Deactivation mechanism of AuCl3 catalyst in acetylene hydrochlorination reaction: a DFT study
Jinli
Zhang
a,
Zhenghua
He
a,
Wei
Li
b and
You
Han
*c
aKey Laboratory of Systems Bioengineering MOE, Tianjin University, Tianjin, 300072, People’s Republic of China
bKey Laboratory for Green Chemical Technology MOE, Tianjin University, Tianjin, 300072, People’s Republic of China
cTianjin Key Laboratory of Membrane Science and Desalination Technology, Tianjin, 300072, People’s Republic of China
First published on 20th March 2012
Abstract
The deactivation mechanism of AuCl3 catalyst in the reaction of acetylene hydrochlorination was studied by using AuCl3 dimer model and the density functional theory (DFT) method. Four possible paths for the acetylene hydrochlorination reaction catalyzed by AuCl3 were illustrated with corresponding transition states. The activation free energies and reaction rate constants of the four paths were also analyzed. It is apparent that when HCl and C2H2 coadsorbed on the AuCl3 dimer, the C2H2 was co-catalyzed by HCl and the AuCl3 dimer to produce C2H3Cl and the reaction energy barrier was as low as 23.35 kcal mol−1. If the HCl in the gas phase could not adsorb on the Au site within the set time, the intermediate chlorovinyl was difficult to desorb from the AuCl3 catalyst as its desorption energy was as high as 41.336 kcal mol−1. As the reaction temperature increased, C2H2 became easier to be adsorbed on the AuCl3 catalyst prior to HCl, which resulted in the side reaction and the rapid deactivation of the AuCl3 dimer due to the loss of Cl atoms. Our calculations are necessary for us to clearly understand the experimental results, which indicate a great dependence of activity and stability of AuCl3 catalysts on the HCl![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :C2H2 ratio as well as the temperature.
:C2H2 ratio as well as the temperature.
1. Introduction
Acetylene hydrochlorination is currently an important industrial process in China for the manufacture of vinyl chloride monomer (VCM) that is polymerized to produce polyvinyl chloride (PVC) plastic. This importance is due to vast domestic coal resources and the relative lack of oil available to China.1 In the current industrial acetylene hydrochlorination reaction process, acetylene is reacted with anhydrous hydrogen chloride gas over a mercuric chloride (HgCl2) catalyst supported on activated carbon to give vinyl chloride.2 The use of mercuric chloride catalyst results in severe environmental pollution problems because of its strong tendency to sublime during the production of VCM.3,4 Hence, it is crucial to explore environmentally friendly non-mercury catalysts for acetylene hydrochlorination for sustainable PVC production via the acetylene-based method in China.In recent decades, many metal complexes have been studied as possible candidates to replace the toxic HgCl2 catalyst. Hutchings and co-workers5–9 found that the catalytic activity of the metal complex correlated with the standard electrode potential of the metal cation, and their experimental studies6,10 confirmed that the catalytic activity of Au3+ was the highest among the selected metal complexes, involving Bi3+,11,12 Pd2+,13 Pt2+14 and Pt4+.15. However, the XPS spectra in their experiments showed that Au-based catalysts are easy to deactivate with increasing reaction time due to the reduction of Au3+ to Au0.16,17 Conte et al.17 studied the effect of the second metallic component, including Pd2+, Pt4+, Rh3+, Ir3+ and Ru3+, on the Au-based catalyst, however, the stability of these bimetallic catalysts provided no significant improvement. Wang et al.18 reported a Au3+-Cu2+/C catalyst with a stability exceeding 200 h on stream reaction. Dai and co-workers19 reported that the component of La3+ on the Au-based catalyst can inhibit the valence change of gold to improve the stability of the catalyst. Conte et al.20 investigated the effect of the HCl:C2H2 ratio on the catalytic activity of Au-based catalysts and suggested that excessive HCl can enhance the activity of Au-based catalysts. Hutchings and coworkers7 reported that the deposition of carbonaceous materials on the catalyst is the predominant deactivation process occurring at temperatures lower than 100 °C, while the reduction of Au3+ to Au0 is the other deactivation pathway at temperatures higher than 120 °C. However, the deactivation mechanism of Au-based catalysts in the acetylene hydrochlorination reaction, which is fundamental to develop highly efficient and environmentally benign non-mercuric catalysts for industrial VCM production, has remained unknown.
With the rapid development of computer technology and quantum chemistry, theoretical studies on the mechanism of the catalytic reaction as well as catalyst deactivation have become an area of great interest.21–26 Studies at the electronic level via density functional theory (DFT) are considered valuable in order to provide theoretical guidance for catalyst design.27–29
In this article, a systemic study of the catalytic mechanism of AuCl3 in the reaction of acetylene hydrochlorination was carried out by using DFT. A better understanding of the deactivation mechanism of the AuCl3 catalyst will not only help to improve the performance of Au-based catalysts, but also shed light on the development of efficient industrial non-mercury catalysts for the reaction of acetylene hydrochlorination.
2. Computational methods
All calculations were carried out with the Gaussian 03 program package.30 The geometrical optimizations of the reactants, products, intermediates and transition states were performed using Beeke’s three-parameter exchange functional31 and the nonlocal correlation functional of Lee, Yang, and Parr32 (B3LYP) with the 6-31G(d) basis set for all atoms except for Au, which has been described with Lanl2dz pseudo-potential basis set. Harmonic vibrational frequency calculations were performed at the same level in order to confirm the various stationary points to be either a minimum or a transition structure (TS). Intrinsic reaction coordinate (IRC)33,34 calculations were carried out to confirm the connection of each TS to its corresponding reactants and products. All the charge analyses reported were calculated by natural bond orbital (NBO) analysis.35,36 The discussed energies are relative Gibbs free energies (ΔG298K). The relative enthalpies (ΔH298K) and ZPE corrected electronic energies (ΔE0K) are also provided for reference.The reaction rate constants (k) of all the discussed reactions were calculated using transition state theory (TST) (equation (1)).37,38
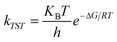 | (1) |
3. Results and discussion
3.1 The adsorption of acetylene and hydrogen chloride on gold trichloride.
The structures of gold chlorides, especially those of trichlorides, differed from the structures of many other metal trichlorides. AuCl3 forms planar dimeric molecules in both the low-temperature gas phase and its crystal structure.39,40 Since the reaction of acetylene hydrochlorination catalyzed by gold trichloride is a gas-solid reaction system, it is more reasonable to use the AuCl3 dimer structure to study its deactivation mechanism here. According to our calculation result, the dimer of gold trichloride has a planar D2h-symmetry chloride-bridged structure, which is in agreement with the 5d8 structure of Au3+. Relativistic effects bring about the contraction of the 6s orbital and the expansion of the 5d orbital. Due to this effect, the 5d orbital is the major contributor to the valence shell. The shapes of these orbitals favor the planar arrangement found in AuCl3.41 In the planar structure, the angle of Clside-Au-Clbridge is 177.6°. The bond lengths of Au-Clbridge and Au-Clside are 2.467 and 2.333 Å, respectively. The lowest unoccupied molecular orbital (LUMO) of this structure exhibits a large lobe on the six chlorines of the complex, especially on the four side chlorines (shown in Fig. 1). | ||
| Fig. 1 The calculated LUMO state for Au2Cl6 at the B3LYP/6-31G(d) (Lanl2dz for Au) level. | ||
The electron affinity of the Au3+ decreased because of the planar four-coordinate structure. Since the electron affinity of the Cl− was low, it is difficult for C2H2 and HCl to form a stable complex structure with the planar AuCl3 dimer. The favored adsorption structures of the C2H2 molecule and the HCl molecule on the AuCl3 dimer are shown in Fig. 2. They differ from the adsorption of C2H2 and HCl on a single AuCl3.17Ead shown in the figure represents the adsorption energy between the reactants and the catalyst. It was calculated using equation (2).
 | ||
| Fig. 2 The adsorption structures of reactant (C2H2, HCl, HCl–C2H2 complex) on Au2Cl6. | ||
| E ad = Eabsorption-state − (Ereactant + Ecatalyst) | (2) |
3.2 Reaction mechanisms of acetylene hydrochlorination over Au2Cl6
In order to better understand the deactivation mechanism of the AuCl3 catalyst in the reaction of acetylene hydrochlorination, the reaction mechanism of acetylene hydrochlorination catalyzed by Au2Cl6 was systematically studied. The reaction paths beginning with the HCl and C2H2 coadsorption structures (shown in Fig. 2 (d,e)) were denoted as path 1 and path 2, respectively. When the gas phase around the Au catalyst is lacking HCl, the co-absorption of HCl with C2H2 on the catalyst surface can not occur, and the acetylene will react with the AuCl3 catalyst first. When C2H2 adsorbed at the Clbridge site, it was difficult for C2H2 to react with the AuCl3 dimer because the Clbridge atoms were very stable. But the adsorbed C2H2 can easily transfer from the Clbridge site to the Autop site and begin to react with the AuCl3 dimer. The reaction paths beginning with adsorbed C2H2 at Autop site and Clside site were denoted as path 3 and path 4, respectively. The reaction mechanism of these paths is discussed in detail below.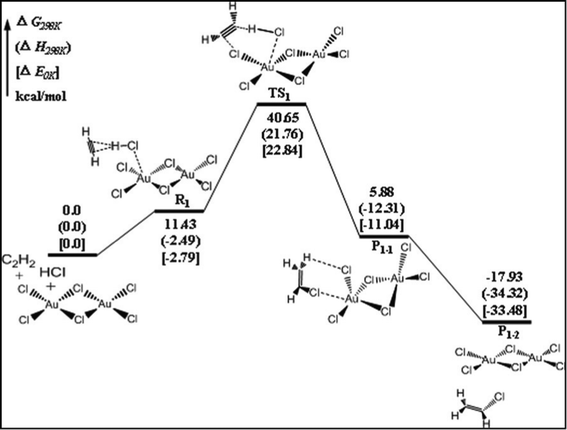 | ||
| Fig. 3 DFT calculated energy surface for C2H2 hydrochlorination through path 1. | ||
 | ||
| Fig. 4 The optimized key structures for C2H2 hydrochlorination through path 1 (the unit of bond length is Å). | ||
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond is weakened and the bond length stretched from 1.205 to 1.225 Å. The planar structure of the AuCl3 dimer catalyst is distorted. The Cl 4-Au 2 bond reverses downward and forms an angle of 85.25° with the plane. Meanwhile, the H atom of HCl interacts with the Cl 4 atom of the AuCl3 dimer to form a H 14–Cl 4 bond (shown in the Fig. 6 R2-1). The distance of Au 2 and Cl 5 is 3.214 Å, implying that the Au 2–Cl 5 bond is broken. The AuCl3 dimer catalyst displays a monomer T-shape feature. The complex of R2-1 can convert into the intermediate P2-1 with a six-membered ring transition structure TS2-1. In this reaction process, nucleophilic attack occurs between the adsorbed HCl and C2H2. The Cl 13 atom (−0.19 e) from HCl and C 10 atom (+0.05 e) from C2H2 begin to interact with each other and their distance is shortened from 4.055 to 2.511 Å in R2-1 and TS2-1, respectively. Thus, a six-membeedr ring is formed in TS2-1, which is similar to that in the TS1 of path 1. The π electron on the Au is transferred back to C2H2 through the six-member ring. A nucleophilic addition reaction finished with producing the intermediate chlorovinyl. Contrary to path 1, the Cl of the intermediate chlorovinyl is supplied by HCl, and then the H 14 combines with Cl 4 from the AuCl3 dimer to form a new adsorbed HCl molecule on the AuCl3 dimer. When the chlorovinyl intermediate is produced, the AuCl3 dimer loses a Cl atom, while a HCl molecule exists before and after the production of chloride vinyl. It seems as though the AuCl3 dimer acts as a reactant and the HCl acts as a catalyst in this reaction pathway. In the intermediate P2-1, the Au catalyst becomes a Au2Cl5 coordinate structure by losing a Cl atom, but it still maintains its planar structure due to the adsorption of chlorovinyl. The TS2-1 transition state requires an activation free energy of 23.35 kcal mol−1, and a 36.57 kcal mol−1 free energy is released during this reaction (shown in Fig. 5).
C bond is weakened and the bond length stretched from 1.205 to 1.225 Å. The planar structure of the AuCl3 dimer catalyst is distorted. The Cl 4-Au 2 bond reverses downward and forms an angle of 85.25° with the plane. Meanwhile, the H atom of HCl interacts with the Cl 4 atom of the AuCl3 dimer to form a H 14–Cl 4 bond (shown in the Fig. 6 R2-1). The distance of Au 2 and Cl 5 is 3.214 Å, implying that the Au 2–Cl 5 bond is broken. The AuCl3 dimer catalyst displays a monomer T-shape feature. The complex of R2-1 can convert into the intermediate P2-1 with a six-membered ring transition structure TS2-1. In this reaction process, nucleophilic attack occurs between the adsorbed HCl and C2H2. The Cl 13 atom (−0.19 e) from HCl and C 10 atom (+0.05 e) from C2H2 begin to interact with each other and their distance is shortened from 4.055 to 2.511 Å in R2-1 and TS2-1, respectively. Thus, a six-membeedr ring is formed in TS2-1, which is similar to that in the TS1 of path 1. The π electron on the Au is transferred back to C2H2 through the six-member ring. A nucleophilic addition reaction finished with producing the intermediate chlorovinyl. Contrary to path 1, the Cl of the intermediate chlorovinyl is supplied by HCl, and then the H 14 combines with Cl 4 from the AuCl3 dimer to form a new adsorbed HCl molecule on the AuCl3 dimer. When the chlorovinyl intermediate is produced, the AuCl3 dimer loses a Cl atom, while a HCl molecule exists before and after the production of chloride vinyl. It seems as though the AuCl3 dimer acts as a reactant and the HCl acts as a catalyst in this reaction pathway. In the intermediate P2-1, the Au catalyst becomes a Au2Cl5 coordinate structure by losing a Cl atom, but it still maintains its planar structure due to the adsorption of chlorovinyl. The TS2-1 transition state requires an activation free energy of 23.35 kcal mol−1, and a 36.57 kcal mol−1 free energy is released during this reaction (shown in Fig. 5).
 | ||
| Fig. 5 DFT calculated energy surface for C2H2 hydrochlorination through path 2. | ||
 | ||
| Fig. 6 The optimized key structures for C2H2 hydrochlorination through path 2 (the unit of bond length is Å). | ||
When the intermediate P1-2 adsorbs a HCl on the Au 2 site (shown in Fig. 6 R2-2), it can generate product P2-2 with a transition state TS2-2. In R2-2, the Au 2–Cl 5 bond is broken due to the adsorption of HCl. The Cl of the adsorbed HCl bonds with Au 2 and the bond length is 2.429 Å. In addition, the H atom forms a weak hydrogen bond with the nearby Cl 5. Then, the H 14–Cl 13 bond rotates due to the H 14–C 8 interaction, which forms a four-membered ring in the transition state TS2-2 structure. In TS2-2, an electrophilic collision occurs between H 14 (+0.3 e) and C 8 (−0.62 e). The H 14–Cl 13 bond is broken and the H 14–C 8 bond is formed in the product P2-2. In P2-2, Cl 13 from HCl bonds to Au 2 and the Au2Cl5 recovers to the Au2Cl6 coordinate structure. The planar structure of Au2Cl6 was distorted after reaction. The activation free energy from R2-2 to P2-2 is 22.56 kcal mol−1 and the process is almost thermodynamically neutral (the exergonic energy is only 1.39 kcal mol−1). The distorted dimer Au catalyst is a non-stable structure,41 and it can convert into the planar structure spontaneously.
C bond is weakened. The distance between C 10 and Cl 4 shortens from 3.734 to 2.745 Å and the bond of Au 2–Cl 4 is stretched to 2.699 Å. The conformation of transition state TS3 through R3 needs an activation free energy of 29.84 kcal mol−1, which is larger than that in path 2. In the intermediate product P3, the Cl 4 from AuCl3 dimer is added to the C2H2 to form the intermediate chlorovinyl, which causes the Au2Cl6 to become reduced to Au2Cl5. In P3, the structure of the adsorbed chlorovinyl on Au2Cl5, had the same structure feature as the intermediate P2-1 in path 2. Therefore, it can convert into the final product P2-2 along the same path shown in Fig. 5, when sufficient HCl adsorbs on the site of Au 2.
 | ||
| Fig. 7 DFT calculated energy surface for C2H2 Hydrochlorination through path 3. | ||
 | ||
| Fig. 8 The optimized key structures for C2H2 hydrochlorination through path 3 (bond length measured in Å). | ||
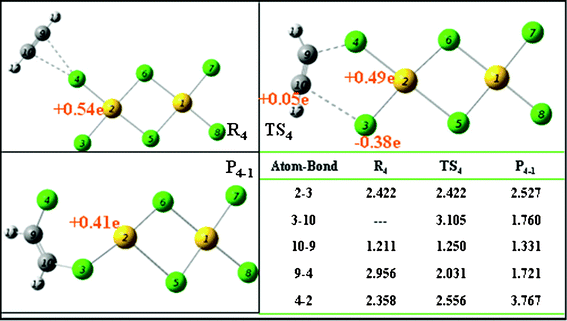
C bond of C2H2 is slightly stretched from 1.205 to 1.211 Å. When the partly activated C2H2 molecule moves close to the AuCl3 catalyst, the interaction of C 9–Cl 4 is enhanced and C 10 forms a bond with Cl 3. Then, the product of P4-1 is generated through the transition state TS4. In the structure of TS4, the distance C 9–Cl 4 is shortened from 2.956 to 2.031 Å. Meanwhile, electrophilic adsorption between C 10 (+0.05 e) and Cl 3 (−0.38 e) occurs and the CC bond length of C2H2 is further stretched to 1.25 Å. The activation free energy for this path is 23.97 kcal mol−1. In P4-1, the bond length of C 10–Cl 3 is 1.760 Å, demonstrating that P4-1 is actually an adsorption structure of 1,2-dichloroethane on Au2Cl4. 1,2-Dichloroethane would need a minimum energy of 17.54 kcal mol−1 to be desorbed from Au2Cl4. In path 4, the Au catalyst would be reduced because of the loss of Cl.
 | ||
| Fig. 9 DFT calculated energy surface for C2H2 hydrochlorination through path 4. | ||
 | ||
| Fig. 10 The optimized key structures for C2H2 hydrochlorination through path 4 (bond length measured in Å). | ||
3.3 Dynamic analysis of the four reaction paths
According to transition state theory (TST), the reaction rate constant is related with the activation free energy. It could be calculated by equation 1 described in section 2, which is simplified as the following (equation 3).| kTST = 2.08 × 1010Te−ΔG/RT | (3) |
Here, the rate constants of the reaction paths discussed in section 3.2 are calculated using equation 3 and the changes of kTST with reaction temperature are shown in Fig. 11.
 | ||
| Fig. 11 Reaction rate constants versus the reaction temperature via TST calculation. | ||
Among the four reaction paths, path 2 and path 3 are two-step reactions. The results in Fig. 11 show that the rate constant of the second step reaction in paths 2 and 3 is far larger than that of the first step reaction, so the first step reaction is the rate-limiting step in paths 2 and 3. It is noted that both the rate constants of paths 1 and 3 are in a magnitude of 10−3, while the rate constants of paths 2 and 4 are in a magnitude of 100. The rate constants of paths 2 and 4 are much larger than those of the paths 1 and 3, indicating that the reaction of acetylene hydrochlorination catalyzed by Au2Cl6 mainly occurs through paths 2 and 4. In addition, the reaction rate constant of path 4 is larger than that of path 2. Furthermore, the difference becomes more apparent as the reaction temperature is increased. It is suggested that a higher temperature causes more rapid deactivation of the AuCl3 catalyst, which is consistent with Hutchings and coworkers’ experimental results.7 They reported that the conversion rate of HCl decreased with a corresponding increase in temperature when the temperature is higher than 100 °C.
3.4 Deactivation mechanism of AuCl3 catalyst during the reaction of acetylene hydrochlorination
The dynamic analysis above shows that the rate constants of paths 2 and 4 are much larger than those of paths 1 and 3. The activation free energy comparison among the four paths gives the same conclusion. The activation free energies of paths 2 and 4 are 23.35 and 23.97 kcal mol−1, respectively, whereas paths 1 and 3 need 29.22 and 29.84 kcal mol−1, respectively. Thus, the reaction of acetylene hydrochlorination catalyzed by AuCl3 would be much easier to occur via paths 2 and 4 rather than paths 1 and 3. Since the activation free energies of paths 2 and 4 are nearly equal, they become the competitive reaction paths.When the reaction of acetylene hydrochlorination occurs via path 2, the AuCl3 catalyst still keeps the Au2Cl6 coordinate structure after the reaction and can recover to the initial planar configuration. Therefore, its catalytic activity would not decrease. If the HCl in the gas phase could not adsorb on the Au 2 site efficiently, the second step of path 2 would be more difficult. In this case, chlorovinyl is harder to be desorbed from the catalyst as its desorption energy is as high as 41.336 kcal mol−1, which causes the catalytic activity of AuCl3 to decrease due to the active Au sites being occupied. Therefore, the sufficient supply of HCl and its efficient adsorption on the Au site is the key to maintain the activity of the AuCl3 catalyst during the reaction of acetylene hydrochlorination. Our theoretical analysis was supported by the experimental works of Hutchings and co-workers.7,20 Their results showed that the activity and stability of the AuCl3 catalyst were sensitive to the ratio of HCl:C2H2. When the HCl:C2H2 ratio was 1:1, the conversion of C2H2 decreased by 10% after 2 h and the activity of the AuCl3 catalyst also decreased. When the HCl:C2H2 ratio is 1.5:1, the activitiy of the AuCl3 catalyst increased and the conversion of C2H2 was still high after 2 h.20 Hutchings and co-workers also reported that purging the reaction system with HCl gas during the reaction process could improve the activity of the AuCl3 catalyst. This phenomenon is also well explained by our theoretical results. When HCl gas is added in the reaction system during the reaction process, HCl is adsorbed at the Au 2 site to form R2-2 (shown in Fig. 5). Then it would react with the adsorbed chlorovinyl through the second step of path 2. Therefore, the additional HCl could help chlorovinyl to be desorbed from the Au site and release the active site, which reactivated the AuCl3 catalyst.
If the gas phase around the catalyst is lacking in HCl, the reaction occurs through path 4 by adsorbing C2H2 and the by-product dichloride ethylene would be generated. What is worse is that the AuCl3 catalyst is deactivated by losing two Cl atoms. The NBO analysis shows that the charge of Au in the planar AuCl3 dimer was +0.54 e, while it reduced to +0.41 e in P4-1, indicating the Au is reduced through path 4. In order to analyze whether the HCl purging method can reactivate the reduced Au2Cl4, the adsorption of HCl on Au2Cl4 is investigated here and the result is shown in Fig. 12. Although the HCl molecule can adsorb on the Au2Cl4, no apparent changes in either the charge or the structure of the Au2Cl4 are observed. The result indicates that the Au2Cl4 can not be reactivated by the simple purging of HCl once it has formed. If the AuCl3 catalyst is pretreated by purging HCl gas and the HCl is kept in at an appropriate excess during the reaction process, the reaction via path 4 will be avoided. This is also proved by the experiments of Hutchings and co-workers.20
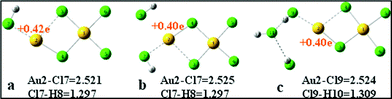 | ||
| Fig. 12 The optimized structures for Au2Cl4 purged by HCl (bond length measured in Å). | ||
4. Conclusions
In this work, the deactivation mechanism of AuCl3 catalyst in the reaction of acetylene hydrochlorination has been studied using DFT calculations. Our results show that there are four paths for the acetylene hydrochlorination reaction catalyzed by AuCl3. The activation energy analysis demonstrated that two of these reaction paths are more likely to occur. One is a main reaction and the other is a side reaction, and they compete with each other due to having similar activation free energy.In the two reaction paths mentioned above, two different deactivation mechanisms are discussed. In the main reaction, HCl and the AuCl3 dimer co-catalyze C2H2 and the intermediate chlorovinyl is produced. If the HCl in the gas phase could not adsorb on the Au site, chlorovinyl is difficult to desorb from the catalyst by converting to C2H3Cl as its desorption energy is as high as 41.336 kcal mol−1. Thus, the catalytic activity of AuCl3 would be decreased owing to the active Au sites being occupied. The timely and sufficient supply of HCl can release the active site to increase the catalytic activity. In the side reaction, the by-product 1,2-dichloroethane, which further produces other by-products, is generated. The AuCl3 catalyst deactivates rapidly by losing Cl atoms and the reduced state of catalyst, Au2Cl4, is formed and can not be reactivated by the HCl purging method.
The dynamic analysis showed that the side reaction occurs more easily when the reaction temperature is higher than 140 °C. Therefore, the appropriate reaction temperature must be selected in the actual experiment to avoid the side reaction occurring. Furthermore, if the AuCl3 catalyst—after modification—can efficiently activate a HCl molecule rather than C2H2, the side reaction will be suppressed, while the main reaction will be promoted. As a result of this, the deactivation of the AuCl3 catalyst may be effectively avoided .
Acknowledgements
This work was supported by the Special Funds for Major State Basic Research Program of China (2012CB720300), and NSFC (21176174, 20836005).References
- J. L. Zhang, N. Liu, W. Li and B. Dai, Front. Chem. Sci. Eng., 2011, 5, 514–520 CrossRef CAS.
- O. N. Temkin, G. K. Shestakov, Y. A. Treger, Acetylene: Chemistry, Reaction Mechanisms and Technology, Khimiya, Moscow, 1991, p.416 (in Russian) Search PubMed.
- G. J. Hutchings and D. T. Grady, Appl. Catal., 1985, 16, 411–415 CrossRef CAS.
- G. J. Hutchings and D. T. Grady, Appl. Catal., 1985, 17, 155–160 CrossRef CAS.
- G. J. Hutchings, J. Catal., 1985, 96, 292–295 CrossRef CAS.
- B. Nkosi, N. J. Coville and G. J. Hutchings, J. Chem. Soc., Chem. Commun., 1988, 1, 71–72 RSC.
- B. Nkosi, N. J. Coville, G. J. Hutchings, M. D. Adams, J. Friedl and F. E. Wagner, J. Catal., 1991, 128, 366–377 CrossRef CAS.
- G. J. Hutchings and M. Haruta, Appl. Catal., A, 2005, 291, 2–5 CrossRef CAS.
- G. J. Hutchings, Top. Catal., 2008, 48, 55–59 CrossRef CAS.
- B. Nkosi, N. J. Coville and G. J. Hutchings, J. Catal., 1991, 128, 378–386 CrossRef CAS.
- D. M. Smith, P. M. Walsh and T. L. Slager, J. Catal., 1968, 11, 113–130 CrossRef CAS.
- X. B. Wei, F. Wei, W. Z. Qian, G. H. Luo, H. B. Shi and Y. Jin, J. Process Eng. (Chinese), 2008, 8, 1218–1222 CAS.
- S. J. Wang, B. X. Shen, J. G. Zhao and J. C. Liu, Petrochem Technol. (Chinese), 2009, 38, 249–254 CAS.
- N. Dan, N.P. Khue, in: Fundamental Research in Homogeneous Catalysis, A. E. Shilov (Ed.), vol. 2, Gordon and Breach, 1986, 675 Search PubMed.
- S. A. Mitchenko, E. V. Khomutov, A. A. Shubin and Y. M. Shul’ga, J. Mol. Catal. A: Chem., 2004, 212, 345–352 CrossRef CAS.
- M. Conte, A. F. Carley and G. J. Hutchings, Catal. Lett., 2008, 124, 165–167 CrossRef CAS.
- M. Conte, A. F. Carley, G. Attard, A. A. Herzing, C. J. Kiely and G. J. Hutchings, J. Catal., 2008, 257, 190–198 CrossRef CAS.
- S. J. Wang, B. X. Shen and Q. L. Song, Catal. Lett., 2010, 134, 102–109 CrossRef CAS.
- H. Y. Zhang, B. Dai, X. G. Wang, L. L. Xu and M. Y. Zhu, J. Ind. Eng. Chem., 2012, 18, 49–54 CrossRef CAS.
- M. Conte, A. F. Carley, C. Heirene, D. J. Willock, P. Johnston, A. A. Herzing, C. J. Kiely and G. J. Hutchings, J. Catal., 2007, 250, 231–239 CrossRef CAS.
- J. G. Wang and B. Hammer, Top. Catal., 2007, 44, 49–56 CrossRef CAS.
- J. G. Wang and B. Hammer, Phys. Rev. Lett., 2006, 97, 136107 CrossRef CAS.
- N. Liu, B. H. Chen, Y. P. Li, R. D. Zhang, X. Liang, Y. X. Li and Z. G. Lei, J. Phys. Chem. C, 2011, 115, 12883–12890 CAS.
- J. J. Liu and Q. F. Ge, J. Chem. Theory Comput., 2009, 5, 3079–3087 CrossRef CAS.
- G. Ciancaleoni, N. Fraldi, P. H. M. Budzelaar, V. Busico, R. Cipullo and A. Macchioni, J. Am. Chem. Soc., 2010, 132, 13651–13653 CrossRef CAS.
- W. P. Zhang, S. T. Xu, X. W. Han and X. H. Bao, Chem. Soc. Rev., 2012, 41, 192–210 RSC.
- J. H. Guo, C. Xie, K. T. Lee, N. Guo, J. T. Miller, M. J. Janik and C. S. Song, ACS Catal., 2011, 1, 574–582 CrossRef CAS.
- M. Badawi, J. F. Paul, S. Cristol, E. Payen, Y. Romero, F. Richard, S. Brunet, D. Lambert, X. Portier, A. Popov, E. Kondratieva, J. M. Goupil, J. El Fallah, J. P. Gilson, L. Mariey, A. Travert and F. Mauge, J. Catal., 2011, 282, 155–164 CrossRef CAS.
- X. Gao, X. S. Du, Y. C. Fu, J. H. Mao, Z. Y. Luo, M. J. Ni and K. F. Cen, Catal. Today, 2011, 175, 625–630 CrossRef CAS.
- M. J. Frisch, Gaussian 03, revision C.02; Gaussian, Inc.: Pittsburgh, PA, 2004.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS.
- C. Gonzalez and H. B. Schlegel, J. Chem. Phys., 1989, 90, 2154–2161 CrossRef CAS.
- C. Gonzalez and H. B. Schlegel, J. Phys. Chem., 1990, 94, 5523–5527 CrossRef CAS.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter and F. Weinhold, NBO 4.M, Theoretical Chemistry Institute, University of Wisconsin, Madison, WI, 1999.
- E. D. Glendening, A. E. Reed, J. E. Carpenter and F. Weinhold, NBO Version Search PubMed , 3.1.
- J. I. Steinfeld, J. S. Francisco, W. L. Hase, Chemical Kinetics and Dynamics; Prentice-Hall, Upper Saddle River, NJ, 1989, p.246–268 Search PubMed.
- A. M. El-Nahas, A. H. Mangood, H. Takeuchi and T. Taketsugu, J. Phys. Chem. A, 2011, 115, 2837–2846 CrossRef CAS.
- P. Schwerdtfeger, P. D. W. Boyd, S. Brienne and A. K. Burrell, Inorg. Chem., 1992, 31, 3411–3422 CrossRef CAS.
- E. S. Clark, D. H. Templeton and C. H. MacGillavry, Acta Crystallogr., 1958, 11, 284–288 CrossRef CAS.
- M. Hargittai, A. Schulz, B. Réffy and M. Kolonits, J. Am. Chem. Soc., 2001, 123, 1449–1458 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |