A novel potential adsorbent for ultra deep desulfurization of jet fuels at room temperature
Yuesong
Shen
ab,
Xinhai
Xu
a and
Peiwen
Li
*a
aDepartment of Aerospace and Mechanical Engineering, The University of Arizona, Tucson, AZ 85721, USA. E-mail: peiwen@email.arizona.edu
bCollege of Materials Science and Engineering, State Key Laboratory of Materials-Oriented Chemical Engineering, Nanjing University of Technology, Nanjing 210009, People's Republic of China. E-mail: sys-njut@163.com
First published on 21st May 2012
Abstract
Using jet fuels in an integrated system by micro fuel processors and SOFC stacks for portable power sources is very attractive. This technology needs ultra-deep desulfurization of jet fuels to maintain high performance of fuel processors and SOFCs. So far, investigations on the adsorbents for direct ultra-deep desulfurization of jet fuels with high sulfur content at room temperature and atmospheric pressure are insufficient, and the achieved desulfurization performance is not satisfactorily high. This article presents the design and analysis of a new adsorbent, Ni–Ce/Al2O3–SiO2, for direct desulfurization of jet-A fuel with total sulfur content of 949.03 mg kg−1 at room temperature and atmospheric pressure without any other assisted reaction conditions. Experimental tests were conducted and high desulfurization efficiency (96.43%) was obtained, which verifies the expected effect. The analysis and experimental data demonstrates broad application prospects of the Ni–Ce/Al2O3–SiO2 adsorbent for room temperature and atmospheric pressure ultra-deep desulfurization of high-sulfur-content jet fuels.
High energy efficiency and energy density, together with rapid refuelling capability, render fuel cells highly attractive for portable power generation,1,2 for serving as backup power units. To provide hydrogen for fuel cell backup power units in military and civilian applications, jet fuels are widely considered by many as an excellent choice.3 Integrated micro fuel processors in combination with solid oxide fuel cell (SOFC) stacks using jet fuels have been viewed as achievable technologies for power generation.4 The real jet fuels contain various organic sulfur compounds with total sulfur content ranging from 300 to 3000 parts per million by weight (ppmw), which can poison the fuel processing catalysts such as reforming and water-shift catalysts significantly. Even a trace of sulfur in the feed can poison the anode catalysts in some fuel cells.4–7 Additionally, stringent emission regulations, rigorous pollution control standards and growing environmental awareness are being imposed on fuels of low sulfur pollution. Therefore, ultra deep desulfurization of jet fuels and low environmental impact are very important in order to obtain synthesis gas (syngas) for SOFCs.
Targeting providing syngas (H2 and CO) in SOFCs for auxiliary power units, the jet fuel desulfurization technology of particular interest in this study must be simple and convenient, with no extra separation processes. It also must be able to obtain sufficient ultra deep desulfurization efficiency at room temperature and atmospheric pressure without hydrogen consumption. Among all desulfurization technologies, selective adsorption for removing sulfur (SARS) has been regarded as the most promising approach for processing of jet fuels in application of SOFCs.8 As the technological core of the SARS process, absorbents have been attracting wide attention. A wide variety of adsorbents such as ion-exchanged zeolites, supported metals, metal oxides, activated carbons, ionic liquids and so on,6,9 are being evaluated for the SARS processes under ambient conditions. However, so far the SARS process still has some remaining issues:9 (a) a limited saturated adsorption capacity and regeneration of the adsorbents make the SARS process difficult to put into practice in large scale applications; (b) a systematic study of the effects of material structure and structural defects on the adsorption performance has not been seen yet, which restricts the improvement of desulfurization performance of adsorbents; (c) the current studies on the mechanism of the SARS reaction are still focused on some specific types of sorbents, and a universal theory for all the adsorbents is still not established.
Developing a novel adsorbent that simultaneously possesses very large sulfur-adsorption capacity and high selectivity for sulfur compounds, especially at ambient temperature (even at room temperature) and atmospheric pressure without hydrogen consumption, and is also easy for regeneration has been an emerging research topic. In the present work, starting from current SARS mechanisms and material structure of adsorbents, we developed a novel adsorbent for room temperature ultra deep desulfurization of jet fuels to be applied in SOFCs for the first time. According to the π-complexation mechanism,10–13 the π-complexation adsorbents, particularly the Y-zeolites, exhibit high sulfur-adsorption capacity, but show low selectivity for sulfur compounds as a result of competitive adsorption of aromatic compounds. Meanwhile, according to the adsorption mechanism of direct S–M interaction,14 the adsorbents possess high selectivity for sulfur compounds, but the steric hindrance makes it difficult to remove sulfur from DMDBT etc. However, the contradiction of the above two mechanisms may be utilized properly. On the one hand, thiophene has two lone pairs of electrons on the sulfur atom: one pair lies on the six-electron π system and the other lies in the plane of the ring. Thiophene can act either as an n-type donor by donating the lone pairs of electrons that lie in the plane of the ring to the adsorbent (direct S–M σ bond) or as a π-type donor by utilizing the delocalized π electrons of the aromatic ring (π bond) to form a π-type complex with the metal ions.14 On the other hand, the sulfur-adsorption capacity and selectivity of adsorbents can be further improved by modifying various types of surface active sites for sulfur adsorption, such as Lewis acid sites, useful functional groups, electronic defect centers, micro-structural defects and so on.
The previously reported ambient temperature SARS processes usually involve physical or chemical adsorptions on adsorbent surface, and the adsorbents include zeolites, activated carbons, ionic liquids and other mesoporous materials, which usually possess high sulfur-adsorption capacity. It is indicated that big specific surface area of solid adsorbents is beneficial to obtain high sulfur-adsorption capacity at ambient temperature by physical or chemical adsorption. According to Lewis acid–base theory, most thiophene sulfur compounds in jet fuels are Lewis base,15 which are easy to be adsorbed at Lewis acid sites. Hence, we can design and select materials that possess strong Lewis acid sites to selectively adsorb thiophene sulfur compounds with the lone pair electrons in jet fuels. The Lewis acid–base adsorption mechanism is the interaction between the acid sites on the surface of adsorbents and thiophene derivatives. Additionally, it is known that sulfur compounds have more affinity to oxidation than their analogue hydrocarbons in jet fuels,16 therefore, perfect redox properties of adsorbents can improve oxidization of sulfur compounds into sulfones and sulfoxides. High conversions of sulfides to sulfones and sulfoxides provide stronger polarities that enhanced selective removal of organic sulfur compounds with solid adsorbents at ambient temperature and atmospheric pressure. As a consequence, perfect redox properties of adsorbents can indirectly increase the sulfur-adsorption capacity and selectivity. Song and coworkers14 found that Ce–exchanged Y zeolites exhibited higher selectivity for sulfur compounds as compared to the selectivity of aromatics in the adsorptive desulfurization of a model jet fuel and a real JP-8 fuel, for which a comparative study indicated that the sulfur compounds were adsorbed over Ce–exchanged Y zeolites via direct sulfur-adsorbent (S–M) interaction rather than via π–complexation. It is suggested that some specific metal ions with strong ionic polarities are able to enhance direct S–M interactions and further improve adsorption selectivity for sulfur compounds at low temperature. Together with the material structure of an advanced solid adsorbent, a mesoporous structure can produce a big specific surface and different size-selective adsorption properties. Extremely dispersed state of active metal ions can avoid the possible aggregation of active species.17 Sometimes a geometric effect is the dominant reason for the strong synergistic adsorption effect rather than any electronic effect.18 As stated above, a novel ideal adsorbent can be depicted as shown in Fig. 1.
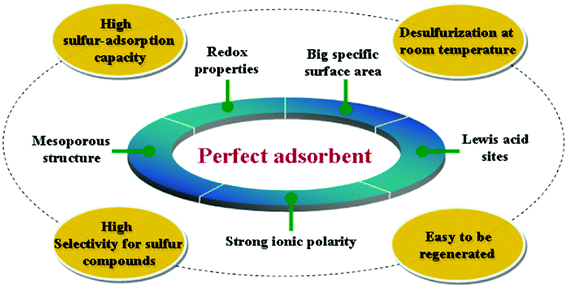 | ||
| Fig. 1 Ideal adsorbent model for ultra deep desulfurization of jet fuels. | ||
At present, there are few investigations and no systematic ones on adsorbents for ultra-deep desulfurization of real jet fuels, and most studies are on the basis of model jet fuels or a single sulfur compound which is very different to real jet fuels. In particular, the desulfurization performance of real jet fuels with high sulfur content at low temperatures, such as room temperature, is still not ideal. Therefore, the current study focused its efforts on improving the room temperature desulfurization performance of adsorbents in SARS of real jet fuels for portable power applications. With this objective we are developing a novel hybrid mesoporous material of Ni2+ and Ce4+ modified Al2O3–SiO2 binary oxide for selectively removing sulfur from real jet-A fuel with high sulfur content at room temperature. The first step was to screen some ideal adsorbents, which are efficient and active components for ultra-deep desulfurization of jet fuels. Examining the intrinsic chemical activities of all the current solid adsorbents, an adsorbent of reducing nickel supported on silica–alumina (Ni/SiO2–Al2O3) was found to exhibit an excellent performance in removing sulfur from jet fuel.3,4 In this approach, sulfur compounds are selectively removed by a direct S–M interaction rather than π-complexation, however, the nickel-based adsorbents usually have much higher capacity for removing sulfur at more than 200 °C.19 The major reason is that reducing nickel is easy to oxidize into nickel oxide, and the Ni-based components are easy to aggregate at higher temperature. Therefore, if we can make the nickel exist in a highly dispersed reducing state and maintain its structural stability, much higher capacity and selectivity for removing sulfur at lower temperature may be obtained. In the current study, we utilize cerium ions to highly disperse the nickel ions as an active component, and cerium also inhibits active nickel phase transformation because cerium ions possess big ion size effect.20 Meanwhile, cerium component possesses perfect redox properties that can improve oxidization of sulfur compounds into sulfones and sulfoxides which are easier to adsorb on adsorbents compared with the original sulfur compounds. Furthermore, the bigger specific surface area is able to induce higher desulfurization efficiency at room temperature. To obtain the effect we design Al2O3–SiO2 binary oxide, with Al2O3 as the major component, to be the adsorbent support because Al2O3 and SiO2 are common catalyst supports and they both possess big specific surface and low cost.
The structure of SiO2–Al2O3 with SiO2 as the major component oxide is shown in Scheme 1a. According to Tanabe's hypothesis,21 the charge difference for each bond is +4/4 − 3/6 = 1/2, and for all the bonds the valence unit of −1/2 × 6 = −3 is excess. In this case, the Brønsted acidity is assumed to appear in the presence of an excess of the negative charge, because three protons are considered to associate with six oxygens to keep the electric neutrality. Although the SiO2–Al2O3 support possesses big specific surface area, the SiO2–Al2O3 binary oxide itself exhibits the Brønsted acidity or Lewis base, which presents the same acid nature to repulse adsorbing thiophene sulfur. Consequently, its big specific surface area was not able to produce high sulfur-adsorption capacity at low temperatures. However, if we change the binary oxide mode into Al2O3–SiO2 with Al2O3 being the major component, the result will be contrary in effect and more interesting. The structure of Al2O3–SiO2 with Al2O3 being the major component oxide is shown in Scheme 1b. According to Tanabe's hypothesis, the charge difference for each bond is +4/4−3/6 = 1/2, and for all the bonds the valence unit of 1/2 × 4 = 2 is excess. In this case, the Lewis acidity is assumed to appear in the presence of an excess of the positive charge. Thereby the big specific surface and the Lewis acidity of the mesoporous Al2O3–SiO2 binary oxide will greatly enhance adsorption capacity of thiophene sulfurs in jet fuels at low temperatures.
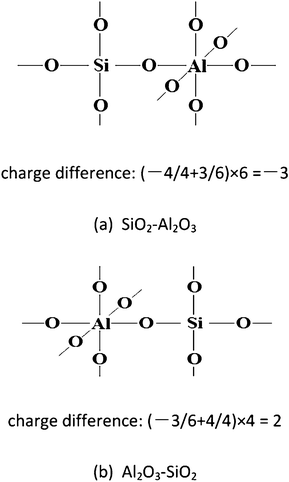 | ||
| Scheme 1 Model structures of SiO2–Al2O3 and Al2O3–SiO2 pictured according to Tanabe's hypothesis. (a) With SiO2 as the major oxide; (b) with Al2O3 as the major oxide. | ||
As indicated in the above discussions, the novel environmentally friendly adsorbent, Ni–Ce/Al2O3–SiO2, not only possesses big specific surface and as the Lewis acidity, but also exhibits perfect redox properties, highly dispersed active nickel components, as well as good structural stability and low cost. All these perfect properties are beneficial to the improvement of sulfur-adsorption capacity and selectivity for sulfur compounds in SARS process at room temperature.
Besides the intrinsic properties determined by chemical composition, the adsorption performances of adsorbent material are mainly dependent on material structure and defects, and the specific material structure is merely obtained by preparation and synthesis. With the objectives of indirectly studying the effect of adsorbent material structure on the sulfur adsorption performance and optimizing the preparation conditions of adsorbents, herein we synthesized the adsorbent of Ni–Ce/Al2O3–SiO2 by three methods: extrusion molding, wet impregnation, and sol–gel. We compared the effect of the three preparation methods on desulfurization efficiency in SARS of real jet-A fuels at room temperature. Fig. 2 shows the synthesis processes of the Ni–Ce/Al2O3–SiO2 adsorbents.
 | ||
| Fig. 2 Ni–Ce/Al2O3–SiO2 adsorbents prepared by three different methods. (a) Extrusion molding; (b) wet impregnation; (c) sol–gel. | ||
The major objective of our investigation is to explore the potential of Ni–Ce/Al2O3–SiO2 for room temperature ultra deep desulfurization of real jet-A fuel, an air-transportation logistic fuel, for solid oxide fuel cell applications. Some of our preliminary results on the desulfurization efficiency of jet-A fuels with high sulfur content over various adsorbents of Ni–Ce/Al2O3–SiO2 under static condition without stirring at room temperature and atmospheric pressure are given in Table 1.
| Ni–Ce/Al2O3–SiO2 | Original concentration (S-mg kg−1) | Concentration after adsorption (S-mg kg−1) | Desulfurization efficiency (%) | ||
|---|---|---|---|---|---|
| Sample | Preparation method | Sintering condition | |||
| 1 | Extrusion | 600 °C × 3 h, under helium | 949.03 | 62.26 | 93.47 |
| 2 | Sol–gel | 600 °C × 3 h, under helium | 949.03 | 79.76 | 91.60 |
| 3 | Wet impregnation | 600 °C × 3 h, under helium | 949.03 | 734.83 | 22.57 |
| 4 | Extrusion | 800 °C × 3 h, under helium | 949.03 | 84.02 | 91.15 |
| 5 | Sol–gel | 800 °C × 3 h, under helium | 949.03 | 196.98 | 79.24 |
| 6 | Wet impregnation | 800 °C × 3 h, under helium | 949.03 | 648.96 | 31.62 |
| 7 | Extrusion | 600 °C × 3 h, under air | 949.03 | 593.74 | 37.44 |
| 8 | Sol–gel | 600 °C × 3 h, under air | 949.03 | 264.25 | 72.16 |
| 9 | Wet impregnation | 600 °C × 3 h, under air | 949.03 | 709.93 | 25.19 |
| 10 | Extrusion | 800 °C × 3 h, under air | 949.03 | 590.85 | 37.74 |
| 11 | Sol–gel | 800 °C × 3 h, under air | 949.03 | 219.97 | 76.82 |
| 12 | Wet impregnation | 800 °C × 3 h, under air | 949.03 | 739.62 | 22.07 |
| 13 | Extrusion | 650 °C × 3 h, under helium | 949.03 | 33.88 | 96.43 |
| 14 | Extrusion | 700 °C × 3 h, under helium | 949.03 | 34.64 | 96.35 |
| 15 | Extrusion | 750 °C × 3 h, under helium | 949.03 | 41.09 | 95.67 |
Fig. 3 and 4 give a clear comparison of the desulfurization efficiencies of jet-A fuels by different adsorbents of Ni–Ce/Al2O3–SiO2 at room temperature and atmospheric pressure. For the adsorbents calcined under helium, the desulfurization efficiency decreases in the order of extrusion > sol–gel > wet impregnation. The adsorbents calcined at 600 °C generally possess higher desulfurization efficiency than that of the samples calcined at 800 °C. An exceptional case is that the adsorbent synthesized by wet impregnation and calcined at 600 °C has slightly lower desulfurization efficiency than that was calcined at 800 °C. This might be due to less interaction between the support material Al2O3–SiO2 and Ni–Ce active agent. However, adsorbent due to wet impregnation process generally has lower desulfurization performance compared to other two methods and may not meet the need of ultra deep desulfurization, which will not be further investigated. The desulfurization efficiency of the adsorbents calcined in air decreases in the order of sol–gel > extrusion > wet impregnation. The adsorbents calcined at 600 °C generally possess slightly lower desulfurization efficiency than that of the ones calcined at 800 °C. Increasing the temperature can accelerate adsorbent sintering, enhance combustion of organic matter, and improve volatilization of physically and chemically combined waters, and can enhance the surface activation energy of the adsorbents.22 The desulfurization efficiency of the adsorbents calcined under helium and air were compared and it was found that the adsorbents calcined in helium possesses much higher desulfurization efficiency than that of the samples calcined in air. The best desulfurization effect attribute to the adsorbent which was synthesized by extrusion molding, calcined at 600 °C for 3 h under He atmosphere, with particle sizes of 0.5–5.0 mm. At room temperature and atmospheric pressure a desulfurization efficiency of 93.47% to real jet-A fuel with a high sulfur content of 949.03 ppmw has been achieved. It confirms that the reducing states of nickel and cerium are able to provide much stronger polarities for adsorbing sulfur at room temperature compared with the oxidizing states. Considering the fact that the specific surface area is dependent on the matrix of Al2O3–SiO2 binary oxide, it is clear that both the adsorbents calcined under helium and air should have a similar specific surface. This further proves that the specific surface area here may not be a major reason that induces the high desulfurization efficiency at room temperature.
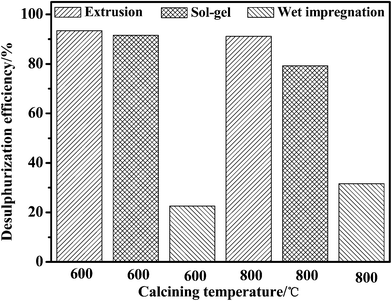 | ||
| Fig. 3 The desulfurization efficiency of jet-A fuels by adsorbents of Ni–Ce/Al2O3–SiO2 synthesized under helium. | ||
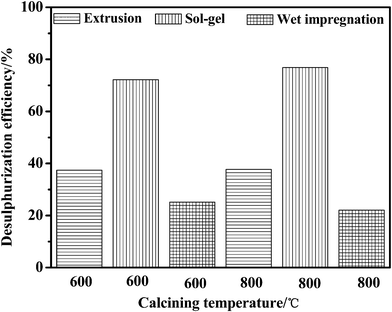 | ||
| Fig. 4 The desulfurization efficiency of jet-A fuels by adsorbents of Ni–Ce/Al2O3–SiO2 synthesized under air. | ||
In order to prove that the adsorbent effect is not random, and also to verify the chemical stability of the desulfurization performance over the adsorbents of Ni–Ce/Al2O3–SiO2 prepared by the extrusion method and calcined under a helium atmosphere, we studied the effect of calcination temperature on the desulfurization efficiency of jet-A fuels over different adsorbents prepared by extrusion and calcined at 600–800 °C. As seen in Fig. 5, the desulfurization efficiency is very stable at room temperature under atmospheric pressure. As the calcination temperature increases, the desulfurization efficiency shows a slight ascent, and then presents a slight decrease. The desulfurization efficiency of more than 95.00% was obtained in the calcination temperature range of 650–750 °C, and the best desulfurization efficiency obtained was 96.43% over the adsorbent calcined at 650 °C for 3 h.
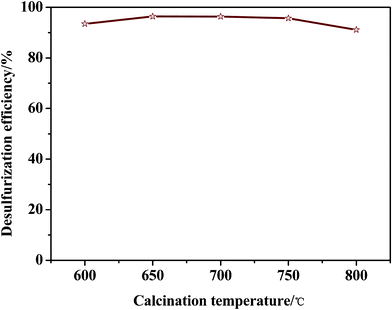 | ||
| Fig. 5 Effect of calcination temperature on desulfurization efficiency of jet-A fuels over Ni–Ce/Al2O3–SiO2 calcined under He. | ||
In conclusion, the high desulfurization efficiency obtained from the current experimental data verifies the designated novel concept of designing the new adsorbent Ni–Ce/Al2O3–SiO2. Due to the very simple and convenient reaction conditions of room temperature and atmospheric pressure without any assisted conditions, the Ni–Ce/Al2O3–SiO2 adsorbent provides broad application prospects for room temperature ultra deep desulfurization of real high-sulfur-content jet fuels without using any fraction or separation processes. The optimizations of the compatibility of Ni–Ce/Al2O3–SiO2 and reaction conditions will be given more insight and systematic study in our future work.
The above discussion reveals that the charge-compensating anion has a very strong effect on the π–complexation or direct S–M interaction by the cation for the sorbents.18,23 In the mean time, the novel adsorbent of Ni–Ce/Al2O3–SiO2 also possesses the same geometric effect as that of metal salts, which is another dominant reason for the strong synergistic adsorption effect.18 According to the edge sites theory, there are a large number of peripheral sites on the edges of the supported Ni2+ and Ce4+ ions which are well dispersed on the Al2O3–SiO2 binary oxide matrix, and the edge sites provide an ideal combination of sites for the thiophene molecule: Ni2+, Ce4+ for the thiophene sulfur and Lewis acid sites on the surface of Al2O3–SiO2 for the thiophene ring. Adsorption of a substituted thiophene molecule on such an edge site is depicted in Fig. 6. In this depiction, thiophene ring is adsorbed strongly at the Lewis acid sites on the surface of Al2O3–SiO2 matrix, while the thiophene sulfur is bonded to the adjacent Ni2+ sites or Ce4+ sites via direct S–M interaction or π–complexation. By binding cooperatively in this manner, stronger adsorption and hence higher adsorbed amounts could be achieved.
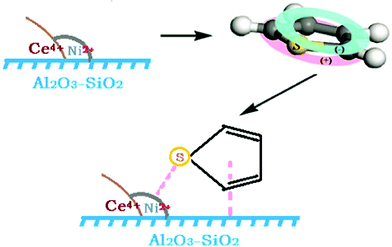 | ||
| Fig. 6 Depiction of synergistic effect in adsorption of thiophene on an adsorbent of Ni–Ce/Al2O3–SiO2. | ||
Experimental
Synthesis of adsorbents
The adsorbents of Ni–Ce/Al2O3–SiO2 (Ni/Ce = 10 in mol, Al/Si = 15 in mol, Al–Si–Ox/Ni–Ce–Oy = 9 in mass) were separately synthesized by three methods of extrusion molding, sol–gel and wet impregnation.All the prepared adsorbents have the same particle size of 0.5–5.0 mm, with the details given in Table 1.
Measurement of desulfurization efficiency
Desulfurization efficiency measurements of the adsorbents for selectively removing sulfur from real jet-A fuel were carried out in a fixed-bed reactor, using a 200 ml beaker under static conditions without stirring. Commercial grade jet-A fuel purchased from a local logistic fuel company in Tucson, Arizona was used for the tests. The jet-A fuel has total sulfur content of 949.03 mg kg−1 as measured in our laboratory. Jet-A fuel has the same flash point as Jet-A1 but a higher freeze point maximum (−40 °C). The additives in the jet-A fuel mainly include tetraethyl lead (TEL), antioxidants (AO–30, AO–31 or AO–37), fuel system icing inhibitor (FSII), chemicals for killing the biological substances, and so on. A certain amount of adsorbents was impregnated into the jet-A fuel for a short time at room temperature under atmospheric pressure. A TS 3000 total sulfur analyzer (Thermo Electron Corporation) with a TS-UV module total sulfur detector was used for the quantitative analysis to determine the total sulfur in the original jet-A fuel and the desulfurized jet-A fuels. The sulfur content expressed in mg kg−1 in this paper is on the basis of weight. The sulfur detection range was 0.02–5000 mg kg−1, the inaccuracy is less than 5%. The desulfurization efficiency of jet-A fuels is expressed by the equation:| XSulfur = ([S]Original − [S]Treated)/[S]Original × 100% | (1) |
Acknowledgements
The support from the Office of Naval Research of the USA and the University of Tennessee SimCenter under the contract #8500011366 is gratefully acknowledged. The first author is also grateful for the support from the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD) for his sabbatical and participation in this research.References
- Z. P. Shao, S. M. Haile, J. Ahn, P. D. Ronney, Z. L. Zhan and S. A. Barnett, Nature, 2005, 435, 795–798 CrossRef CAS.
- A. Heinzel, C. Hebling, M. Muller, M. Zedda and C. Muller, J. Power Sources, 2002, 105, 250–255 CrossRef CAS.
- X. L. Ma, S. Velu, L. Sun and C. S. Song, Prepr. Am. Chem. Soc., Div. Fuel Chem., 2003, 48, 688–689 CAS.
- S. Velu, X. L. Ma, C. S. Song, M. Namazian, S. Sethuraman and G. Venkataraman, Energy Fuels, 2005, 19, 1116–1125 CrossRef CAS.
- T. Fukunaga, H. Katsuno, H. Matsumoto, O. Takahashi and Y. Akai, Catal. Today, 2003, 84, 197–200 CrossRef CAS.
- C. S. Song, Catal. Today, 2003, 86, 211–263 CrossRef CAS.
- K. Liu, C. S. Song and S. Velu, Hydrogen and Syngas Production and Purification Technologies, John Wiley & Sons, Inc., Hoboken, New Jersey, 2009 Search PubMed.
- X. L. Ma, L. Sun and C. S. Song, Catal. Today, 2002, 77, 107–116 CrossRef CAS.
- Y. S. Shen, P. W. Li, X. H. Xu and H. Liu, RSC Adv., 2012, 2, 1700–1711 RSC.
- R. T. Yang, A. Takahashi and F. H. Yang, Ind. Eng. Chem. Res., 2001, 40, 6236–6239 CrossRef CAS.
- A. J. Hernandez-Maldonado, F. H. Yang, G. Qi and R. T. Yang, Appl. Catal., B, 2005, 56, 111–126 CrossRef CAS.
- A. Takahashi, F. H. Yang and R. T. Yang, Ind. Eng. Chem. Res., 2002, 41, 2487–2496 CrossRef CAS.
- A. J. Hernandez-Maldonado, G. S. Qi and R. T. Yang, Appl. Catal., B, 2005, 61, 212–218 CrossRef CAS.
- S. Velu, X. L. Ma and C. S. Song, Ind. Eng. Chem. Res., 2003, 42, 5293–5304 CrossRef CAS.
- M. Xue, R. Chitrakar, K. Sakane, T. Hirotsu, K. Ooi, Y. Yoshimura, Q. Feng and N. Sumida, J. Colloid Interface Sci., 2005, 285, 487–492 CrossRef CAS.
- O. Etemadi and T. F. Yen, Energy Fuels, 2007, 21, 2250–2257 CrossRef CAS.
- X. L. Song, L. B. Sun, G. S. He and X. Q. Liu, Chem. Commun., 2011, 47, 650–652 RSC.
- Y. Wang and R. T. Yang, Langmuir, 2007, 23, 3825–3831 CrossRef CAS.
- P. Zeuthen, K. G. Knudsen and D. D. Whitehurst, Catal. Today, 2011, 65, 307–314 CrossRef.
- S. M. Zhu, Y. S. Shen, W. F. Li and Y. Y. Liu, J. Rare Earths, 2006, 24, 234–238 CrossRef.
- K. Tanabe, T. Sumiyoshi, K. Shibata, T. Kiyoura and J. Kitagawa, Bull. Chem. Soc. Jpn., 1974, 47, 1064–1066 CrossRef CAS.
- Y. S. Shen, Y. F. Ma and S. M. Zhu, Catal. Sci. Technol., 2012, 2, 589–599 CAS.
- R. T. Yang, Adsorbents: Fundamentals and Applications, Wiley, New York, 2003 Search PubMed.
| This journal is © The Royal Society of Chemistry 2012 |