Intermatrix synthesis of monometallic and magnetic metal/metal oxide nanoparticles with bactericidal activity on anionic exchange polymers†
Amanda
Alonso
*a,
Xavier
Muñoz-Berbel
b,
Núria
Vigués
c,
Rosalía
Rodríguez-Rodríguez
d,
Jorge
Macanás
e,
Jordi
Mas
b,
María
Muñoz
a and
Dmitri N.
Muraviev
a
aDepartment of Chemistry, Universitat Autonòma de Barcelona (UAB), Bellaterra, Barcelona, Spain. E-mail: amanda.alonso@uab.cat; Fax: +34-(0)5812379; Tel: +34 935811017
bCentre Nacional de Microelectrònica (IMB-CNM, CSIC), Bellaterra, Barcelona, Spain
cDepartment of Genetics and Microbiology, UAB, Barcelona, Spain
dDepartment of Pharmacology, Universidad de Sevilla, Sevilla, Spain
eDepartment of Chemical Engineering, Universitat Politècnica de Catalunya (UPC), Terrassa, Spain
First published on 28th March 2012
Abstract
In this communication, the synthesis of nanoparticles on anionic exchange polymers by the Intermatrix Synthesis method is reported. Monometallic (Ag) and core–shell metal/metal oxide (Ag@Fe3O4) nanocomposites were synthesized and characterized. Their magnetic and bactericidal activities were evaluated.
Intermatrix Synthesis (IMS) is a simple and fast protocol for the synthesis of nanoparticles (NPs) on polymeric matrices based on the loading of ion metal precursors and their chemical reduction generally under soft experimental conditions.1,2 Additionally, this method is also advantageous in terms of NP distribution since the difficulty of the ionic precursors to penetrate inside the matrix favours NP-formation on the polymer surface3,4 where they are highly accessible and functional. All these reasons make IMS one of the most promising routes to produce polymer stabilized metal or metal oxide NPs (PS-MNPs or PS-MONPs), as demonstrated by the number of publication involving NP synthesis on polymeric matrices by IMS.5,6
It is noteworthy that, even if amines are known to stabilize NPs against aggregation without disturbing their properties, most of the work reported up to now have been based on cationic exchange polymers and little attention has been paid to anion exchange polymers (i.e. amine-type, quaternary ammonium-type). This is basically due to the fact that in the case of anion exchangers both the polymeric matrix and ion precursors are positively charged. This limitation has been overcome using different strategies. The first consisted of changing the sign of the charge of either the NPs or the polymeric matrix. Hence, Yonezawa and Kunitake7a and Praharaj et al.7b modified Au-NPs with negatively charged molecules (3-mercaptopropionate or citrate, respectively) to favour their immobilization on positively charged polymeric matrices. Although being a good alternative, these approaches required strong experimental conditions (high temperature, reflux, etc.) and could not be directly used to synthesize other MNPs or MONPs. Another strategy was proposed by Sarkar et al.8. In this case, the anionic exchange polymer was initially loaded with a negatively charged oxidizing agent (e.g., NaOCl) that in situ oxidized Fe2+ ions to Fe3+ which, in the presence of hydroxyl anions, precipitated generating hydrated ferric oxide NPs. This approach was limited to insoluble metal oxide NPs and it was not possible to obtain metallic NPs.
Conversely, this communication reports an IMS method for the synthesis of metal and core–shell metal/metal oxide NPs on positively charged matrices. Particularly, Ag- and superparamagnetic Ag@Fe3O4-NPs with bactericidal activity were synthesized on anionic exchange polymers for the reagent-free disinfection of water. The granulated resin A520E9 (Purolite), consisting of a poly(vinylbenzyl chloride) backbone cross-linked with divinylbenzene and containing quaternary ammonium functional groups (–NR3+), was used as the polymeric matrix. The total ion exchange capacity (IEC) of the material was 1.4 milliequivalent of functional group per polymer gram (meq g−1).
The synthesis of NPs on this material was performed as follows. Initially, the polymer was pre-treated with 1.0 M NaCl for 1 h to neutralize the counter ions of the NR3+ groups in the polymer by Cl−. After washing with deionized water (3 times) and drying for 24 h at 80 °C, the polymer particles were sieved to obtain a homogeneous bead size close to 500 μm. From that point, the synthesis protocol depended on the NP type.
For Ag-NPs, the raw polymer was firstly loaded with 20 mL of the reducing agent solution (Na2S2O4 or NaBH4) for 1 h. Four Na2S2O4 concentrations ranging from 0.025 to 0.5 M and a single concentration of NaBH4 (0.5 M) were used. The material was then washed with deionized water (3 times) to eliminate the excess of reducing agent and loaded with 10 mL of AgNO3, the ionic precursor of the Ag-NPs. The AgNO3 concentration depended on the experiment, varying from 0.01 to 0.5 M. During this step, the Ag+ ions were in situ reduced, as illustrated in eqn (1) (R′ = organic chain). This resulted in the formation of the polymer–metal nanocomposites (NCs) containing Ag-NPs.
| (R′–NR3+)2 (S2O42−) + 4Ag+ + 4OH− → (R′–NR3+)2(S2O62−) + 4Ag0 + 2H2O | (1) |
Finally, samples were washed (3 times) with deionized water and dried for 24 h at 80 °C. It is worthy to note that the synthetic protocol for anionic exchange polymers is advantageous when compared with conventional IMS methods for cationic exchange materials. The main advantage is that, in this case, the loading and the reduction of the ionic precursor takes place simultaneously, thus avoiding metal leaking during the synthetic process. This is especially relevant for expensive metals.
On the other hand, the synthesis of Ag@Fe3O4-NPs required the combination of a co-precipitation method, commonly used for ferrite NPs preparation,10,11 with the IMS method, as described below. Initially, the raw material was pre-treated with 1.0 M trisodium citrate at 70 °C for 1 h to exchange the Cl− ions (eqn (2), where cit = citrate).
| 3(R′–NR3+)(Cl−) + (Na+)3(cit3−) → (R′–NR3+)3(cit3−) + 3NaCl | (2) |
After washing with deionized water (3 times), 0.2 g of polymer were incubated in 100 mL of a solution containing 26 mM FeCl2 and 41 mM FeCl3 (Fe2+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Fe3+ molar ratio = 1:2) for 1 h at 80 °C, with continuous stirring and under Ar atmosphere (eqn (3)).
:Fe3+ molar ratio = 1:2) for 1 h at 80 °C, with continuous stirring and under Ar atmosphere (eqn (3)).
| 4(R′–NR3+)3(cit3−) + Fe2+ + 2Fe3+ → (R′–NR3+)4(cit3−)4 (Fe2+, 2Fe3+) + 8R′–NR3+ | (3) |
Next, 125 mL of 0.5 M NaOH were slowly added with continuous stirring into the suspension containing iron salts and the polymeric material (final pH = 9). The suspension was incubated for 1 h at 80 °C. During the incubation, the polymer became black/brown in colour due to the formation of magnetite NPs by the following reaction:
| (R′–NR3+)4(cit3−)4 (Fe2+, 2Fe3+) + 8NaOH → 4(R′–NR3+)(cit3−)(Na+)2 + Fe3O4 + 4H2O | (4) |
The polymer beads containing magnetic NPs were washed with deionized water (4–5 times), collected with a magnet and dried for 24 h at 80 °C. The dried polymeric material containing Fe3O4-NPs was then loaded with Ag+ ions by incubation in 10 mL of 0.1 M AgNO3 for 1 h at room temperature. The polymeric material was washed with deionized water (3 times) and the immobilized Ag+ ions were reduced by incubation in 10 mL of 0.5 M NaBH4 (1 h, room temperature) to obtain Ag@Fe3O4-NPs. The final polymeric material was collected with a magnet and dried for 24 h at 80 °C.
The metal content (Table 1) on Ag and Ag@Fe3O4-NC, respectively, was determined by Inductively Coupled Atomic Emission Spectrometry (ICP-AES) or ICP Mass Spectroscopy (ICP-MS) as detailed in the ESI† (S.I.1).
| mgM/gNC | mmol/meq | |||||
|---|---|---|---|---|---|---|
| NaBH4/M | Na2S2O4/M | AgNO3/M | Ag | Fe | Ag | Fe |
| 0.50 | — | 0.10 | 16.1 | — | 0.11 | — |
| — | 0.50 | 0.10 | 86.2 | — | 0.57 | — |
| — | 0.25 | 0.10 | 78.0 | — | 0.52 | — |
| — | 0.10 | 0.10 | 18.4 | — | 0.12 | — |
| — | 0.025 | 0.10 | 10.00 | — | 0.07 | — |
| — | 0.10 | 0.50 | 23.2 | — | 0.15 | — |
| — | 0.10 | 0.25 | 21.3 | — | 0.14 | — |
| — | 0.10 | 0.10 | 18.4 | — | 0.12 | — |
| — | 0.10 | 0.01 | 18.2 | 0.12 | ||
| 0.50 | — | 0.10 | 260 | 83 | 1.7 | 1.1 |
In the case of the Ag-NC, the Ag content (from the NPs) was found to be extremely dependent on the reducing agent. Samples prepared with NaBH4 showed a lower metal content than those prepared with the same concentration of Na2S2O4 (Table 1). This fact was also confirmed by Scanning Electron Microscopy (SEM). Ag-NC cross-sections were prepared for SEM imaging as detailed in S.I.2†. In the images, the Ag-NPs appeared as a more or less intense bright area, depending on the concentration of NPs. Hence, samples prepared using NaBH4 (Fig. 1A) showed lower intensities (and a lower NPs concentration) than those prepared with Na2S2O4 (Fig. 1C), even when using a lower reducing agent concentration (Fig. 1B). Although the reasons for this are still controversial, the Donnan Exclusion Effect12 was identified to play a relevant role in the loading of the reducing agent. In this sense, molecules with a higher charge (e.g., S2O42−) would be easily loaded on the polymeric structure, thus favouring the formation of NPs. Besides, the metal content was also found to be very sensitive to the reducing agent concentration. The Ag content increased when increasing the reducing agent concentration until 0.25 M Na2S2O4, when it reached a plateau (Table 1 and Fig. 2A). This fact was confirmed by SEM imaging (Fig. 1B and 1C).
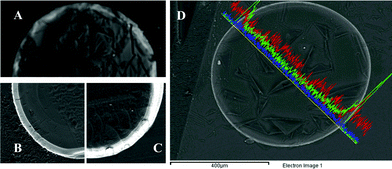 | ||
| Fig. 1 SEM images of cross-sections of Ag-NC samples prepared using (A) 0.5 M NaBH4, (B) 0.1 M Na2S2O4 and (C) 0.5 M Na2S2O4. (D) SEM image of the cross-section of Ag@Fe3O4-NC. The line scans (by EDS) show the distribution of the metal ions across the particles' diameter. Blue = Ag, red = Fe and green = O. | ||
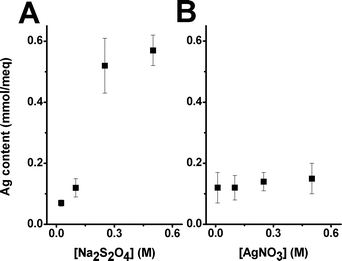 | ||
| Fig. 2 Representation of the variation of the Ag content with the concentration of (A) the reducing agent (Na2S2O4) and (B) the metal ionic precursor (AgNO3) during the loading step. | ||
On the contrary, the metal content in the sample did not significantly change with the ionic precursor concentration (Table 1 and Fig. 2B). This suggested that the most limiting step in the synthetic protocol was the initial loading of the polymeric matrix with the reducing agent.
For the Ag@Fe3O4-NC, the Ag content was higher than that obtained in the Ag-NCs, indicating that the magnetite core did not affect either the loading or the deposition of Ag. Moreover, Ag and Fe were found to co-localize in the Ag@Fe3O4-NC matrix (Fig. 1D).
The particles distribution in the NC was analyzed by SEM and Transmission Electron Microscopy (TEM), as detailed in S.I.2†. SEM images showed that, in all cases, the spots corresponding to NPs were mainly found concentrated on the polymer surface (Fig. 1D). In fact, TEM images demonstrated that the density of NP decreased when moving from the polymer surface to the center (Fig. 3A and 3B).
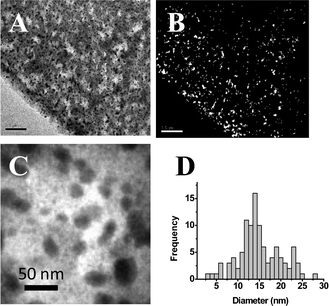 | ||
| Fig. 3 (A) TEM image of the external part of Ag-A520E-NC samples and (B) the corresponding mask (NP = white spots) obtained using Image J software. (C) Magnified TEM image of Ag-NPs in the anionic matrix. (D) NPs size histogram for Ag-A520E-NC samples. | ||
This non-homogeneous distribution of the NPs may be attributed to the Donnan Exclusion Effect. That is, the positive charge of the material impeded a deep diffusion of the positively charged ionic metal precursors into the polymeric matrix. This fact was even more accentuated in the case of Ag@Fe3O4-NPs (Fig. 1), where NPs were concentrated in a very thin layer on the polymer bead surface. Thus, even in the presence of citrate to compensate the charge of the polymer, the penetration of Fe2+ and Fe3+ ions was impeded.
TEM images were also used to determine the NP’s size. After the analysis of more than 100 isolated Ag-NPs, an average size of 14.4 ± 0.3 nm was obtained (Fig. 3D). The determination of the particles size was made easier than when cationic exchange fibres were used4 due to the low degree of aggregation of the NPs (Fig. 3C). This may be due to the presence of amino groups in the polymeric matrix since they are well known to stabilize NPs against aggregation, without modifying their properties.14
The magnetic properties of the Ag@Fe3O4-NCs were determined with a Superconducting Quantum Interference Device (SQUID) and compared with those obtained by polymeric structures only containing Fe3O4-NPs (see S.I.3†).
Similar magnetic hysteresis curves and saturation values were obtained when comparing both NCs, suggesting that the presence of Ag did not affect the magnetic properties of the material (Fig. 4). This was especially relevant when considering the final application of the NCs to reagent-free water purification. In particular, Ag-NPs have been found much more toxic than bulk Ag metal,13 limiting their application to real life environments. Thus, the possibility of collecting Ag@Fe3O4-NPs accidentally released from the polymeric matrix with a simple magnetic trap would be extremely desirable for water purification. Moreover, the low level of toxicity (probed in S.I.4†) of the superparamagnetic ferric oxides made them very convenient for biological applications.
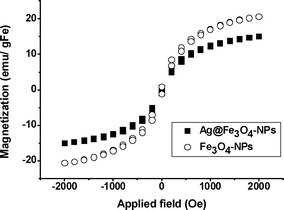 | ||
| Fig. 4 SQUID hysteric curve of (○) Fe3O4-NC and (■) Ag@Fe3O4-NC. | ||
The capacity of the NCs to inhibit bacterial proliferation was evaluated by using the Minimum Inhibitory Concentration (MIC) test (as detailed in S.I.5.1†) and continuous flow analysis. The MIC of both Ag- and Ag@Fe3O4-NCs was determined and compared with that obtained by the raw material without NPs or containing Fe3O4-NPs. The results are plotted in Fig. 5A.
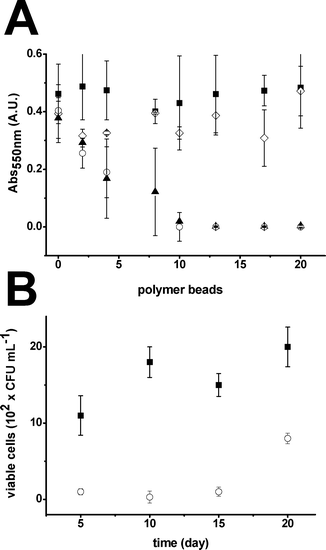 | ||
| Fig. 5 (A) Representation of the variation of the absorbance at 550 nm (indicative of bacterial proliferation) with the number of polymer beads for (■) the raw material, (◇) Fe3O4-NC, (○) Ag-NC and (▲) Ag@Fe3O4-NC (n = 3). (B) Representation of the number of viable cells with the treatment time for (■) the raw material and (○) Ag-NC. | ||
Both Ag- and Ag@Fe3O4-NCs showed high inhibitory activities with a deep decrease in the absorbance magnitude at 550 nm (Abs550) when increasing the number of NC beads in the suspension. In contrast, the raw material and the Fe3O4-NC did not present significant inhibitory activity at this concentration range, with a constant Abs550 value around 0.4 A.U. in all cases. This result indicated that Ag-NPs were responsible for the inhibition of bacteria proliferation and it was not affected by the presence of iron oxides. In the continuous flow analysis, the Ag-NC performance was evaluated with time over 20 days of continuous operation, as described in S.I.5.2†. The raw polymer was used as a control. According to Fig. 3B, the Ag-NC showed high bactericidal activity (between 80–90% killing efficiency) for at least 15 days but, after that, it quickly decreased. Several mechanisms may be involved in the decrease of bactericidal activity with time. The Ag release from the NC matrix and the formation of bacterial biofilms were identified as the most plausible causes. In this sense, the amount of Ag released from the NC matrix (determined by ICP-AES) reached 25% of the total immobilized after 2 weeks of continuous operation. Additionally, bacterial formations were also found attached to the NCs (see Fig. S.I.5.3†). Thus, both metal leaking and biofilm formation were found to contribute in the bactericidal activity decrease. Hence, the material demonstrated good performance for quite long operation periods (i.e. similar to water purification jars).
In this work, a protocol for the synthesis of monometallic and metal/metal oxide NPs on anionic exchange polymers was presented. Ag and Ag@Fe3O4-NPs were synthesized on granulated resins by following a protocol based on the IMS method. In both cases, NPs were mainly found on the polymer surface favouring their contact with bacteria. In fact, both NCs presented excellent bactericidal activity, which combined with the magnetic properties of ferrite, made them excellent candidates for reagent-free water purification.
Acknowledgements
This work was supported by Research Grant MAT2006-03745, CSD2006-00044 TRAGUA (CONSOLIDER-INGENIO2010) and CTQ2009-14390-C02-02 from the Ministry of Science and Technology of Spain and by ACC1Ó for VALTEC 09-02-0057 Grant within FEDER Program. A. Alonso and X. Muñoz-Berbel respectively acknowledge the FI grant (AGAUR) and the Spanish Ministry of Science and Education for the award of a Ramón y Cajal contract.References
- P. Walter, E. Welcomme, P. Hallégot, N. J. Zaluzec, C. Deeb, J. Castaing, P. Veyssière, R. Bréniaux, J. L. Lévêque and G. Tsoucaris, Nano Lett., 2006, 6, 2215 CrossRef CAS.
- D. N. Muraviev, J. Macanás, J. Parrondo, M. Muñoz, A. Alonso, S. Alegret, M. Ortueta and F. Mijangos, React. Funct. Polym., 2007, 67, 1612 CrossRef CAS.
- M. Auffan, H. J. Rsoe, J. Y. Bottero, G. V. Lowry, J. P. Jolivet and M. R. Wiesner, Nat. Nanotechnol., 2009, 4, 634 CrossRef CAS.
- A. Alonso, N. Vigues, X. Munoz-Berbel, J. Macanás, M. Muñoz, J. Mas and D. M. Muraviev, Chem. Commun., 2011, 47, 10464 RSC.
- B. A. Rozenberg and R. Tenne, Prog. Polym. Sci., 2008, 33, 40–112 CrossRef CAS.
- P. Ruiz, M. Muñoz, J. Macanás and D. M. Muraviev, Chem. Mater., 2010, 22(24), 6616 CrossRef CAS.
- (a) T. Yonezawa and T. Kunitake, Colloids Surf., A, 1999, 149, 193 CrossRef CAS; (b) S. Praharaj, S. Nath, S. Kumar Ghosh, S. Kundu and T. Pal, Langmuir, 2004, 20, 9889 CrossRef CAS.
- S. Sarkar, L. M. Blaney, A. Gupta, D. Ghosh and A. K. SenGupta, React. Funct. Polym., 2007, 67, 1599 CrossRef CAS.
- A-520E Macroporous Strong Base Anion Exchange Resin Technical Data, PUROLITE S.A.
- A. J. Amali and R. K. Rana, Green Chem., 2009, 11, 1781–1786 RSC.
- S. Laurent, D. Forge, M. Port, A. Roch, C. Robic, L. Vander and R. N. Muller, Chem. Rev., 2008, 108(6), 2064 CrossRef CAS.
- V. Vatanpour, S. S. Madaeni, R. Moradian, S. Zinadini and B. Astinchap, J. Membr. Sci., 2011, 375, 284–284 CrossRef CAS.
- N. R. Panyala, E. M. Peña-Méndez and J. Havel, J. Appl. Biomed., 2008, 6, 117 CAS.
- K. S. Soppimatha, T. M. Aminabhavia, A. R. Kulkarni and W. E. Rudzinski, J. Controlled Release, 2001, 70(1–2), 1–2 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2ra20216f |
| This journal is © The Royal Society of Chemistry 2012 |