In vitro conversion of chanoclavine-I aldehyde to the stereoisomers festuclavine and pyroclavine controlled by the second reduction step†
Marco
Matuschek
a,
Christiane
Wallwey
a,
Beate
Wollinsky
a,
Xiulan
Xie
b and
Shu-Ming
Li
*a
aPhilipps-Universität Marburg, Institut für Pharmazeutische Biologie und Biotechnologie, Deutschhausstraβe 17A, D-35037 Marburg, Germany. E-mail: shuming.li@Staff.uni-Marburg.de; Fax: 0049-6421-2825365; Tel: 0049-6421-2822461
bPhilipps-Universität Marburg, Fachbereich Chemie, Hans-Meerwein-Straβe, 35032, Marburg, Germany
First published on 6th February 2012
Abstract
Ergot alkaloids are mycotoxins produced mainly by the genera Claviceps, Aspergillus and Penicillium. Natural and semisynthetic ergot alkaloids are used as drugs in modern medicine. Chanoclavine-I aldehyde was found to be the branch point in the biosynthesis of different ergot alkaloid types. In Claviceps purpurea, it was converted to agroclavine by agroclavine synthase EasG in the presence of reduced glutathione, while festuclavine synthase FgaFS and the old yellow enzyme FgaOx3 from Aspergillus fumigatus catalysed the formation of festuclavine by two reduction steps. In this study, pyroclavine was detected as a minor product in the incubation mixture with enzymes from A. fumigatus under optimised HPLC conditions. Furthermore, we demonstrated that the homologues of FgaFS and FgaOx3 from Penicillium commune, FgaFSpc and FgaOx3pc, also catalysed the formation of pyroclavine and festuclavine, but with a significantly higher yield for pyroclavine. In addition, EasG was also shown to convert chanoclavine-I aldehyde to pyroclavine and festuclavine in the presence of the old yellow enzyme FgaOx3 or FgaOx3pc. More importantly, the ratio of pyroclavine to festuclavine was controlled by the reduction of an imine intermediate catalysed by FgaFS, FgaFSpc or EasG, which was proven by using different combinations of these enzymes in the presence of FgaOx3 or FgaOx3pc. The structure of pyroclavine was unequivocally elucidated by NMR and MS analyses.
Introduction
Ergot alkaloids with the characteristic tetracyclic ergoline ring system are produced by different fungi of Ascomycota and can be classified, based on their structures, as clavine-type alkaloids and derivatives of D-lysergic acid, termed also ergoamides and ergopeptines.1–3 The clavine-type alkaloids, e.g. fumigaclavines A and C (Fig. 1), consisting merely of the ergoline ring and lacking an amide or a peptide moiety, are mainly produced by the fungal family Trichocomaceae, e.g. Aspergillus fumigatus (A. fumigatus) and Penicillium commune (P. commune).2,4 In contrast, derivatives of lysergic acid, e.g. ergotamine (Fig. 1), are mainly found in fungi of the family Clavicipitaceae, e.g. Claviceps purpurea (C. purpurea).3,5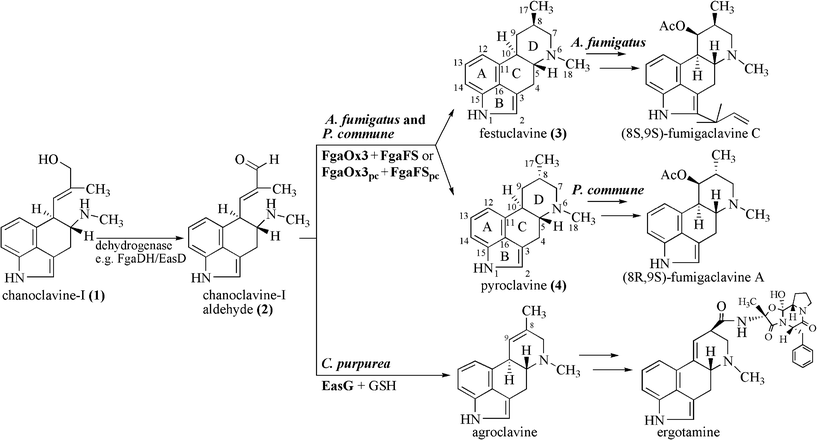 | ||
| Fig. 1 Proposed pathways from chanoclavine-I (1) to fumigaclavines in A. fumigatus and P. commune and to ergotamine in C. purpurea. | ||
The biosynthetic gene cluster of ergot alkaloids in C. purpurea has been proposed to consist of 14 genes, whereas 11 genes in A. fumigatus and 9 in P. commune NRRL2033 were predicted for the biosynthesis of fumigaclavines (Fig. 2).6–8 Comparison of the ergot alkaloid biosynthetic gene clusters in C. purpurea, A. fumigatus and P. commune revealed the presence of seven orthologous/homologous genes, which have been proven to be responsible for the formation of the tetracyclic ergoline scaffold.3,8–10 The early steps of ergot alkaloid biosynthesis are shared in all three fungi, whereas later steps differ from each other.3 Chanoclavine-I (1) was oxidised to its aldehyde (2), which was found to be a branch point for ergot alkaloid biosynthesis in these fungi (Fig. 1). In C. purpurea, 2 is converted to agroclavine by EasG in the presence of reduced glutathione without a requirement of the old yellow enzyme EasA.9 Agroclavine serves as a precursor of D-lysergic acid in the formation of ergopeptines, e.g. ergotamine.3 In A. fumigatus, 2 is converted to festuclavine (3) by festuclavine synthase FgaFS in the presence of the old yellow enzyme FgaOx3.11,12 The end products of the biosynthetic pathways in A. fumigatus and P. commune were found to be fumigaclavine C and fumigaclavine A, respectively.13–15 The structural difference of these ergot alkaloids in the two fungi is perfectly reflected by the presence of an additional prenyltransferase gene fgaPT1 in A. fumigatus.14 An additional difference between the structures of fumigaclavine A from Penicillium, and fumigaclavines from Aspergillus is the stereochemistry at C-8 (Fig. 1). The 8S-configuration was reported for fumigaclavines A, B and C from A. fumigatus,16,17 which has the same stereochemistry at this position as 3 obtained from enzyme assay with FgaFS and FgaOx3 from A. fumigatus.12 From P. crustosum18 and P. commune,14 fumigaclavine A with an 8R-configuration was reported, which would correspond to the stereochemistry of pyroclavine (4) at this position. To understand the molecular basis of the formation of fumigaclavines from different fungi with different stereochemistry at C-8, we proved the structure of fumigaclavine A from P. commune NRRL2033 and cloned and expressed the homologues of fgaOx3 and fgaFS from this fungus, termed fgaOx3pc and fgaFSpc in this study. In addition, EasA and EasG from C. purpurea were also investigated for production of 3 and 4, because Blaney et al.19 reported the accumulation of 3 in Claviceps africana. Enzyme assays of different combinations of the three imine reductases FgaFS, FgaFSpc and EasG with the three old yellow enzymes FgaOx3, FgaOx3pc and EasA provided evidence for the step determining the stereochemistry at C-8 of fumigaclavine precursors.
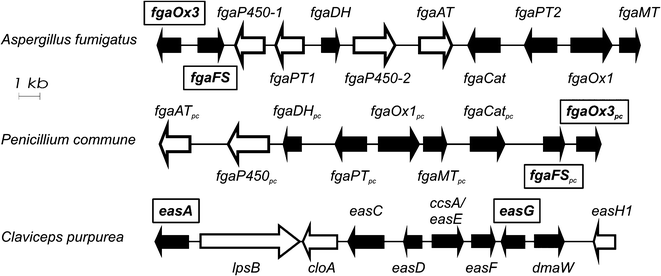 | ||
| Fig. 2 Biosynthetic gene clusters of ergot alkaloids from A. fumigatus Af293, P. commune NRRL2033 and C. purpurea (only parts shown). Homologous genes are indicated as black arrows and the genes investigated in this study, are placed in frames. | ||
Results and discussion
Confirmation of (8R,9S)-fumigaclavine A after isolation from P. commune NRRL2033
As mentioned before, the stereochemistry of fumigaclavine A from Penicillium spp. at C-8 differs from that of fumigaclavines from A. fumigatus.15–18,20 To confirm the configuration of fumigaclavine A from P. commune NRRL2033, the fungus was cultivated on wheat medium and extracted with ethyl acetate (see Experimental Section†). HPLC analysis of the extract showed the presence of one predominant peak at 19.4 min (Fig. S1, ESI†). No peak with same retention times as 1, 3 or 4 was detected (Fig. S1B, ESI†). For structure determination, 1.8 mg of fumigaclavine A were isolated on HPLC and subjected to NMR (Table S1 and S2 and Figs. S2.1–S2.4, ESI†) and MS analyses. NOESY, HSQC and HMBC spectra confirmed the isolated compound unequivocally as (8R,9S)-fumigaclavine A. NOE contact of the protons of the methyl group at C-8 with H-10α as well as with H-7α proved the α-configuration of this methyl group, which was also confirmed by the observation that no NOE contact was detected between H-8β and H-10α. It seems that the distance between H-8β and H-5β is too long, so that no NOE contact was detectable, as in the case of pyroclavine (see below). NOE contacts of H-9α with H-10α and α-methyl group at C-8 proved the β-configuration of the acetoxy group. The stereoisomer (8S,9S)-fumigaclavine A was not detectable in the HPLC chromatograms (Fig. S1B, ESI†).Sequence analysis and cloning of fgaOx3pc and fgaFSpc from P. commune NRRL2033
The genomic DNA sequence of P. commune NRRL2033 was available from a cosmid library constructed by Unsöld in 2006.21 The putative old yellow enzyme gene fgaOx3pc consists of one exon of 1140 bp and the deduced gene product has a length of 379 amino acids with a calculated molecular mass of 42.0 kDa. The putative festuclavine synthase gene fgaFSpc contains three exons of 418, 194 and 249 bp, interrupted by two introns of 67 and 65 bp, respectively. The deduced gene product comprises 286 amino acids with a calculated molecular mass of 31.5 kDa. An alignment with the program BLAST 2 SEQUENCES (http://blast.ncbi.nlm.nih.gov) showed on the amino acid level a sequence identity of 66% for FgaOx3pc and its homologue FgaOx3 (EAL94095.1) from A. fumigatus Af293. FgaFSpc shares a sequence identity of 62% with its homologue FgaFS (EAL94096.1) from A. fumigatus Af293. The sequence identities of FgaOx3pc and FgaFSpc with the putative old yellow enzyme EasA (CAG28312.1) and the agroclavine synthase EasG (AAW57089.1) from C. purpurea were found to be 56% and 42%, respectively.For cloning, fgaOx3pc was amplified by PCR from a cosmid 28D721 and cloned into pGEM-T easy vector. After sequence confirming by sequencing (MWG Biotech AG), the coding region was cloned into the expression vector pQE70 at the SphI and BamHI sites, resulting in the plasmid pMM050. The coding sequence of fgaFSpc was synthesised chemically by MWG Biotech AG and cloned into the pQE60 expression vector at the NcoI and BamHI sites, resulting in the plasmid pMM048. The predicted coding sequence was later also confirmed by PCR amplification from cDNA prepared from mRNA of P. commune NRRL2033.
Overproduction and purification of His6-FgaOx3pc and His6-FgaFSpc
The plasmids pMM050 and pMM048 were transferred into E. coli and cultivated at 22 °C and 30 °C, respectively. Induction of gene expression was achieved by addition of 0.1 mM IPTG to the bacterial cultures (see Experimental Section†). For purification of the His6-tagged FgaOx3pc and FgaFSpc a combination of Ni-NTA agarose and HisPur Cobalt resin was used. In the case of pMM050, a major protein band with migration below the 45 kDa size marker was observed (Fig. S3, ESI†), as expected for His6-FgaOx3pc with a calculated mass of 43.2 kDa. For pMM048, a major protein band with migration at the 30 kDa size marker was detected (Fig. S3, ESI†), as expected for the His6-FgaFSpc with a calculated mass of 32.6 kDa. For both purified enzymes, low yields of 50 μg l−1 of culture were obtained.Enzyme activity and biochemical properties of FgaOx3pc and FgaFSpc
We have previously shown that FgaDH catalyzed the conversion of 1 to 2,22 which was then converted to 3 by FgaFS in the presence of the old yellow enzyme FgaOx3, all from A. fumigatus (Fig. 3B and 4A).12,22 Detailed inspection of the HPLC chromatogram of the incubation mixture with 1, FgaDH, FgaOx3 and FgaFS under the optimised HPLC conditions (Fig. 3B) revealed the presence of a minor product with a retention time of 39 min. The product was not observed previously and identified in this study as pyroclavine (4) (see below). The ratio of 3 to 4 was estimated to be 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5.
:5.
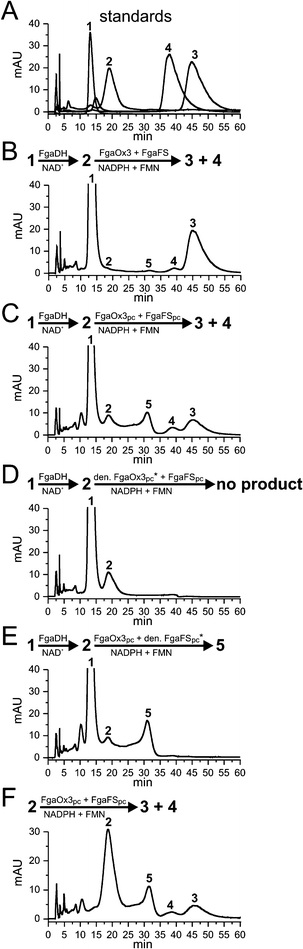 | ||
| Fig. 3 HPLC chromatograms of incubation mixtures of chanoclavine-I and chanoclavine-I aldehyde with different enzyme combinations. The reaction mixtures contained 5 mM of the cofactors NAD+, FMN, NADPH, 1 mM chanoclavine-I, or chanoclavine-I aldehyde and 5 μg of the recombinant enzymes and were incubated at 30 °C for 15 h. The substances were detected with a Photodiode Array detector and illustrated for absorption at 282 nm. 1: chanoclavine-I; 2: chanoclavine-I aldehyde; 3: festuclavine; 4: pyroclavine; 5: shunt product (* heat-inactivated FgaOx3pc or FgaFSpc by boiling for 60 min) | ||
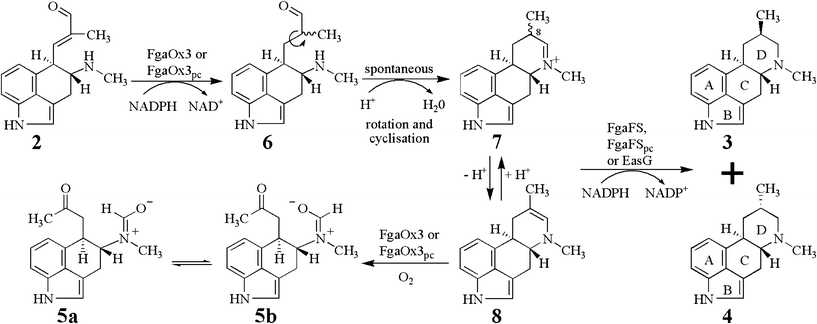 | ||
| Fig. 4 Proposed mechanism of the conversion of chanoclavine-I aldehyde (2) to festuclavine (3) and pyroclavine (4) in P. commune and A. fumigatus by two reduction steps. | ||
In analogy to FgaFS and FgaOx3 from A. fumigatus, FgaOx3pc and FgaFSpc from P. commune were assayed in the presence of FgaDH from A. fumigatus with 1 by using NAD+, FMN and NADPH as cofactors. As shown in Fig. 3C, product peaks with same retention times to those of 3 and 4 were detected after incubation for 15 h. Formation of the peaks 3 and 4 from 1 was strictly dependent on the presence of all of the three active enzymes. Neither 3 nor 4 was detected in the incubation mixtures with heat-denatured FgaOx3pc (Fig. 3D) or FgaFSpc (Fig. 3E). Formation of 3 and 4 was also observed, when 2 was isolated from an enzyme assay of 1 with FgaDH and NAD+ and incubated with FgaOx3pc and FgaFSpc (Fig. 3F). Therefore, the presence of FgaDH was only necessary for the conversion of 1 to 2.
As observed for FgaOx3 from A. fumigatus, an additional peak (5) with a retention time of 31 min was detected in the incubation mixture of FgaOx3pc and FgaFSpc. This peak had been identified as a shunt product consisting of two isomers (5a and 5b), in which the aldehyde group had migrated to form a formamide with the amino group (Fig. 4).12,23 Formation of 5 was dependent on the presence of active FgaOx3pc but not of active FgaFSpc (Fig. 3D and 3E) proving that FgaOx3pc from P. commune also catalysed, in analogy to its homologue FgaOx3 from A. fumigatus, the formation of the shunt product peak 5.
To determine the native molecular mass of FgaOx3pc and FgaFSpc, size exclusion chromatography was used. For His6-FgaOx3pc, a molecular mass of 40 kDa was determined, suggesting that the native FgaOx3pc is a monomer. In the case of the recombinant His6-FgaFSpc, the molecular mass was calculated to be 64 kDa, suggesting that the native FgaFSpc is a dimer.
EasG catalysed in the absence of reduced glutathione also the formation of 3 and 4
We have recently reported that the homologue of FgaFS from C. purpurea, i.e. EasG, catalysed the conversion of 2 to agroclavine in the presence of reduced glutathione (GSH).9 The old yellow enzyme EasA from C. purpurea was inactive and not involved in this conversion. We have proposed that EasG catalysed, in analogy to FgaFS, also a reduction step of an imine intermediate, which differs from that of FgaFS just by the presence of one additional double bond.9 We proposed in this study, that EasG would also catalyse the conversion of 2 to 3 and/or 4 in the presence of an active old yellow enzyme. Therefore, EasG was incubated with 1 in the presence of FgaDH, FgaOx3 or FgaOx3pc. As shown in Figs. S6C and S6F,† formation of 3 and 4 was clearly observed. This proved that EasG can catalyse the same reaction as its homologues in the absence of GSH.Identification of the enzyme product 4 as pyroclavine
To elucidate the structures of the enzyme products 3 and 4, 1 was incubated with FgaDH, FgaOx3 and EasG in the presence of NAD+, NADPH and FMN. Because the ratio of 3 to 4 in the incubation mixture with EasG was higher than those with FgaFS or FgaFSpc (Fig. S6† and Table 1), EasG instead of FgaFS or FgaFSpc was chosen as the imine reductase for this experiment. 1.7 mg of 3 and 0.7 mg of 4 were isolated by preparative thin layer chromatography (TLC) followed by HPLC purification in order to remove silica gel impurities.| Enzyme combination | Incubation with NADPH | Incubation with NADH | ||||
|---|---|---|---|---|---|---|
| Ratio of products | Ratio of products | |||||
| chanoclavine-I consumption (%) | festuclavine (3) | pyroclavine (4) | chanoclavine-I consumption (%) | festuclavine (3) | pyroclavine (4) | |
| FgaOx3pc + FgaFS | 43 | 95 | 5 | 23 | 88 | 12 |
| FgaOx3pc + FgaFSpc | 18 | 79 | 21 | 3 | 72 | 28 |
| FgaOx3pc + EasG | 35 | 70 | 30 | 8 | 63 | 37 |
| FgaOx3 + FgaFS | 26 | 96 | 4 | 48 | 90 | 10 |
| FgaOx3 + FgaFSpc | 25 | 79 | 21 | 9 | 74 | 26 |
| FgaOx3 + EasG | 22 | 70 | 30 | 19 | 65 | 35 |
After HPLC purification, NMR spectra were taken for 3 and 4 in their protonated form. 3 was unequivocally identified as festuclavine by comparison of its NMR data with those published previously.12 The NMR Data and spectra of 4 are supplied as Table S3 and Fig. S4 as well as Figs. S5.1–S5.6 in ESI,† respectively. Assignments of 1H and 13C signals have been proven by DQF-COSY, NOESY, HSQC and HMBC experiments.
We initially speculated that 4 could be pyroclavine, i.e. the stereoisomer of 3. Indeed, 4 has the same molecular formula C16H20N2 as 3 by detection of a [M]+ ion at m/z 240.1609 (calculated value 240.1626). In the 1H-NMR spectrum of 4, the signals of the aldehyde and olefinic protons of 222 were absent. Comparison of the 1H-NMR spectrum of 4 with that of 3 revealed the presence of similar signals. Clear differences were found for H-7β, H-9β, H-8β and H-17 (for numbering please see Fig. 1). Slight differences were obtained for H-7α, H-9α, H-5β and H-10α. Almost no differences were observed for signals of aromatic protons. The HMBC contacts between H-18 and C-7 as well as H-7α and C-5 (Table. S3, ESI†) proved unambiguously the presence of the ring D (Fig. 1). This demonstrated that very likely 3 and 4 differ from each other by the substitution at position C-8. The stereochemistry of 4 was confirmed by NOE contacts of the protons within rings C and D. The configurations at C-5 (H-5β) and C-10 (H-10α) were used as reference (Fig. S4, ESI†). Medium NOE correlation was observed between H-17 and H-10α, proving that H-17 is α-substituted and H-8 β-substituted, respectively. This was supported by the medium NOE contacts between H-17α and H-7α as well as H-17α and H-9α. Both H-7α and H-9α showed no NOEs with H-5β, which showed medium NOEs with H-7β and H-9β. Therefore, 4 was unequivocally identified as pyroclavine. In summary, FgaFS and its homologues FgaFSpc and EasG catalyzed in the presence of FgaOx3 or FgaOx3pc the conversion of 2 to 3 and 4 by using the cofactors FMN and NADPH (Fig. 1).
Stereoselectivity of the conversion of 2 to 3 and 4
As shown in Fig. 1 and 4, conversion of 2 to 3 and 4 needs two reduction steps, i.e. reduction of a C–C double bond and an imine intermediate. The presence of the latter one in the incubation mixture was proven by MS analysis.24 During the conversion of 2, a new chiral centre at C-8 was formed resulting in the formation of two isomers 3 and 4 (see above). We have speculated that the reduction step by FgaFS from A. fumigatus controlled the stereochemistry and 3 was identified as predominant enzyme product (95%).12 It is however also possible, even more plausible, that the configuration at this position was already determined during the reduction of the double bond by FgaOx3. To provide evidence that the reductase FgaFS or its homologues FgaFSpc or EasG was responsible for the stereochemistry, we carried out incubations with different enzyme combinations in the presence of NAD+, FMN and NADPH or NADH as cofactors. In all of these enzyme assays, 1 was converted to 2 by FgaDH from A. fumigatus. Each of the three old yellow enzymes, FgaOx3 from A. fumigatus, FgaOx3pc from P. commune or EasA from C. purpurea, was assayed with one of the three imine reductases, i.e. FgaFS, FgaFSpc or EasG. The incubation mixtures were analysed on HPLC and illustrated in Fig. S6†. As shown in Figs. S6G, S6H and S6I, neither 3, nor 4 or 5 was detected in the combinations of EasA with all of the three imine reductases, proving that EasA was inactive and confirming our previously reported results.9 In contrast, product formation was clearly detected for the incubation mixtures of FgaOx3 and FgaOx3pc with all the three imine reductases (Figs. S6A–S6F†), indicating the compatibility of these enzymes. More importantly, ratio of 3 to 4 was dependent on the presence of the imine reductase FgaFS, FgaFSpc or EasG. For example, in the incubation mixtures of FgaFS with NADPH (Figs. S6A and S6D†), 3 was the predominant product (about 95%). Replacement of FgaFS by FgaFSpc resulted in the formation of 3 and 4 in a ratio of about 79:21 (Table 1). In the incubation mixture with EasG, the content of 3 was higher than those with FgaFS and FgaFSpc. From the obtained results, it is also obvious that the old yellow enzyme FgaOx3 or FgaOx3pc had almost no influence on the product ratio of 3 to 4. As cofactor for FgaFS, FgaFSpc and EasG, NADH was also tested with 5 μg of each recombinant enzyme (Table 1). With an exception for FgaOx3 and FgaFS, NADPH was a better electron donor than NADH, concluded from the consumption of 2. As observed with NADPH, the ratio of 3 to 4 was dependent on the presence of the used imine reductase, not on the old yellow enzyme. Similar results were obtained by using 10 μg of each recombinant enzyme (Table S4, ESI†), although in most cases the conversion yields with 10 μg were higher than with 5 μg protein.
The in vitro incubation of FgaOx3 with FgaFS resulted in the formation of festuclavine as main enzyme product (about 95%), which is consistent with the stereochemistry of the reported (8S,9S)-fumigaclavines from A. fumigatus.16,17,20 FgaOx3pc and FgaFSpc from P. commune NRRL2033 catalysed the conversion of chanoclavine-I aldehyde (2) to festuclavine (3) and pyroclavine (4). Coexistence of 3, 4 and fumigaclavine A and B was reported for P. commune VKM F-3088 (= NRRL2033).15 Unfortunately, no structures and therefore no information on the stereochemistry for C-8 were given for fumigaclavines in that work.15 As confirmed in this study, only (8R,9S)-fumigaclavine A accumulated in P. commune NRRL2033. The stereochemistry at C-8 would correspond to that of pyroclavine (4). However, the main product of the in vitro incubation with FgaOx3pc and FgaFSpc was identified as its stereoisomer festuclavine (3) (about 79%, Fig. S6E and Table 1). Therefore, (8S,9S)-fumigaclavine A, which has the same configuration at C-8 as 3, would be expected as the main ergot alkaloid in P. commune NRRL2033. The reason for this inconsistent result is unknown. One possible explanation could be that of the two products of FgaOx3pc and FgaFSpc only 4 is hydroxylated at C-9. The subsequent acetylation of the hydroxyl group by FgaAT25 would result in the formation of fumigaclavine A with the (8R,9S)-configuration. The stereoisomer 3 could not be accepted by the hydroxylase and would accumulate in the fungal cultures. Inspection of the HPLC chromatogram of the cultural extract (Fig. S1, ESI†), however, did not indicate the presence of 3 and therefore excluded this possibility. Another possible explanation could be that both 3 and 4 were accepted by the hydroxylase as substrates and the chirality at C-8 were however changed during the hydroxylation step. It can be speculated that the hydroxylase from A. fumigatus controls the formation of fumigaclavine B with a (8S,9S)-configuration and its homologue from P. commune the formation of a product with a (8R,9S)-configuration. Experiments to support or exclude this hypothesis are under investigation. It is also possible that in vitro conversion of 2 differs from the in vivo conditions, e.g. by involvement of additional cofactors or enzymes under controlled regulation, so that 3 or/and 4 would be produced and immediately metabolized.
Proposed mechanism for the formation of 3 and 4
From the results obtained in this study, we postulate following reaction mechanism (Fig. 4) for the conversion of 2 to 3 and 4 catalysed by FgaOx3pc or FgaOx3 with FgaFS, FgaFSpc or EasG. The 8,9-alkene of 2 would be reduced by FgaOx3 or FgaOx3pc resulting in the formation of the intermediate 6, which would undergo single bond rotation and formation of the imine 7 spontaneously by an intramolecular cyclisation. 7 will be reduced by FgaFS, FgaFSpc or EasG to the two stereoisomers 3 and 4. The ratio of 3 and 4 would be controlled by the stereoselectivity of these imine reductases. Equilibration between 7 and 8, which is an isomer of agroclavine, ensures the existence of sufficient 8α- and 8β-isomers of 7 to be reduced by FgaFS, FgaFSpc, or EasG stereoselectively. EasG from C. purpurea showed relatively poor stereoselectivity in comparison to FgaFS and FgaFSpc (Fig. S6† and Table 1). This sounds plausible, because its natural product agroclavine (Fig. 1) has no chirality centre at C-8 due to the presence of a double bond between C-8 and C-9. In the absence of an imine reductase, 8 was oxidised to 5a and 5b by the old yellow enzyme FgaOx3 or FgaOx3pc.Conclusions
In this study, we demonstrated clearly that the conversion of chanoclavine-I aldehyde (2) to festuclavine (3) and pyroclavine (4) was achieved by two reduction steps catalysed by an old yellow enzyme and an imine reductase, respectively. For this conversion, the old yellow enzyme FgaOx3 from A. fumigatus and FgaOx3pc from P. commune were successfully combined with the festuclavine synthase FgaFS from A. fumigatus and FgaFSpc from P. commune as well as with the agroclavine synthase EasG from C. purpurea. Interpretation of the ratio of 3 to 4 in enzyme assays revealed that the second reduction step catalysed by the imine reductase FgaFS, FgaFSpc or EasG determined the stereochemistry at C-8 of the products.Experimental section
Computer-assisted sequence analysis
Alignments of amino acid sequences were performed by using the programs “BLAST 2 SEQUENCES” (www.ncbi.nlm.nih.gov) and ClustalW2 (http://www.ebi.ac.uk/Tools/msa/clustalw2). The program GENEius (www.genius.de) was used for codon optimisation for expression of fgaFSpc in E. coli XL1 Blue MRF' cells.Chemicals
NAD+, FMN, NAPH and NADPH were obtained from Sigma-Aldrich (Munich, Germany) and Carl Roth (Karlsruhe, Germany). Chanoclavine-I was kindly provided by Prof. Leistner (Bonn, Germany).Bacterial strains, plasmids and cultural conditions
The expression vectors pQE60 and pQE70 were obtained from Qiagen (Hilden, Germany). Escherichia coli XL1 Blue MRF' (Stratagene, Amsterdam, The Netherlands) was used for cloning and expression experiments and grown in liquid or on solid Luria-Bertani or Terrific-Broth medium with 1.5% (w/v) agar at 22 °C, 30 °C or 37 °C.26 Carbenicillin (50 μg ml−1) was used for the selection of recombinant E. coli strains.Fungal cultivation and isolation of (8R,9S)-fumigaclavine A
P. commune NRRL2033 was cultivated at 25 °C in darkness on wheat medium for 15 days and extracted with ethyl acetate. The ergot alkaloids in the ethyl acetate phase were then converted to their protonated form by extraction with 0.01 N HCl. The pH value of the aqueous phase was adjusted to 8–9 with 25% ammonia and the ergot alkaloids were again extracted with ethyl acetate. After evaporation of the solvent, the residue was dissolved in 300 μl methanol and purified on HPLC described below.Chemical DNA synthesis, DNA isolation and cloning
Standard procedures for DNA isolation and manipulation were performed as described.26 PCR amplification was carried out on a MJ MiniCycler from BioRad (Munich, Germany). A PCR fragment of 1151 bp containing the entire coding sequence of fgaOx3pc, was amplified from the cosmid 28D7 from P. commune NRRL203321 by using the Expand High Fidelity Kit (Roche Diagnostics GmbH, Mannheim, Germany) and primers PrMM049-for (5′-TG![[G with combining low line]](https://www.rsc.org/images/entities/b_char_0047_0332.gif)
![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif)
![[A with combining low line]](https://www.rsc.org/images/entities/char_0041_0332.gif)
![[T with combining low line]](https://www.rsc.org/images/entities/char_0054_0332.gif)
![[G with combining low line]](https://www.rsc.org/images/entities/char_0047_0332.gif)
![[C with combining low line]](https://www.rsc.org/images/entities/b_char_0043_0332.gif) CAAAAACCACCCA-3′) and PrMM049-rev (5′-AC
CAAAAACCACCCA-3′) and PrMM049-rev (5′-AC![[A with combining low line]](https://www.rsc.org/images/entities/b_char_0041_0332.gif)
![[T with combining low line]](https://www.rsc.org/images/entities/b_char_0054_0332.gif) CGTTCCTCGAC-3′). Bold letters represent mutations inserted in comparison to the original genome sequence. The underlined letters indicated the restriction site SphI at the
CGTTCCTCGAC-3′). Bold letters represent mutations inserted in comparison to the original genome sequence. The underlined letters indicated the restriction site SphI at thesta
rt codon in PrMM049-for and the BamHI site at the predicted stop codon in PrMM049-rev. The PCR product was cloned into pGEM-T Easy resulting in plasmid pMM049, which was sequenced by MWG Biotech AG, Ebersberg, Germany. To create the expression construct pMM050, the plasmid pMM049 was digested with SphI and BamHI resulting in a fragment of 1139 bp, which was isolated on a preparative agarose gel and ligated into the SphI and BamHI sites of pQE70.
The gene fgaFSpc was synthesized chemically and cloned into pCR2.1 (MWG Biotech AG, Ebersberg, Germany), resulting in plasmid pMM047. To create the expression construct pMM048, pMM047 was digested with NcoI and BamHI. The resulted NcoI-BamHI fragments of 860 bp was isolated and subsequently ligated into pQE60, which had also been digested with NcoI and BamHI, previously.
Overproduction and purification of His6-FgaOx3pc and His6-FgaFSpc
fgaOx3 pc in pMM050 and fgaFSpc in pMM048 were expressed in E. coli XL1 Blue MRF'. The cells were cultivated in 2000 ml Erlenmeyer flasks containing 1000 ml of liquid Terrific-Broth medium supplemented with 50 μg ml−1 of carbenicillin at 30 °C and 22 °C, respectively, to an absorption at 600 nm of 0.6. Gene expression was induced by addition of IPTG to a final concentration of 0.1 mM. Both bacterial cultures were further cultivated for 15 h at 22 °C and then centrifuged to harvest the cells. The pellets were resuspended in lysis buffer (10 mM imidazole, 50 mM NaH2PO4, 300 mM NaCl, 2% Tween 20, pH 8.0) at 2 ml per gram wet weight. 1 mg ml−1 of lysozyme, 10 μg ml−1 of RNaseI and 5 μg ml−1 of DNaseI were added to the suspension for cell lysis. The mixtures were incubated on ice for 30 min and then 6 times sonicated for 10 s each at 200 W. For separation of the cellular debris from the soluble proteins, the lysate was centrifuged at 14000 x g for 30 min at 4 °C. For FgaOx3pc, a two-step purification of the recombinant His6-tagged fusion protein by affinity chromatography with Ni-NTA agarose resin (Qiagen, Hilden, Germany) and subsequently with HisPur Cobalt resin (Thermo Fisher Scientific, Rockford, USA) was carried out according to each manufacturer's instructions. In the case of FgaFSpc, better results were obtained by using HisPur Cobalt resin as first and Ni-NTA agarose as second purification step. Proteins were eluted with 250 mM imidazole in 50 mM NaH2PO4, 300 mM NaCl, pH 8.0. To change the buffer after the first purification step, the protein fractions were passed through a PD-10 column (GE Healthcare, Freiburg, Germany), which had been equilibrated with 50 mM Tris-HCl, pH 7.5, previously. After the second purification step, 50 mM Tris-HCl, 15% (v/v) of glycerol, pH 7.5 was used as buffer for storage of the frozen proteins at −80 °C.
Protein analysis and determination of the molecular mass of active His6-FgaOx3pc and His6-FgaFSpc
Proteins were analysed on SDS-PAGE according to the method of Laemmli27 and stained with Coomassie Brilliant Blue R-250. The molecular mass of the recombinant His6-FgaOx3pc and His6-FgaFSpc was determined on a HiLoad 16/60 Superdex 75 column (GE Health Care, Freiburg, Germany) by using 50 mM Tris-HCl (pH 7.5) containing 150 mM NaCl as elution buffer. The column was calibrated with blue dextran 2000 (2000 kDa), conalbumin (75 kDa), ovalbumin (43 kDa), ribonuclease A (13.7 kDa), aprotinin (6.5 kDa) (GE Healthcare, Freiburg, Germany) and chymotrypsinogen A (25 kDa) (Boehringer, Mannheim, Germany).HPLC analysis of enzyme assays
The enzyme assays contained 50 mM Tris-HCl, pH 7.5 and 1.2–9.0% (v/v) of glycerol. The reaction mixtures contained 5 mM of the cofactors NAD+, FMN, NADPH, 1 mM chanoclavine-I and 5 μg or 10 μg of the recombinant enzymes and were incubated at 30 °C for 15 h. After incubation, the pH value was adjusted to 9.0 with 1 M sodium hydroxide and extracted twice with two volumes of dichloromethane. The combined organic phase was evaporated to dryness and dissolved in 100 μl methanol and analysed on an Agilent HPLC Series 1200 by using a Multospher 120 RP18 column (250 × 4 mm, 5 μm, Chromatographie Service, Langerwehe, Germany) at a flow rate of 1 ml min−1. Water (solvent A) and acetonitrile (solvent B), each containing 0.5% (v/v) trifluoroacetic acid, were used as solvents. The assays were analysed with 21% B over 60 min. The column was then washed with 100% solvent B for 5 min and equilibrated with 21% solvent B for 5 min. The substances were detected with a Photodiode Array detector.Isolation of festuclavine and pyroclavine from an enzyme assay as well as of fumigaclavine A from fungal extracts
For isolation of 3 and 4, a reaction mixture of 20 ml contained 1 mM chanoclavine-I, 2 mg (1 μM) of purified FgaDH, 2 mg (2.4 μM) FgaOx3, 2 mg (3 μM) EasG, 5 mM NAD+, 5 mM FMN and 5 mM NADPH. After incubation for 4 h, the pH value of the reaction mixture was adjusted to pH 9.0 with 1 M sodium hydroxide and extracted twice with ethyl acetate. The combined organic phase was evaporated on a rotation evaporator at 30 °C to dryness and the remaining residue was dissolved in 800 μl methanol. The sample was separated on TLC silica gel 60 F254 plates (Merck, Darmstadt, Germany) by using a mixture of 70% (v/v) chloroform, 30% (v/v) methanol and 0.2% (v/v) ammonia as developing solvent.The festuclavine and pyroclavine spots were taken from the TLC plates by a scalpel and extracted with methanol. The methanol extracts were evaporated on a rotation evaporator at 30 °C to dryness and the remaining residue was dissolved in 300 μl methanol and injected onto an Agilent HPLC Series 1200 with a Multospher 120 RP18 column (250 × 10 mm, 5 μm, Chromatographie Service) at a flow rate of 2.5 ml min−1. For separation, a gradient from 25 to 42% B over 28 min and a washing phase with 100% solvent B for 5 min was used. The column was equilibrated with 38% solvent B for 5 min. The collected fractions containing the enzyme products after HPLC purification were evaporated to dryness and subjected to MS and NMR analyses.
For isolation of fumigaclavine A from the fungal extract mentioned above, the same column and flow rate as for the isolation of 3 and 4 were used. For separation, a gradient from 30 to 100% B over 25 min and a washing phase with 100% solvent B for 5 min was used. The column was equilibrated with 30% solvent B for 5 min.
HR-EI-MS and NMR analyses of the enzyme products
The positive high resolution electron impact mass spectrometry (HR-EI-MS) was carried out with an AutoSpec instrument (Micromass Co. UK Ltd).NMR spectra were recorded at room temperature on a Bruker Avance 600 MHz spectrometer equipped with an inverse probe with z gradient. The DQF-COSY and HSQC spectra were recorded with standard methods.28 Gradient-selected NOESY experiment was performed in phase-sensitive mode.29 All spectra were processed with Bruker TOPSPIN 2.1.
Nucleotide sequence accession numbers
The genomic sequences of fgaFSpc and fgaOx3pc from P. commune NRRL2033 were deposited at GenBank under the accession number JQ085282 and JQ085283, respectively.Acknowledgements
This work was supported by a grant from the Deutsche Forschungsgemeinschaft (Li844/3-1 to S.-M. Li). We thank Inge Unsöld for cosmid library construction and Prof. E. Leistner for providing chanoclavine-I.References
- M. Flieger, M. Wurst and R. Shelby, Folia Microbiol., 1997, 42, 3–30 CrossRef CAS.
- C. L. Schardl, D. G. Panaccione and P. Tudzynski, Alkaloids: Chem. Biol., 2006, 63, 45–86 CrossRef CAS.
- C. Wallwey and S.-M. Li, Nat. Prod. Rep., 2011, 28, 496–510 RSC.
- D. Gröger and H. G. Floss, Alkaloids: Chem. Biol., 1998, 50, 171–218 CrossRef.
- H. G. Floss, Tetrahedron, 1976, 32, 873–912 CrossRef CAS.
- P. Tudzynski, K. Holter, T. Correia, C. Arntz, N. Grammel and U. Keller, Mol. Gen. Genet., 1999, 261, 133–141 CrossRef CAS.
- S.-M. Li and I. A. Unsöld, Planta Med., 2006, 72, 1117–1120 CrossRef CAS.
- I. A. Unsöld and S.-M. Li, Microbiology, 2005, 151, 1499–1505 CrossRef.
- M. Matuschek, C. Wallwey, X. Xie and S.-M. Li, Org. Biomol. Chem., 2011, 9, 4328–4335 CAS.
- K. E. Goetz, C. M. Coyle, J. Z. Cheng, S. E. O'Connor and D. G. Panaccione, Curr. Genet., 2011, 57, 201–211 CrossRef CAS.
- C. M. Coyle, J. Z. Cheng, S. E. O'Connor and D. G. Panaccione, Appl. Environ. Microbiol., 2010, 76, 3898–3903 CrossRef CAS.
- C. Wallwey, M. Matuschek, X.-L. Xie and S.-M. Li, Org. Biomol. Chem., 2010, 8, 3500–3508 CAS.
- D. G. Panaccione and C. M. Coyle, Appl. Environ. Microbiol., 2005, 71, 3106–3111 CrossRef CAS.
- I. A. Unsöld and S.-M. Li, ChemBioChem, 2006, 7, 158–164 CrossRef.
- N. G. Vinokurova, S. M. Ozerskaya, B. P. Baskunov and M. U. Arinbasarov, Microbiology (Moscow), 2003, 72, 149–151 CAS.
- S. Ohmomo, M. Kaneko and P. Atthasampunna, MIRCEN J. Appl. Microbiol. Biotechnol., 1989, 5, 5–13 CrossRef CAS.
- H. M. Ge, Z. G. Yu, J. Zhang, J. H. Wu and R. X. Tan, J. Nat. Prod., 2009, 72, 753–755 CrossRef CAS.
- K. Kawai, K. Nozawa, T. Yamaguchi, S. Nakajima and S. Udagawa, Mycotoxins, 1992, 36, 19–24 CAS.
- B. Blaney, S. Chakraborty and S. A. Murray, Aust. J. Agric. Res., 2006, 57, 1023–1028 CrossRef CAS.
- R. J. Cole, J. W. Kirksey, J. W. Dorner, D. M. Wilson, J. C. Johnson, Jr., A. N. Johnson, D. M. Bedell, J. P. Springer, K. K. Chexal, J. C. Clardy and R. H. Cox, J. Agric. Food Chem., 1977, 25, 826–830 CrossRef CAS.
- I. A. Unsöld, Dissertation Universität Tübingen, 2006 Search PubMed.
- C. Wallwey, M. Matuschek and S.-M. Li, Arch. Microbiol., 2010, 192, 127–134 CrossRef CAS.
- X. Xie, C. Wallwey, M. Matuschek, K. Steinbach and S. M. Li, Magn. Reson. Chem., 2011, 49, 678–681 CrossRef CAS.
- J. Z. Cheng, C. M. Coyle, D. G. Panaccione and S. E. O'Connor, J. Am. Chem. Soc., 2010, 132, 12835–12837 CrossRef CAS.
- X. Liu, L. Wang, N. Steffan, W.-B. Yin and S.-M. Li, ChemBioChem, 2009, 10, 2325–2328 CrossRef CAS.
- J. Sambrook and D. W. Russell, Molecular Cloning: a Laboratory Manual, Cold Spring Harbor Laboratory Press, New York, 2001 Search PubMed.
- U. K. Laemmli, Nature, 1970, 227, 680–685 CrossRef CAS.
- S. Berger and S. Braun, 200 and More NMR Experiments. A Practical Course, Wiley-VCH, Weinheim, Germany, 2004 Search PubMed.
- T.-L. Hwang and A. J. Shaka, J. Am. Chem. Soc., 1992, 114, 3157–3159 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2ra20104f |
| This journal is © The Royal Society of Chemistry 2012 |