following a drop-cast method from a very dilute sample in DMF, revealed the presence of palladium particles having nanospheric dimension (Fig. 1A,B) in between the range 5 to 14 nm (Fig. 1C) and EDX spectrum collected from TEM confirmed the presence of palladium in the SWNT–PdNPs sample (see ESI†). The SEM study revealed the presence of carbon nanotubes in the sample (Fig. 2). AFM images recorded on a glass cover slip slide after spotting a drop from a very dilute solution of SWNT–PdNPs in DMF revealed the existence of spherical PdNPs on the surface of carboxylic acid functionalized SWNTs (Fig. 3) while the height of the nanoparticles was determined as ∼20 nm calculated from the height profile diagram considering large shiny metallic nanoclusters (see ESI†). The loading of palladium (wt%) in the present catalytic material was investigated through inductively coupled plasmon atomic emission spectroscopy (ICP-AES) where the analysis revealed 33% Pd loading in the SWNT–PdNPs sample.
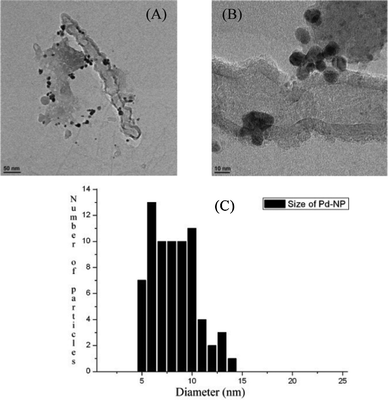 | ||
| Fig. 1 (A) TEM image of SWNT–PdNPs recorded on a carbon–copper grid; (B) magnified TEM image of SWNT–PdNPs in 10 nm scale revealing the presence of palladium nanoparticles attached to single walled carbon nanotubes; (C) size distribution histogram of palladium nanoparticles decorated on single walled carbon nanotubes. | ||
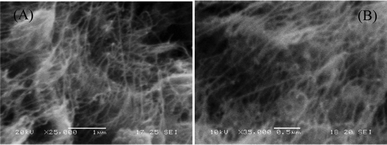 | ||
| Fig. 2 SEM images of SWNT–PdNPs (A) in 1 μm scale and (B) in 0.5 μm scale. | ||
 | ||
| Fig. 3 (A) AFM image of SWNT–PdNPs showing palladium nanoparticles concentrated on the single walled carbon nanotubes and (B) magnified AFM image of SWNT-PdNPs. | ||
To have a better insight into the present material, we analyzed SWNT-PdNPs further by a combination of optical (UV-vis-NIR) and resonance Raman spectroscopic studies. UV-vis-NIR spectroscopy is recognized as a significant method for the characterization of electronic band structures of SWNTs. It has been well documented that although the electronic structure of SWNTs is reminiscent of 2-D graphene sheets, it produces a series of spikes due to van Hove singularities in its density of states (DOS).39 Electronic transitions between these singularities in the SWNT valence and conduction bands give rise to prominent features in the UV, visible, and NIR spectral regions (Fig. 4).40 The semiconducting SWNTs typically show three absorbance bands, termed S11–S33 (where S11 lies in between 1700–2100 nm region, S22 lies in between 850–1200 nm region and S33 lies in between 400–500 nm region), while metallic SWNTs show one absorbance band, termed M11 (600–800 nm region). However, the principal point of interest lies in the change of optical spectra of SWNTs after palladium metal incorporation. Smalley and co-workers showed a significant enhancement in conductivity of SWNTs bundles after doping with potassium metal41 while Haddon and co-workers had shown that this type of doping leads to a decrease in intensity of the first semiconducting interband transition (S11) that occurs in the near-infrared (NIR) region (1700–2000 nm) of the spectrum (Fig. 4B), which may be attributed to the presence of holes into the valence band of the semiconducting SWNTs.42 In recent years, this piece of evidence was further supported by Star and co-workers where they located a ∼30% decrease in the absorbance of the SWNT (S11 band) after deposition of gold nanoparticles onto SWNTs43 due to the removal of electronic density from the SWNT valence band.42a,44 Additionally, they experienced similar trends in the S11 band absorbance for platinum and rhodium nanoparticles also.43 In the present study, we also observed a sharp decrease in intensity of the S11 absorbance band (Fig. 4B) after incorporation of palladium nanoparticles onto carboxylic acid functionalized SWNT bundles while the M11 transitions are relatively unaffected as revealed by previous studies (Fig. 4A).45
 | ||
| Fig. 4 UV-vis-NIR spectra of carboxylic acid functionalized SWNTs (black line) and SWNT–PdNPs (red line) recorded after dispersing in N,N-dimethylformamide in the absorbance range (A) 300–1500 nm and (B) 1700–2000 nm. | ||
In addition to the optical spectroscopy, resonance Raman spectroscopy also provides useful information regarding the interaction of PdNPs and carboxylic acid functionalized SWNTs. To notice any observable difference in the case of SWNT–PdNPs from its precursor material (SWNT-COOH), a 488 nm laser source (Fig. 5) was utilized. Chemically functionalized SWNTs exhibit the following characteristic Raman modes: the diameter-dependent radial breathing mode (RBM, observable in between 100–300 nm range) in which all the carbon atoms are moving in-phase in the radial direction, the higher frequency tangential mode (G-band, observable in between 1500–1600 nm range) and the disorder band (D-band, observable in between 1250–1400 nm region), which includes the presence of sp3-hybridized carbon atoms in SWNTs, and normally represented as a proof of the disruption of the aromatic system of π-electrons on the nanotube sidewalls by the attached functional groups.46 Thus the presence of D-bands in SWNT-COOH as well as in the catalyst material (SWNT-PdNPs) upon excitation with the laser source (Fig. 5) has been rationalized. The ratio of the intensities of the D- and G-bands (ID/IG) remains as a useful index to provide information about the degree of graphitization of SWNTs. The lower the value higher will be the graphitic character of SWNTs. Upon excitation with 488 nm laser source we found a slight decrease in the ID/IG value for SWNT–PdNPs (0.36) compared to the SWNT-COOH material (0.42), which may be attributed to the fact that the palladium nanoparticles utilize the carboxylic acid groups as its anchoring point without affecting the π-electrons network of the SWNT-COOH, and a similar observation was noted by Tai and co-workers in the case of carboxylic acid functionalized few-walled carbon nanotubes upon palladium nanoparticles decoration.47
 | ||
| Fig. 5 Resonance Raman spectra of palladium nanoparticles decorated carboxylic acid functionalized SWNT sample (red solid line) and carboxylic acid functionalized SWNT sample (black solid line) recorded using a 488 nm laser source (note that the scales along Y-axis in Fig. 5 have been modified for the sake of clarity). | ||
To focus prominently on the interaction between PdNPs and carboxylic acid functionalized SWNTs in the present material (SWNT–PdNPs) we have carried out a detailed XPS study. XPS of Pd(3d) core level in the SWNT–PdNPs sample is shown in Fig. 6. Pd(3d) spectral envelop indicates that PdNPs are present in various oxidation states. The Pd(3d5/2,3/2) peaks could be resolved into two sets of spin–orbit doublets. Pd(3d5/2,3/2) peaks were fitted by constraining the spin–orbit separation of 5.2 eV and ratio of doublet intensities at 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2. Accordingly, Pd(3d5/2,3/2) peaks observed at 335.2 and 340.4 eV could be assigned to Pd metal only, whereas peaks at 337.4 and 342.4 eV as seen in the deconvoluted spectrum correspond to Pd(3d5/2,3/2) peaks of Pd2+ species. All these values for Pd metal and Pd2+ species are close to the values reported in the literature.48 Comparing the intensity of peaks assigned to Pd metal and Pd2+ species in the deconvoluted spectrum, it reveals that around 60% of total Pd is in a metallic state and the rest of the Pd in the sample is present as a Pd2+ species. Thus, XPS analysis demonstrates that the SWNT–PdNPs sample contains mainly Pd metal with a certain amount of Pd2+ species.
:2. Accordingly, Pd(3d5/2,3/2) peaks observed at 335.2 and 340.4 eV could be assigned to Pd metal only, whereas peaks at 337.4 and 342.4 eV as seen in the deconvoluted spectrum correspond to Pd(3d5/2,3/2) peaks of Pd2+ species. All these values for Pd metal and Pd2+ species are close to the values reported in the literature.48 Comparing the intensity of peaks assigned to Pd metal and Pd2+ species in the deconvoluted spectrum, it reveals that around 60% of total Pd is in a metallic state and the rest of the Pd in the sample is present as a Pd2+ species. Thus, XPS analysis demonstrates that the SWNT–PdNPs sample contains mainly Pd metal with a certain amount of Pd2+ species.
 | ||
| Fig. 6 XPS of the Pd(3d) core level in SWNT–PdNPs. | ||
Fig. 7A displays deconvoluted C(1s) spectra. Spectral envelop of C(1s) core level of the SWNT–PdNPs sample is broad and asymmetrical indicating that several carbon components are present in the sample. Accordingly, peaks at 284.5, 285.4, 286.4, 287.5, and 288.9 eV observed in the sample could stand for C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, C–C, C–O or C–OH, CO, and O–CO, respectively. The peak at 291.0 eV could be attributed to the characteristic satellite for the π–π* transition observed in these materials. The peak assignments agree well with the previous work done for similar kinds of materials.49 Furthermore, the analysis of the O(1s) spectrum could complement the information obtained from the C(1s) spectral analysis. Broad asymmetrical spectral features of O(1s) indicate that there are different oxygen species in the SWNT–PdNPs sample. Deconvoluted O(1s) core level spectra are shown in Fig. 7B. The peak observed at 531.2 eV could be assigned to the contributions from CO or O–CO groups, whereas a higher binding energy peak at 533.1 eV corresponds to C–O or C–OH groups.50 The origin of the feature at 535.4 eV might be due to loosely bound CO species. The decomposition or fragmentation of O–CO groups present on the surface could result in loosely bound CO species.50b
C, C–C, C–O or C–OH, CO, and O–CO, respectively. The peak at 291.0 eV could be attributed to the characteristic satellite for the π–π* transition observed in these materials. The peak assignments agree well with the previous work done for similar kinds of materials.49 Furthermore, the analysis of the O(1s) spectrum could complement the information obtained from the C(1s) spectral analysis. Broad asymmetrical spectral features of O(1s) indicate that there are different oxygen species in the SWNT–PdNPs sample. Deconvoluted O(1s) core level spectra are shown in Fig. 7B. The peak observed at 531.2 eV could be assigned to the contributions from CO or O–CO groups, whereas a higher binding energy peak at 533.1 eV corresponds to C–O or C–OH groups.50 The origin of the feature at 535.4 eV might be due to loosely bound CO species. The decomposition or fragmentation of O–CO groups present on the surface could result in loosely bound CO species.50b
 | ||
| Fig. 7 XPS of (A) C(1s) core level and (B) O(1s) core level in SWNT–PdNPs. | ||
XPS studies of SWNT–PdNPs demonstrate the presence of Pd metal as well as Pd2+ species and different carbon–oxygen groups on the surface. In some studies, the formation of the palladium carbide (PdCx) type of species at the Pd–C interface has been reported.51 There would be a C(1s) core level peak around 282.0 eV if PdCx species gets formed in these materials.52 However, in the present study, there is no peak in this region of the C(1s) core level (see Fig. 7A) related to Pd–C bonds, indicating that Pd is not bound to carbon on the surface. Therefore, the possible anchoring of Pd nanoparticles could take place through the oxygen of the carboxylic acid groups present on SWNT.
Thus it may be anticipated that the carboxylic acid groups (–COOH) act as nucleation centers for PdNPs rendering strong interaction for metal nanoparticles. As a result, the carboxylic acid functionalized SWNTs have a strong grip over the palldium nanoparticles during catalytic cycles holding them tightly on their surface. To comprehend such an interaction we carried out preliminary density functional theory (DFT) calculations on a model system using a Pd3 cluster on carboxylic acid functionalized metallic as well as semiconducting single walled carbon nanotubes. The calculation indicated that the palladium metal trimer (Pd3) experiences bonding interactions with the –CO oxygen of the carboxylic acid groups attached to the SWNTs (see ESI† for details).
As a part of our ongoing interest to develop catalysts based on palladium nanoparticles for C–C coupling reactions,29,38 we have tested this fresh material (SWNT–PdNPs) for its efficacy as a catalyst towards copper free acyl Sonogashira reactions. Initial screening of conditions was examined using thiophene-2-carbonyl chloride (1a) and phenyl acetylene (2a) as substrates to obtain quantitative yields of 3-phenyl-1-(thiophen-2-yl)prop-2-yn-1-one (3aa) (Table 1) using 20 mg SWNT–PdNPs as the catalyst. Among three solvents (namely, toluene, THF, and acetonitrile), acetonitrile provided the desired product quantitatively even with 5 mg amount of the catalyst loading (entry 6, Table 1).
|
|
|||
|---|---|---|---|
| Entry | Catalyst amount (mg) | Solvent | Yield (%)b |
| a Reaction condition: SWNT–PdNPs, thiophene-2-carbonyl chloride (1 mmol), phenyl acetylene (1 mmol), and dry triethylamine (1 mmol) in 5 mL of dry solvent were used. b Yield was calculated after isolating the product through short column chromatography. | |||
| 1 | 20 mg | Toluene | 81 |
| 2 | 20 mg | THF | 87 |
| 3 | 20 mg | Acetonitrile | ∼99 |
| 4 | 15 mg | Acetonitrile | ∼99 |
| 5 | 10 mg | Acetonitrile | ∼99 |
| 6 | 5 mg | Acetonitrile | ∼99 |

To evaluate the substrate scope using the present catalyst, we inspected the catalytic behavior of SWNT–PdNPs for a range of substrates including various aromatic/aliphatic acid chlorides as well as aromatic/aliphatic terminal alkynes to construct the whole library of ynones, which is summarized in Table 2. We used both 10 mg and 5 mg catalyst loading to carry out these catalytic reactions and to our delight, most of the acid chloride–alkyne combinations endowed products in very good to excellent yields under mild reaction conditions. The catalytic results with 10 mg catalyst loading are discussed here and that with 5 mg catalyst loading is presented in the parenthesis of Table 2 (under Yield column). After we obtained the quantitative yield of 3-phenyl-1-(thiophen-2-yl)prop-2-yn-1-one (3aa) under the optimized reaction condition as mentioned in entries 5 and 6 of Table 1, the oxygen analogue of thiophene-2-carbonyl chloride (1b) was first tested with phenyl acetylene and the coupling product (3ba) was obtained quantitatively (entry 1, Table 2). In next step, 1- and 2-naphthoyl chlorides (1c and 1d, respectively) were successfully coupled with aromatic alkyne (2a) at slightly elevated temperature (entries 2 and 3, Table 2). Similarly, an α,β-unsaturated acid chloride (cinnamoyl chloride, 1e) also furnished the corresponding coupling product of phenyl acetylene (3ea) in quantitative amount (entry 4, Table 2). Inspired by these flourishing coupling results of aromatic acid chlorides with alkyne, we then checked the catalytic efficiency of SWNT–PdNPs on comparatively less reactive aliphatic acid chlorides as well as aliphatic alkynes directed to the acyl Sonogashira reaction. To initiate this investigation, an aliphatic acid chloride (1-adamantane carbonyl chloride, 1f) was first assessed to couple with the aromatic alkyne (phenyl acetylene, 2a) and the desired coupled product was obtained in 84% yield at 50 °C (entry 5, Table 2). In addition to this, new ynones had been productively synthesized when the heteroaryl acid chlorides (1a and 1b, respectively) were allowed to couple with an aliphatic alkyne, namely, 3,3-dimethylbut-1-yne, 2b (entries 6 and 7, Table 2). To generalize this trend, the heteroaryl acid chloride, 1b was further allowed to react with another two different aliphatic alkynes, (1-hexyne, 2c and 1-octyne, 2d, respectively) delivering the resultant ynones in very good yields (80% and 76%, respectively; entries 8 and 9, Table 2). In a reverse combination, new ynones were synthesized in excellent yields when the aliphatic acid chloride, 1f, was subjected to two different aromatic alkynes, namely 4-ethynylbiphenyl, 2e and 1-ethynyl-4-fluoro-benzene, 2f (entries 10 and 11, Table 2). The catalytic performance of SWNT–PdNPs was then scrutinized when the aliphatic acid chloride, 1f, was allowed to react with the aliphatic alkynes (2b and 2d) and a new alkynone (3fb) was synthesized in 81% yield along with the other one (3fd) in 83% yield (entries 12 and 13, Table 2). A hitherto unknown ynone, 3-cyclohexyl-1-(thiophen-2-yl)prop-2-yn-1-one (3ag) was formed quantitatively at room temperature (25 °C) when thiophene-2-carbonyl chloride (1a) was allowed to couple with an aliphatic alkyne (ethynylcyclohexane, 2g; entry 14, Table 2). The scope of the ynone synthesis was further utilized by using 2-methoxybenzoyl chloride, 1g, as the coupling partner resulting in the formation of (2-methoxyaryl)-substituted ynone, 3ga (entry 15, Table 2) in 98% yield. This particular ynone can be employed as a starting material for the preparation of chromenones, potential intermediates in the synthesis of bioactive molecules.53 Additionally, we also carried out these catalytic reactions with lower catalyst loading of 5 mg and the results are presented in Table 2. The results indicate that 5 mg catalyst loading is adequate for some of these substrates leading to comparable yields as observed with 10 mg catalyst loading (Table 2). Nevertheless, by comparing the results presented in Table 2, the catalyst loading of 10 mg has been considered as the optimum for further studies.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Acid chloride, 1 | Alkyne, 2 | Ynone, 3 | Temp. (°C) | Yield (%)b |
| a Reaction condition: SWNT–PdNPs (10 mg) was used together with acid chlorides 1 (1 mmol), terminal acetylenes 2 (1 mmol) and dry triethylamine (1 mmol) in 5 mL dry acetonitrile. b Isolated yields of the products after purification through column chromatography; isolated yields of 10 mg and 5 mg (in the parenthesis) catalyst loading are presented. | |||||
| 1 |
|

|

|
25 | 98 (98) |
| 2 |

|

|

|
50 | 94 (95) |
| 3 |
|

|

|
50 | 91 (57) |
| 4 |

|
|

|
50 | 98 (58) |
| 5 |

|

|

|
50 | 84 (60) |
| 6 |

|
|

|
50 | 98 (61) |
| 7 |

|

|

|
50 | 84 (43) |
| 8 |

|

|

|
50 | 80 (43) |
| 9 |

|

|

|
50 | 76 (43) |
| 10 |

|

|
|
50 | 87 (64) |
| 11 |
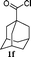
|

|

|
50 | 93 (81) |
| 12 |

|
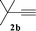
|

|
50 | 81 (67) |
| 13 |

|

|
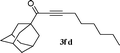
|
50 | 83 (69) |
| 14 |

|

|

|
25 | 98 (53) |
| 15 |

|

|

|
25 | 98 (67) |

Recyclability test of the catalyst
Encouraged by the highly efficient catalytic outcome of SWNT–PdNPs depicted in Table 2, we further tested its ability to perform as a recyclable catalyst for this particular transformation. An enviable heterogeneous nanocatalyst should possess long catalyst lifetime and recycling ability for industrial applications. The recycling efficiency of the catalyst was investigated for the coupling reaction of thiophene-2-carbonyl chloride (1a) with phenyl acetylene (2a). After each catalytic cycle, the catalyst was recovered by filtering through a 0.22 μm membrane filter paper, thoroughly washed with polar organic solvents and finally dried under high vacuum for 12 h before performing the next cycle with equal amount of substrate loading. We checked the recyclability test up to seven consecutive cycles where the first four cycles resulted the quantitative formation of 3-phenyl-1-(thiophen-2-yl)prop-2-yn-1-one (3aa) without any loss of catalytic efficiency, however, from the 5th cycle onwards a slight decrease of catalytic efficiency was observed as shown in Fig. 8. This high recycling efficiency of SWNT–PdNPs for the acyl Sonogashira reaction might be attributed to the presence of a strong interaction between palladium nanoclusters and carboxylic acid groups residing on the surface of SWNT-COOH. We also carried out TEM, AFM, and resonance Raman studies of the catalyst after the 1st and 4th catalytic cycles to understand any change of morphology of the catalyst during the catalytic cycles. TEM images of SWNT–PdNPs recorded after the 4th catalytic cycle revealed that after several catalytic runs, palladium nanoparticles still remain on the surface of SWNT-COOH with slight increase in size (25 nm average diameter, Fig. 9). | ||
| Fig. 8 Recyclability chart of the catalyst, SWNT–PdNPs for seven consecutive catalytic runs. | ||
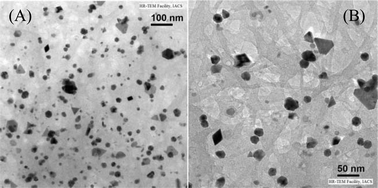 | ||
| Fig. 9 TEM images of SWNT–PdNPs after the 4th catalytic cycle (A) presented with 100 nm scale and (B) presented with 50 nm scale. | ||
AFM images of SWNT–PdNPs were taken in semi-contact mode after the 1st (Fig. 10A) as well as the 4th (Fig. 10B) catalytic cycles. In both cases, it is clearly visible that the palladium nanoparticles are attached onto the surface of the carboxylic acid functionalized SWNTs. Height profile diagrams along the horizontal lines showed that the average height of the nanoclusters remained almost unaltered as compared to the original (see ESI† for additional AFM images with height profile diagrams).
 | ||
| Fig. 10 AFM images of SWNT-PdNPs after (A) the 1st catalytic cycle and (B) after the 4th catalytic cycle. | ||
Also resonance Raman spectra, recorded after first and fourth catalytic cycles upon excitation with the 488 nm laser source (Fig. 11) did not show any appreciable change in the materials when compared to the as prepared SWNT–PdNPs. All these results support that the PdNPs remain quite stable under catalytic condition owing to its strong interaction with the carboxylic acid functionalized SWNT. In addition, the ICP-AES study of the organic filtrate part obtained after the first catalytic cycle revealed only 0.03 wt% of palladium in the filtrate indicating minimal leaching during catalysis which may be attributed to the presence of a strong gripping effect of carboxylic acid groups on PdNPs.
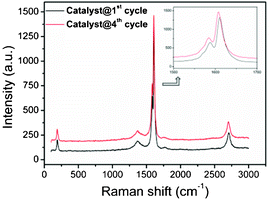 | ||
| Fig. 11 Resonance Raman spectra of SWNT–PdNPs samples after the 1st catalytic run (black solid line) and after the 4th catalytic run (red solid line) recorded using 488 nm laser sources (note that the scales along Y-axis in Fig. 11 have been modified for the sake of clarity). | ||
Furthermore, we checked the advantage of the anchored SWNT-COOH over physically mixed PdNPs and SWNT-COOH as catalysts (see ESI† for details). The physically mixed catalyst was tested on four different acyl Sonogashira substrates (substrates of Table 1 and substrates of entries 5, 13, and 14, Table 2). The catalytic results on these substrates indeed reveal the catalytic activity of physically mixed catalyst (see ESI† for details). However, the physically mixed catalyst does not work in the recycling experiment when compared to the similar recycling experiment using the anchored catalyst SWNT–PdNPs under the identical reaction conditions in the presence of thiophene-2-carbonyl chloride (1a) and phenyl acetylene (2a) as substrates. The physically mixed catalyst after filtration and separation from the first catalytic cycle did not show any catalytic activity in the second catalytic cycle during the recycling experiment. This result indicates that in the case of a physically mixed catalyst the palladium species is leached out during the first catalytic cycle; an observation that was not noticed in the case of catalysis using the anchored SWNT–PdNPs catalyst.
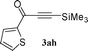
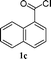
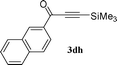
To expand the scope of the copper free Sonogashira catalysis by SWNT–PdNPs, we next examined the coupling reaction of an acid chloride with trimethylsilyl (TMS) acetylene providing trimethylsilyl-ynones (TMS-ynones). This particular type of ynones can be utilized in the synthesis of terminal alkynylketones as reported by Walton and Waugh54 and later on the utility of TMS-ynones has been increased including in the synthesis of 1,3-butadiynes via alkylidene carbenoid rearrangements,55 1,4-disubstituted 1,2,3-triazoles via click chemistry,56 as well as 2,4-disubstituted pyrimidines via a one-pot multicomponent reaction.29,33 In this connection, one-pot multicomponent reactions are the most useful synthetic methods to obtain improved yields of pharmaceutically important compounds by avoiding the handling and isolation of intermediates.57 Herein, we have explored the synthetic feasibility of TMS-ynones employing the present catalyst (SWNT–PdNPs) in a copper free condition at room temperature (25 °C) based on the previously optimized reaction conditions for the synthesis of ynones (entry 5, Table 1). We further extended this protocol towards the synthesis of 2,4-disubstituted pyrimidines in quantitative yields following one-pot multicomponent approach. Catalytic results for the synthesis of TMS-ynones are presented in Table 3 where most of the products were obtained in good to excellent isolated yields (60–95%). This particular catalytic transformation was successfully applied for a range of substrates starting from heteroaryl acid chlorides (1a and 1b; entries 1 and 2, Table 3) to the simplest aryl acid chloride (benzoyl chloride, 1h; entry 6, Table 3). Further, the scope of this reaction was extended to the coupling of naphthoyl chlorides (1c and 1d; entries 3 and 4, Table 3) and an α,β-unsaturated acid chloride (1e; entry 5, Table 3).
|
|
|||
|---|---|---|---|
| Entry | Acid chloride, 1 | TMS-ynone, 3 | Yield (%)b |
| a Reaction condition: the catalyst SWNT–PdNPs (10 mg) was used together with acid chlorides 1 (1 mmol), trimethylsilylacetylene 2h (1 mmol) and dry triethylamine (1 mmol) in 5 mL dry acetonitrile. b Isolated yields of the products after purification through short column chromatography. | |||
| 1 |

|
|
60 |
| 2 |

|

|
60 |
| 3 |
|

|
94 |
| 4 |

|
|
73 |
| 5 |

|
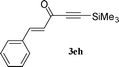
|
71 |
| 6 |
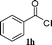
|

|
95 |

With convenient access to TMS-ynones we next moved onto our goal directed to the synthesis of 2,4-disubstituted pyrimidines via a one-pot multicomponent reaction. The pyrimidine nucleus is important in the medicinal field since the presence of such moiety renders various biological molecules having copious pharmacological action ranging anticancer, antiprotozoal, antiviral activities.58 In the present study, it was anticipated that the heterogeneous nature of the catalyst bearing chemically inert carboxylic acid functionalized SWNTs will not affect the subsequent steps of the one-pot reaction, which might result in the synthesis of pyrimidine derivatives including aromatic/aliphatic substituents in improved yields than those reported earlier.33,59 The heteroaryl acid chlorides (1a and 1b) resulted in the synthesis of 2,4-disubstituted pyrimidines with very good to excellent yields of corresponding N-heterocyclic ring systems (entries 1 and 2, Table 4). In the case of 1-naphthoyl chloride as the substrate, 4-(naphthalen-1-yl)pyrimidine-2-amine was obtained in 82% yield (entry 3, Table 4) while analogous 2-naphthoyl chloride furnished the corresponding pyrimidine derivative in 44% isolated yield (entry 4, Table 4). However, (E)-4-styrylpyrimidin-2-amine was achieved in 70% yield (entry 5, Table 4) while 4-phenylpyrimidin-2-amine was obtained in quantitative yield when benzoyl chloride (1h) was used as a substrate (entry 6, Table 4). In the same fashion, derivatives of pyrimidine bearing aliphatic substituents in the 4-position were successfully accomplished employing this particular catalytic system (entries 7 and 8, Table 4).
|
|
|||
|---|---|---|---|
| Entry | Acid chloride, 1 | Pyrimidine, 5 | Yield (%)b |
| a Reaction condition: 10 mg of the SWNT–PdNPs was used together with 1 mmol each of the acid chloride, trimethylsilylacetylene and dry triethylamine in 5 mL of dry acetonitrile; after completion of the first step guanidine hydrochloride (2.5 mmol) and Na2CO3 (3.5 mmol) were added into the reaction medium followed by the addition of 5 mL methanol. b Yields after isolation of the product using column chromatography. | |||
| 1 |

|

|
93 |
| 2 |

|

|
77 |
| 3 |

|

|
82 |
| 4 |

|
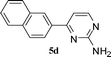
|
44 |
| 5 |
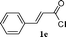
|
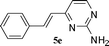
|
70 |
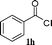
| 6 |
|

|
97 |
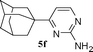
| 7 |

|
|
51 |
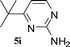
| 8 |

|
|
52 |

Conclusions
In summary, a suitable approach for the synthesis of heterogeneous palladium nanoparticles (PdNPs) decorated onto carboxylic acid functionalized SWNTs via the thermolysis of palladium acetate has been documented. The presence of PdNPs on the SWNT-COOH has been revealed from TEM, EDX and AFM studies while the interaction between PdNPs and carboxylic acid functionalized SWNTs was investigated by UV-vis-NIR spectroscopy, resonance Raman spectroscopy and XPS studies. All these investigations, coupled with DFT calculations, indicated that PdNPs use the carboxylic acid groups as anchoring points to be immobilized tightly onto SWNTs surface. We outlined for the first time that beyond conventional catalytic applications of such heterogeneous nanocomposites, carbon nanotube supported palladium nanoparticles (SWNT–PdNPs) can be utilized as an efficient heterogeneous nanocatalyst for the acyl Sonogashira coupling reaction under copper free mild reaction conditions creating a library of ynones in excellent yields. Additionally, SWNT–PdNPs could be recycled up to seven consecutive cycles without losing its catalytic property to a great extent which could be attributed to the strong gripping ability of carboxylic acid functionalized single walled carbon nanotubes on the PdNPs during successive catalytic cycles. The presence of this strong interaction between PdNPs and carboxylic acid functionalized SWNTs was also supported by TEM, AFM and resonance Raman spectroscopy after catalytic cycles. This protocol was extended in the synthesis TMS-ynones which were further explored in the one-pot multicomponent synthesis of 2,4-disubstituted pyrimidines with improved yields and efficiency than those reported earlier.Experimental section
Materials and instruments
Carboxylic acid functionalized SWNTs (having 5–6 wt% COOH groups) were purchased from Carbon solutions, Inc., USA. All acid chlorides [except pivaloyl chloride (Spectrochem, India)], terminal alkynes and palladium acetate were used directly as received from Sigma-Aldrich. The solvents (toluene, THF, and acetonitrile) were dried following standard protocols under a nitrogen atmosphere and used. Hexane and ethyl alcohol were purchased from Merck, India and distilled before work-up process and column chromatography. The 0.22 μm PTFE membrane filter papers were purchased from Millipore. The remaining chemicals were purchased from Merck, India and used without further purification.1H NMR and 13C NMR spectra were recorded on a JEOL ECS 400 MHz spectrometer and on a Bruker Avance III 500 MHz spectrometer. Chemical shifts (δ) downfield from the reference standard were assigned positive values. Mass spectra were recorded on a micromass Q-Tof micro™, Waters. TEM and EDX analyses were performed either with a Philips CM200-FEG-UT operated at 200 kV or on a JEOL, JEM 2010/111. Samples for transmission electron microscopy (TEM) imaging were prepared by placing a 5 μL drop of dispersed solution in N,N-dimethylformamide containing SWNT–PdNPs on a carbon-coated copper grid and allowing it to dry in the air. UV-vis-NIR spectral studies were carried out on a HITACHI UV4100 spectrophotometer. AFM study was carried out on NT-MDT instrument; model no AP-0100 in semi-contact mode after placing a 5 μL drop of dispersed solution in N,N-dimethylformamide containing SWNT–PdNPs on a cleaned glass cover slip slide and allowing it to dry in the air. Raman spectroscopy was performed using the 2.54 eV (488 nm) line of an Ar ion laser in Horiba Jobin Yvon LabRAM HR 800 instrument by placing a pinch of the solid samples on a cleaned glass slide. The samples were platinum coated and observed through a FESEM instrument (JEOL, JSM 6700F) operating at 5 KV. ICP-AES data were recorded on a Perkin Elmer Optima 7000 DV instrument. All products were isolated by short chromatography on a silica gel (100–200 mesh) column using the required ratio of hexane and ethyl acetate. The known compounds were characterized by comparing their 1H and 13C NMR spectra to the reported data and the relevant spectra are presented in the ESI.† The syntheses of palladium nanoparticles as well as all catalytic reactions were carried out under an atmosphere of dry nitrogen using standard Schlenk line techniques.
XPS study
XPS of the SWNT–PdNPs sample was recorded with a Thermo Fisher Scientific Multilab 2000 (England) spectrometer using non-monochromatic Mg-Kα radiation (1253.6 eV) run at 15 kV and 10 mA as the X-ray source. The binding energies reported here were calculated with reference to C(1s) peak at 284.5 eV with a precision of ± 0.1 eV. For XPS analysis, powder samples were mounted on the sample holder and placed into an ultrahigh vacuum (UHV) chamber at 10−9 Torr housing the analyzer. Before placing the samples into an analyzing chamber, they were kept in the preparation chamber at UHV (10−9 Torr) for 5 h in order to desorb any volatile species present on the surface. All the spectra were obtained here in the digital mode with Avantage software on a personal computer with 30 eV pass energy across the hemispheres of the electron analyzer and 0.05 eV step increment. The experimental data were curve fitted with Gaussian peaks after subtracting a linear background employing PeakFit v4.11 program.Synthesis of palladium nanoparticles decorated onto single walled carbon nanotubes
Carboxylic acid functionalized SWNTs were preheated at 130 °C for 12 h and cooled to 25 °C under high vacuum prior to use. Palladium acetate (100 mg, 0.45 mmol) and P3-SWNTs (100 mg) were kept in a 100 mL Schlenk flask under vacuum (∼10−2 mbar) for one hour. Dry DMF (∼35 mL) was added under nitrogen atmosphere. To carry out the thermal decomposition of palladium acetate, the mixture was heated at 95 °C for 4 h, and then cooled to 25 °C under nitrogen atmosphere, filtered through a 0.22 μm PTFE membrane filter paper. The black residue was washed several times with ethanol (15 mL × 4) until the filtrate became colorless and finally twice with acetone (15 mL × 2). The palladium nanoparticles (SWNT-PdNPs) were finally dried under vacuum (∼10−2 mbar) for 12 h before performing any catalytic reaction.Catalytic acyl Sonogashira reaction
10 mg of the as-synthesized SWNT–PdNPs was kept in a 50 mL Schlenk flask under vacuum (∼10−2 mbar) for 0.5 h. Dry solvent (5 mL) was added to it followed by the addition of acid chloride (1 mmol), alkyne (1 mmol) and dry triethylamine (1 mmol) consecutively under nitrogen atmosphere. Then the catalytic reactions were performed under appropriate temperature as mentioned in Table 2. After completion, the reaction mixture was cooled to 25 °C, filtered through a 0.22 μm PTFE membrane filter paper, and washed several times with ethyl acetate (15 mL × 4). The combined filtrate was completely evaporated under reduced pressure free from any residual solvent and the residue was purified through column chromatography (silica gel 100–200 mesh) using the appropriate ratio of hexane and ethyl acetate (see ESI† for more details).Synthesis of the pyrimidine system
2,4-Disubstituted pyrimidines were obtained in good yields from TMS-ynones applying a one-pot three-component synthesis by a coupling–addition–cyclocondensation sequence. 10 mg of SWNT–PdNPs was kept in a 50 mL Schlenk flask under vacuum (∼10−2 mbar) for 0.5 h. The TMS-ynone moiety was synthesized using acid chloride (1 mmol), trimethylsilylacetylene (1 mmol) and dry triethylamine (1 mmol) in dry acetonitrile (∼5 mL) following the acyl Sonogashira procedure mentioned above. After completion of the first step, the reaction mixture was cooled to 25 °C. Sodium bicarbonate (3.5 mmol) was added to it followed by the addition of guanidine hydrochloride (2.5 mmol) and methanol (5 mL) at 25 °C. The reaction mixture was then refluxed for 14–16 h until the ynone was consumed completely (monitored by TLC). After completion it was cooled to 25 °C, filtered through a 0.22 μm PTFE membrane filter paper, washed several times with ethyl acetate (15 mL × 4). After complete evaporation of the combined filtrate under reduced pressure, the residue was then purified through column chromatography (silica gel 100–200 mesh) using the appropriate ratio of hexane and ethyl acetate (see ESI† for more details).Acknowledgements
Financial support from the Department of Science and Technology (DST), India (Grant No. SR/FT/CS-020/2008) is highly acknowledged. SS thanks CSIR, India for a research fellowship. We gratefully acknowledge Mr. Sibaprasad Maity and Mr. Abhisek Basu for collecting AFM images and Raman data respectively. We are thankful to Dr Suhrit Ghosh group at IACS, India and Prof. Michael Seibt group from University of Goettingen, Germany for using TEM, EDX and HRMS analysis. SKM thanks NMR facility of IISER-Kolkata.References
- D. L. Feldheim, Science, 2007, 316, 699–700 CrossRef CAS.
- (a) M. Moreno-Mañas and R. Pleixats, Acc. Chem. Res., 2003, 36, 638–643 CrossRef; (b) G. A. Somorjai, H. Frei and J. Y. Park, J. Am. Chem. Soc., 2009, 131, 16589–16605 CrossRef CAS; (c) D. Astruc, F. Lu and J. R. Aranzaes, Angew. Chem., 2005, 117, 8062–8083 ( Angew. Chem., Int. Ed. , 2005 , 44 , 7852 ) CrossRef.
- (a) H. Park, J. Park, A. K. L. Lim, E. H. Anderson, A. P. Alivisatos and P. L. McEuen, Nature, 2000, 407, 57–60 CrossRef; (b) S. D. Franceschi and L. Kouwenhoven, Nature, 2002, 417, 701–702 CrossRef; (c) N. Toshima, Macromol. Symp., 2008, 270, 27–39 CrossRef CAS; (d) M. Prato, K. Kostarelos and A. Bianco, Acc. Chem. Res., 2008, 41, 60–68 CrossRef CAS.
- (a) M. T. Reetz and J. G. de Vries, Chem. Commun., 2004, 1559–1563 RSC; (b) D. Astruc, Inorg. Chem., 2007, 46, 1884–1894 CrossRef CAS.
- W. Huang, J. H.-C. Liu, P. Alayoglu, Y. Li, C. A. Witham, C.-K. Tsung, F. D. Toste and G. A. Somorjai, J. Am. Chem. Soc., 2010, 132, 16771–16773 CrossRef CAS.
- S. Mandal, D. Roy, R. V. Chaudhari and M. Sastry, Chem. Mater., 2004, 16, 3714–3724 CrossRef CAS.
- H. Choi, S. R. Al-Abed, S. Agarwal and D. D. Dionysiou, Chem. Mater., 2008, 20, 3649–3655 CrossRef CAS.
- N. Zheng and G. D. Stucky, J. Am. Chem. Soc., 2006, 128, 14278–14280 CrossRef CAS.
- Y. Yu, T. Hu, X. Chen, K. Xu, J. Zhang and J. Huang, Chem. Commun., 2011, 47, 3592–3594 RSC.
- S. Ogasawara and S. Kato, J. Am. Chem. Soc., 2010, 132, 4608–4613 CrossRef CAS.
- R. W. J. Scott, A. K. Datye and R. M. Crooks, J. Am. Chem. Soc., 2003, 125, 3708–3709 CrossRef CAS.
- (a) P. Singh, S. Campidelli, S. Giordani, D. Bonifazi, A. Bianco and M. Prato, Chem. Soc. Rev., 2009, 38, 2214–2230 RSC; (b) S. Niyogi, M. A. Hamon, H. Hu, B. Zhao, P. Bhowmik, R. Sen, M. E. Itkis and R. C. Haddon, Acc. Chem. Res., 2002, 35, 1105–1113 CrossRef CAS.
- B. C. Satishkumar, E. M. Vogl, A. Govindaraj and C. N. R. Rao, J. Phys. D: Appl. Phys., 1996, 29, 3173–3176 CrossRef CAS.
- (a) B. Zhao, H. Hu, S. K. Mandal and R. C. Haddon, Chem. Mater., 2005, 17, 3235–3241 CrossRef CAS; (b) H. Hu, Y. Ni, S. K. Mandal, V. Montana, B. Zhao, R. C. Haddon and V. Parpura, J. Phys. Chem. B, 2005, 109, 4285–4289 CrossRef CAS.
- (a) L. Lattuada, A. Barge, G. Cravotto, G. B. Giovenzana and L. Tei, Chem. Soc. Rev., 2011, 40, 3019–3049 RSC; (b) C. N. R. Rao, S. Natarajan and R. Vaidhyanathan, Angew. Chem., 2004, 116, 1490–1521 ( Angew. Chem., Int. Ed. , 2004 , 43 , 1466 ) CrossRef.
- J. Kong, M. G. Chapline and H. Dai, Adv. Mater., 2001, 13, 1384–1386 CrossRef CAS.
- S. Rather, R. Zacharia, S. W. Hwang, M. Naik and K. S. Nahm, Chem. Phys. Lett., 2007, 441, 261–267 CrossRef CAS.
- T. Xu, J. Yang, J. Liu and Q. Fu, Appl. Surf. Sci., 2007, 253, 8945–8951 CrossRef CAS.
- X. R. Ye, Y. Lin and C. M. Wai, Chem. Commun., 2003, 642–643 RSC.
- M. J. O'Connell, P. Boul, L. M. Ericson, C. Huffman, Y. Wang, E. Haroz, C. Kuper, J. Tour, K. D. Ausman and R. E. Smalley, Chem. Phys. Lett., 2001, 342, 265–271 CrossRef CAS.
- Z. Tan, H. Abe, M. Naito and S. Ohara, Chem. Commun., 2010, 46, 4363–4365 RSC.
- J. L. Bahr, J. Yang, D. V. Kosynkin and M. J. Bronikowski, J. Am. Chem. Soc., 2001, 123, 6536–6542 CrossRef CAS.
- C. Burda, X. Chen, R. Narayanan and M. A. El-Sayed, Chem. Rev., 2005, 105, 1025–1102 CrossRef CAS.
- Y. Lin, K. A. Watson, M. J. Fallbach, S. Ghose, J. G. Jr. Smith, D. M. Delozier, W. Cao, R. E. Crooks and J. W. Connell, ACS Nano, 2009, 3, 871–884 CrossRef CAS.
- W. Li, C. Liang, W. Zhou, J. Qiu, Z. Zhou, G. Sun and Q. Xin, J. Phys. Chem. B, 2003, 107, 6292–6299 CrossRef CAS.
- N. Mackiewicz, G. Surendran, H. Remita, B. Keita, G. Zhang, L. Nadjo, A. Hagège, E. Doris and C. Mioskowski, J. Am. Chem. Soc., 2008, 130, 8110–8111 CrossRef CAS.
- (a) Y. Sun and H. H. Wang, Adv. Mater., 2007, 19, 2818–2823 CrossRef CAS; (b) T. H. Kim, B. Y. Lee, J. Jaworski, K. Yokoyama, W.-L. Chung, E. Wang, S. Hong, A. Majumdar and S.-W. Lee, ACS Nano, 2011, 5, 2824–2830 CrossRef CAS; (c) J. Y. Kim, J. Lee, S. Hong and T. D. Chung, Chem. Commun., 2011, 47, 2892–2894 RSC.
- (a) N. Karousis, G.-E. Tsotsou, F. Evangelista, P. Rudolf, N. Ragoussis and N. Tagmatarchis, J. Phys. Chem. C, 2008, 112, 13463–13469 CrossRef CAS; (b) J. Y. Kim, Y. Jo, S.-K. Kook, S. Lee and H. C. Choi, J. Mol. Catal. A: Chem., 2010, 323, 28–32 CrossRef CAS; (c) A. Corma, H. Garcia and A. Leyva, J. Mol. Catal. A: Chem., 2005, 230, 97–105 CrossRef CAS; (d) J. Guerra and M. A. Herrero, Nanoscale, 2010, 2, 1390–1400 RSC; (e) L. Zhou and H.-F. Jiang, Tetrahedron Lett., 2007, 48, 8449–8452 CrossRef CAS.
- S. Santra, K. Dhara, P. Ranjan, P. Bera, J. Dash and S. K. Mandal, Green Chem., 2011, 13, 3238–3247 RSC.
- A. V. Kel'in, A. W. Sromek and V. Gevorgyan, J. Am. Chem. Soc., 2001, 123, 2074–2075 CrossRef CAS.
- D. B. Grotjahn, S. Van, D. Combs, D. A. Lev, C. Schneider, M. Rideout, C. Meyer, G. Hernandez and L. Mejorado, J. Org. Chem., 2002, 67, 9200–9209 CrossRef CAS.
- J. P. Waldo and R. C. Larock, Org. Lett., 2005, 7, 5203–5205 CrossRef CAS.
- (a) A. S. Karpov and T. J. J. Müller, Org. Lett., 2003, 5, 3451–3454 CrossRef CAS; (b) D. M. D'Souza and T. J. J. Müller, Nat. Protoc., 2008, 3, 1660–1665 CrossRef.
- M. C. Bagley, D. D. Hughes, R. Lloyd and V. E. C. Powers, Tetrahedron Lett., 2001, 42, 6585–6588 CrossRef CAS.
- (a) A. S. Karpov, T. Oeser and T. J. J. Müller, Chem. Commun., 2004, 1502–1503 RSC; (b) A. S. Karpov, F. Rominger and T. J. J. Müller, Org. Biomol. Chem., 2005, 3, 4382–4391 RSC.
- P. R. Likhar, M. S. Subhas, M. Roy, S. Roy and M. L. Kantam, Helv. Chim. Acta, 2008, 91, 259–264 CrossRef CAS.
- (a) M. S. M. Ahmed and A. Mori, Org. Lett., 2003, 5, 3057–3060 CrossRef CAS; (b) A. Fusano, T. Fukuyama, S. Nishitani, T. Inouye and I. Ryu, Org. Lett., 2010, 12, 2410–2413 CrossRef CAS.
- S. Santra, P. Ranjan, S. K. Mandal and P. K. Ghorai, Inorg. Chim. Acta, 2011, 372, 47–52 CrossRef CAS.
- R. Saito, M. Fujita, G. Dresselhaus and M. S. Dresselhaus, Appl. Phys. Lett., 1992, 60, 2204–2206 CrossRef CAS.
- J. Chen, M. A. Hamon, H. Hu, Y. Chen, A. M. Rao, P. C. Eklund and R. C. Haddon, Science, 1998, 282, 95–98 CrossRef CAS.
- R. S. Lee, H. J. Kim, J. E. Fischer, A. Thess and R. E. Smalley, Nature, 1997, 388, 255–257 CrossRef CAS.
- (a) M. E. Itkis, S. Niyogi, M. E. Meng, M. A. Hamon, H. Hu and R. C. Haddon, Nano Lett., 2002, 2, 155–159 CrossRef CAS; (b) H. Hu, B. Zhao, M. A. Hamon, K. Kamaras, M. E. Itkis and R. C. Haddon, J. Am. Chem. Soc., 2003, 125, 14893–14900 CrossRef CAS.
- D. R. Kauffman, D. C. Sorescu, D. P. Schofield, B. L. Allen, K. D. Jordan and A. Star, Nano Lett., 2010, 10, 958–963 CrossRef CAS.
- (a) D. R. Kauffman and A. Star, J. Phys. Chem. C, 2008, 112, 4430–4434 CrossRef CAS; (b) E. Bekyarova, I. Kalinina, M. E. Itkis, L. Beer, N. Cabrera and R. C. Haddon, J. Am. Chem. Soc., 2007, 129, 10700–10706 CrossRef CAS.
- M. S. Strano, C. A. Dyke, M. L. Usrey, P. W. Barone, M. J. Allen, H. Shan, C. Kittrell, R. H. Hauge, J. M. Tour and R. E. Smalley, Science, 2003, 301, 1519–1522 CrossRef CAS.
- J. L. Bahr and J. M. Tour, J. Mater. Chem., 2002, 12, 1952–1958 RSC.
- Y.-A. Li, N.-H. Tai, S.-K. Chen and T.-Y. Tsai, ACS Nano, 2011, 5, 6500–6506 CrossRef CAS.
- (a) T. Pillo, R. Zimmermann, P. Steiner and S. Hüfner, J. Phys.: Condens. Matter, 1997, 9, 3987–3999 CrossRef CAS; (b) M. Brun, A. Berthet and J. C. Bertolini, J. Electron Spectrosc. Relat. Phenom., 1999, 104, 55–60 CrossRef CAS; (c) C. D. Wagner, W. M. Riggs, L. E. Davis and J. F. Moulder, Handbook of X-Ray Photoelectron Spectroscopy; Editor: Muilenberg, G. E.Perkin-Elmer, Eden Prairie, Minnesota, 1979 Search PubMed; (d) J. Y. Kim, K. Park, S. Y. Bae, G. C. Kim, S. Lee and C. C. Choi, J. Mater. Chem., 2011, 21, 5999–6005 RSC.
- (a) X. Song, Y. Yang, J. Liu and H. Zhao, Langmuir, 2011, 27, 1186–1191 CrossRef CAS; (b) C. Mattevi, G. Eda, S. Agnoli, S. Miller, K. A. Mkhoyan, O. Celik, D. Mastrogiovanni, G. Granozzi, E. Garfunkel and M. Chhowalla, Adv. Funct. Mater., 2009, 19, 2577–2583 CrossRef CAS.
- (a) D. Yang, A. Velamakanni, G. Bozoklu, S. Park, M. Stoller, R. D. Piner, S. Stankovich, I. Jung, D. A. Field, C. A. Ventrice Jr. and R. S. Ruoff, Carbon, 2009, 47, 145–152 CrossRef CAS; (b) Y. Xing, L. Li, C. C. Chusuei and R. V. Hull, Langmuir, 2005, 21, 4185–4190 CrossRef CAS.
- B. Wang, H. Q. Wang, Y. Q. Li, S. Y. Zhang and J. G. Hou, Mater. Res. Bull., 2000, 35, 551–557 CrossRef CAS.
- Y.-F. Han, D. Kumar, C. Sivadinarayana, A. Clearfield and D. W. Goodman, Catal. Lett., 2004, 94, 131–134 CrossRef CAS.
- N. Ahmed, C. Dubuc, J. Rousseau, F. Bénard and J. E. van Lier, Bioorg. Med. Chem. Lett., 2007, 17, 3212–3216 CrossRef CAS.
- D. R. M. Walton and F. Waugh, J. Organomet. Chem., 1972, 37, 45–56 CrossRef CAS.
- A. L. K. S. Shun, E. T. Chernick, S. Eisler and R. R. Tykwinski, J. Org. Chem., 2003, 68, 1339–1347 CrossRef.
- F. Friscourt and G.-J. Boons, Org. Lett., 2010, 12, 4936–4939 CrossRef CAS.
- J.-C. Wasilke, S. J. Obrey, T. Baker and G. C. Bazan, Chem. Rev., 2005, 105, 1001–1020 CrossRef CAS.
- (a) K. Undheim and T. Benneche, in Comprehensive Heterocyclic Chemistry II, ed. A. R. Katritzky, C. W. Rees, E. F. V. Scriven, A. McKillop, Pergamon, Oxford, 1996, 6, 93 Search PubMed; (b) A. W. Erian, Chem. Rev., 1993, 93, 1991–2005 CrossRef CAS; (c) E. S. Al-Abdullah, A.-R. M. Al-Obaid, O. A. Al-Deeb, E. E. Habib and A. A. El-Emam, Eur. J. Med. Chem., 2011, 46, 4642–4647 CrossRef CAS and references therein.
- (a) N. V. Makarova, M. N. Zemtsova and I. K. Moiseev, Chem. Heterocycl. Compd., 2001, 37, 840–843 CrossRef CAS; (b) J. F. Djung, R. J. Mears, C. A. G. N. Montalbetti, T. S. Coulter, A. Golebiowski, A. N. Carr, O. Barker, K. D. Greis, S. Zhou, E. Dolan and G. F. Davis, Bioorg. Med. Chem., 2011, 19, 2742–2750 CrossRef CAS; (c) G. B. Bennett, R. B. Mason, L. J. Alden and J. B. Roach, Jr., J. Med. Chem., 1978, 21, 623–628 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: 1H and 13C NMR data with spectra of organic compounds, HRMS data, EDX spectra and additional AFM images. See DOI: 10.1039/c2ra20281f/ |
| This journal is © The Royal Society of Chemistry 2012 |