Antimicrobial macromolecules: synthesis methods and future applications
Peng
Li†
,
Xiang
Li†
,
Rathi
Saravanan
,
Chang Ming
Li
and
Susanna Su Jan
Leong
*
School of Chemical and Biomedical Engineering, 62 Nanyang Drive, Nanyang Technological University, 637459, Singapore. E-mail: SLeong@ntu.edu.sg; Fax: +65 6794 7553; Tel: +65 6316 8775
First published on 31st January 2012
Abstract
This review article presents important and recent progress in the manufacture and application of antimicrobial macromolecules. Microbial infections continue to endanger human health and pose a great economic burden to society. To resolve this crisis, huge efforts to improve or develop macromolecules that can inhibit pathogens without incurring pathogen resistance are required and actively ongoing. Synthetic antimicrobial macromolecules which include antimicrobial peptides (AMPs), polymers and peptide–polymer hybrids represent a huge class of molecules which can incur effective antimicrobial therapy due to their unique biochemical properties. The use of these antimicrobial macromolecules which target the cytoplasmic membrane of microbes, is a promising approach to lower the propensity of pathogen resistance development. Therefore, huge efforts to synthesize these molecules at scales and purities that enable their structure-function and clinical studies are actively underway. Due to the high cost involved in extracting AMPs from natural sources, biological processes are being developed to economically manufacture AMPs at large scale. Synthetic AMP analogs are also being engineered to further improve antimicrobial potency and lower synthesis cost. Synthetic polymers have also been found to exhibit excellent antimicrobial properties which are comparable to those of natural AMPs. Various antimicrobial polymers have been synthesized based on the amphiphilicity of natural AMPs. Although the facile synthesis of polymers poses no cost problems, numerous synthetic antimicrobial polymers are disadvantaged by high toxicity to mammalian cells due to their non-selectivity. To combine the advantages of AMPs and antimicrobial polymers, peptide-polymer hybrid macromolecules are actively being developed, with a few effective and strongly microbicidal models recently demonstrated. With the advancement of biochemical engineering tools and chemical synthesis methods, these antimicrobial macromolecules can be specifically designed to be highly selective, broad spectrum and biocompatible. In this review, we summarize the recent advances and challenges in the manufacture of these antimicrobial macromolecules. Based on their antimicrobial mechanisms, their applications in addressing challenges associated with infectious disease and antibiotic-resistance are also discussed.
1 Introduction
Prolonged antibiotic misuse in human therapeutics, agriculture and veterinary has provided selective pressure favouring the survival and spread of antibiotic resistant microbes. A rapid rise in drug resistant pathogens is attributed to the accumulation of resistance genes on plasmids that are independently replicated and passed between microbial cells and species. Such multi-drug resistant (MDR) microbes are termed as “superbugs”, where they exhibit a plethora of antibiotic resistant mechanisms to strike back and nullify antibiotic actions including reduced drug uptake, active pumping of drugs extracellularly, enzymatic alteration of antibiotics, modification or overproduction of targets, drug sequestering by protein binding, and metabolic bypass of the targeted pathway.1 Almost 70% of current day infections are suspected to be due to drug resistance,2 where bacterial infections now ranks as the 4th leading cause of death in the U.S alone. Over 50% of hospital bloodstream infections are incurred by methicillin resistant Staphylococcus aureus (MRSA) and vancomycin resistant enterococci (VRE). Among the Escherichia coli (E. coli) isolates from China and India, less than 50% remain susceptible to commonly used antibiotics (e.g., Cephalosporins and Ciprofloxacin).3 Despite the alarming rise in MDR pathogens and the severity of their infections, many major pharmaceutical companies have had to abandon antibiotic discovery research due to high regulatory barriers and a low chance of clinical success, leaving only a small number of companies to meet this challenge.4,5 Moreover, most antibiotic compounds in the pipeline are still chemical derivatives of existing antibiotics with known resistance.4 Collectively, the drug resistance problem and the declining number of drugs approved by the US Food and Drug Administration (FDA) in recent years reflect an unresolved crisis for the post-antibiotic era. This review presents alternative antimicrobial molecules including antimicrobial peptides (AMPs), polymers and AMP–polymer hybrids that show promise in alleviating the pathogen drug resistance problem. The state-of-the-art synthesis methods and engineering challenges of these macromolecules are reviewed, and their applications in addressing microbial resistance are discussed.2 Antimicrobial peptides and their chemical and biological production methods
AMPs are a promising class of molecules which have shown to be able to reduce infections without easily incurring pathogen resistance. Cationic AMPs have broad spectrum antimicrobial activity, even against highly resistant bacteria like MDR Pseudomonas aeruginosa and MRSA.6 Besides AMPs' robust antimicrobial activity, they play prominent roles as key effector molecules in innate immune responses such as inflammation, immune activation, host immune homeostasis and wound healing, and hence are aptly known as host defence peptides (HDPs).7–10 AMP-based therapeutics have several advantages which render them superior alternatives to conventional antibiotics in the era of MDR pathogens. The diverse therapeutic potentials of AMPs include their (i) role as natural host defense alternates with immunomodulatory properties, (ii) broad spectrum antimicrobial activity against MDR pathogens, (iii) non-specific mechanism of killing which reduces the emergence of resistance, (iv) potential usage in “combination therapy” with existing drugs for synergistic effect, and (v) potent lipopolysaccharides (LPS) binding and neutralizing property which is advantageous for anti-sepsis drugs.11,12 The therapeutic implications of AMPs could be largely grouped into three main areas: (i) anti-infectives based on their antimicrobial activity; (ii) anti-inflammatory agents, and (iii) adjuvants due to their immunomodulatory properties. The diverse and promising therapeutic potentials of AMPs have provided a large arena for the development of AMP-based anti-infective drugs. Past and ongoing studies clearly demonstrate the plausible application of an assortment of AMPs in treating several infectious diseases (Table 1).| Activity | Peptide | Structure | Source | Pathogen | Reference |
|---|---|---|---|---|---|
| Antibacterial | Cecropin | α-helix | Silk moth | G +ve, G −ve | 13 |
| Magainin | α-helix | Frog | G +ve, G −ve | 14 | |
| Protegrin | β-sheet | Human, Porcine | G +ve, G −ve | 15 | |
| Bactenicin | Cyclic | Bovine neutrophils | G +ve, G −ve | 6 | |
| PR-39 | Extended | Porcine | G +ve, G −ve | 16 | |
| Antiviral | Melittin | α-helix | Honey bee | HSV | 17 |
| Cecropin | α-helix | Insect | HSV, HIV | 18 | |
| Dermaseptin | β-sheet | Frog | HSV, HIV | 19, 20 | |
| Lactoferrin | β-turn | Bovine, Human | HSV, HIV, Papilloma | 21 | |
| Defensin | β-sheet | Human, Rabbit | Adeno, HIV, HSV, HCMV | 21 | |
| Antiparasitic | Magainin2 | α-helix | Frog | P. caudatum | 22 |
| PMAP-23 | α-helix | Porcine | C. elegans | 23 | |
| Defensin | β-sheet | Mussel | L. major | 21 | |
| Antifungal | Melittin | α-helix | Honey bee | C. albicans | 24 |
| Indolicidin | Extended | Bovine | T. beigelii | 25 | |
| Cecropin | α-helix | Insect | A. fumigatus | 26 |
In the past 20 years, hundreds of AMPs have been identified, but further studies to confirm their roles by detailed understanding of the peptide's structure–function have been slow due to the difficulty in obtaining sufficient amounts of pure AMPs.27 AMP production by isolation and purification from natural sources such as epithelial or plants cells is an extremely tedious task, giving low yields and is prohibitively expensive to scale up.28–30 To address this problem, chemical synthesis and biological production methods have been employed to synthesize AMPs at scales and purity values that allows their direct use for characterization or structure–function studies. Advancements in these techniques will be critical to extend the use of AMPs to counter infections. Recent technical progress made in both types of synthesis methods is reviewed in the following sections.
2.1 AMP production by chemical synthesis
Solid phase chemical synthesis, invented by Robert Bruce Merrifield,31 is a mature technique widely employed for peptide production including AMPs at small production scales.32 Compared to biological recombinant methods, peptide production by chemical synthesis has two significant advantages; first, artificial modifications of peptides such as the incorporation of unnatural amino acids, peptide backbone modification, amidation and acetylation can be readily achieved. Second, chemically-synthesized peptides do not carry over cellular contaminants such as LPS, proteins or DNA fragments which may be difficult to purify, leading to biocompatibility problems. The disadvantage of chemical synthesis is the tedious protonation and deprotonation steps necessitated in chain synthesis, low efficiency in cysteine oxidation and therefore the need for cysteine-rich peptides with conformations constrained by disulfide bonds to be subsequently refolded in vitro.Chemical synthesis methods have facilitated the engineering of AMPs for enhanced properties. For example, the susceptibility of AMPs to protease in inflammation areas has been addressed by introducing D-amino acids to natural AMPs through chemical synthesis to render the peptides unrecognizable by these proteases.33,34 D-amino acid substitution has also been reported to enhance both the antimicrobial activity and selectivity of AMPs.35–37 The systematic study of D-amino acid substitution on AMP secondary structure, stability and activity showed that partial D-amino acid substitution can improve the activity of AMPs due to the difference in membrane interaction between natural and D-amino acid substituted AMPs.35,38 Other studies reported that AMPs comprising of D-amino acids alone are also active against HIV-1 and possess in vivo activity against plant pathogenic bacteria, which expands the applications of AMPs.39,40 Amidation and acetylation by chemical synthesis have also been reported to modify natural AMPs for bioactivity evaluation. The role of acetylation and charge in human beta defensin 3 (hBD3) was recently reported, where acetylation of the C-terminus portion of hBD3 led researchers to conclude that peptide net charge is a major factor in antimicrobial activity determination.41 The Dennison group also reported the effect of C-terminal amidation on the efficacy and selectivity of antimicrobial and anticancer peptides,42 where amidation was found to stabilize helix formation upon binding to the E. coli membrane, thus generating higher local concentrations of the peptide at the bilayer interface, and hence greater levels of membrane disruption.43 Other chemical modification approaches like backbone modification by cyclization, and the synthesis of multivalent AMPs by linking two or more monomers for activity enhancement can also be readily achieved by chemical synthesis.44,45
The use of chemical synthesis for peptide production is most suitable for peptides that are active in the linear and unfolded form. Should the peptides contain disulfide bonds, incubation in a refolding–enhancing environment would be necessary to promote correct oxidation and pairing of the cysteines. Some common obstacles that are often encountered in AMP folding include (i) low solubility of AMPs which results in peptide precipitation in the folding buffer, and (ii) the formation of incorrect disulfide connectivity which can significantly reduce peptide yield. Although some studies have shown that disulfide connectivity does not affect the in vitro antimicrobial activity for many AMPs,46,47 there may be other undesirable impacts on the peptide's other functions which cannot be directly determined. To overcome these problems, factors like peptide concentration, buffer polarity, ionic strength, pH and the ratio of oxidative and reducing agents which can influence peptide folding must be properly optimized to maximise yields.
2.2 Biological production of AMPs
Compared with chemical synthesis methods, bio-production of AMPs in microbial cell factories can be more cost-effective, requires a simpler process and is more environmentally friendly.48 Biological production of AMPs eliminates the need for the multiple protection and de-protection steps which increases the process complexity and solvent usage in the chemical synthesis route. By employing readily available genetic engineering techniques, many AMPs have been successfully produced through recombinant expression in different types of microbial hosts.49 Both bacteria and yeast account for over 95% of reported studies on biological production of AMPs. Due to the well-characterised genome, easy availability of cloning vectors and cheap medium for cell growth, E. coli is a common microbial platform for AMP production.30,49,50 Several important considerations in developing an AMP production platform in the bacteria expression system are:27,51,52 (i) AMP toxicity to the host cell post-expression, (ii) intracellular degradation by proteolytic enzymes, as well as (iii) peptide purity, integrity and bioactivity.35,52,53 Sections 2.2.1 to 2.2.2 review bioprocess considerations for the biological production of AMPs.There is no universal fusion tag that can effectively resolve solubility, yield or purification issues for all peptides due to the different advantages and disadvantages of each tag which influences the expression and purification of different proteins to varying extents. Therefore, the choice of fusion tags must be optimized with respect to individual peptide properties.
| Protease | Molecular weight (kDa) | Recognition site | Suggested cleavage buffer | Expression host | AMP candidates |
|---|---|---|---|---|---|
| Enterokinase | ∼26 | DDDK↓ | 50 mM Tris; pH 7.2; 50 mM NaCl; 2.5 Mm CaCl2 | E. coli | hBD4,26,27; Dermaseptin B2; Cecropin |
| Factor Xa | ∼43 | LVPR↓GS | 50 mM Tris; pH 7.5; 150 mM NaCl; 1 mM CaCl2 | Plasma | Carnocin CP52; Pediocin ACH; Brevinin-2GU |
| HEK 293 cells | |||||
| Thrombin | ∼37 | LVPR↓GS | 50 mM Tris; pH 7.5; 150 mM NaCl; 2.5 mM CaCl2 | Plasma | Cecropin; LF cin B; LL-37 variant |
| CHO cells | |||||
| TEVp | ∼27 | ENLYFQ↓G | 50 mM Tris; pH 8; 5 mM DTT | E. coli | hBD25,28; Hepcidin |
Thorough understanding of the characteristics of different proteins in the post-cleavage mixture is important to rationally design AMP purification methods. Chromatography is commonly reported to recover AMPs after the cleavage step (Table 3). Besides chromatography methods, the use of other methods such as solvent-, acid-, and heat-mediated precipitation has also been effective.39,72 For example, the difference in properties of the fusion protein components in MBP-hBD28 allowed the use of hydrochloric acid to purify hBD28 from the other cleavage components, where by optimizing the addition of acid, the MBP fusion tag and cleavage protease were precipitated, leaving the hBD28 peptide in the soluble form.
| Chromatography purification method | Advantages | Disadvantages | Critical parameters for optimisation | AMP candidate |
|---|---|---|---|---|
| Ion exchange | High purity, concentrated product, high loading capacity. | High salt concentration environment is needed (i.e. AMP must be salt-tolerant) | Ion exchange column; salt concentration in elution buffer; | Perinerin; hBD4,5,26,27 |
| Affinity | High purity; time saving | Low loading capacity | Loading speed, quantity. | Plectasin; Scygonadin |
| Size exclusion | High purity; integration of purification and buffer exchange | Low loading capacity; diluted product; low productivity | Loading quantity, buffer composition; elution speed. | hBD25; Cathelicidin LL-37 |
2.3 Engineering AMPs for improved properties
Natural AMPs have limitations such as (i) susceptibility to salt, protease, and serum, (ii) cytotoxicity to mammalian cells, and (iii) high production costs, which pose roadblocks to their wide usage in clinical applications as anti-infective agents. These problems could be addressed by engineering AMPs through rational methods to overcome these limitations and enhance AMP activity. AMP engineering strategies which have been recently reported include developing (i) peptide analogs and hybrids through rational amino acid substitution, the deletion of inactive domains or combination of a few active regions from different natural AMPs for enhanced antimicrobial characteristics; (ii) synthetic AMP mimics with desirable characteristics of natural AMPs; (iii) combinatorial libraries to screen peptide structure–activity relationships for improved understanding;73 (iv) homology models with structurally characterised AMP templates.74 hBDs, for example, are a class of AMPs which have been extensively engineered for improved biological functions. The tertiary structure of hBDs is constrained by three intramolecular disulfide bonds. Reducing the disulfide bonds of hBD1 increased its antimicrobial activity against several pathogens.75 Disulfide linkage in hBD3 appeared to be important for some aspects of the peptide's biological activity; changing disulfide patterning, substitution of the cysteines with other amino acids or complete reduction of the disulfide bonds eliminated the chemotactic cytotoxcity of hBD3 but did not compromise the peptide's in vitro antimicrobial activity.76–80 Structure–function correlation studies of hBD3 analogs showed that the N-terminus of the peptide is not necessary for antimicrobial activity while the C-terminus (R36-K45), which is rich in positively charged amino acids, has higher antimicrobial activity and lower cytotoxicity toward zwitterionic mammalian cells than wild type hBD3.81 Comparative structure–activity studies of different hBD1 and hBD3 analogs showed that the internal (or middle) region of hBD1 and the C-terminus of hBD3 are responsible for antimicrobial activity retention at high salt concentration, which is a unique characteristic of hBD3 not found in other wild type hBD variants.82 Engineering of hBD28 at the C-terminus to enhance positive charge accumulation resulted in improved salt-tolerance and antimicrobial activity both in the presence and absence of salt. The engineering of other AMPs for improved function is summarised in Table 4.| Modification method | AMP candidate | Reference |
|---|---|---|
| Positive charge enhancement by amino acid substitution or addition | Chicken avian beta-defensin-8; hexapeptide PAF26; Gratisin | 95–97 |
| Cysteine substitution | hBD1 ; hBD3; Thanatin | 98–100 |
| Truncation to retain short and active domains | BMAP-27; Pleurocidin; Melittin; Defb14-derived peptides; hBD3 C-terminus | 101-105 |
| Combination of active domains from different AMPs (Chimeric AMPs) | hBD2 and hBD3; Lactoferricin and Lactoferrampin; | 106, 107 |
| D-amino acid substitution | Chicken cathelicidin-2; BP100; | 34,108 |
| Cyclization | Chicken cathelicidin-2; Histatin | 34, 109–111 |
In designing AMP analogs, a good understanding of the effect of engineering on the complete biological function of the AMPs is critical. Since many AMPs have multiple modes of antimicrobial activity exhibition and host immune system modulation, any changes in the sequence of natural AMPs may cause significant changes in their physiochemical properties which would subsequently affect their biological function or cause other side effects.
2.4 AMP-based drugs: challenges and current status of clinical development
Owing to the challenge posed by the rapid spread of resistant bacteria, AMP-based drug development has intensified in the last 25 years, which is reflected by the number of AMP candidates advancing into clinical trials (Table 5).83 Although many of these products demonstrated high efficacy in preclinical trials, they had to be abandoned at various stages of clinical trials owing to failure in meeting stringent requirements by regulatory agencies. These products include (i) Pexiganan (MSI-78) (Magainin Pharmaceutical Inc), a 22 amino acid synthetic analog based on magainin2 for the treatment of diabetic foot ulcers; (ii) Omiganan (MBI-226) (Microbiologix Biotech) a synthetic analog of indolicidin, for prevention of catheter linked infections; (iii) Iseganan (IB-367, Intrabiotics Pharmaceuticals, Inc) a synthetic derivative of protegrin1 from pig leucocytes developed as a local mouthwash for oral mucositis treatment and the only systemic application designed Neuprex (rBPI21), and (iv) N-terminus portion of bactericidal permeability increasing protein (BPI), with potent endotoxin neutralising ability.Despite these high profile failures, AMPs continue to be the “mother lode” of templates for the design of new anti-infectives. Table 6 lists some examples of AMP development programs in the last 2–3 years.84,85 Some major challenges in the successful development of AMP-based drugs are toxicity, in vivo efficacy, stability and cost of production. Active research is underway to address these challenges by various strategies including the incorporation of unusual amino acids,86 C-terminus modifications,87 the addition of acyl chains for lipopeptides,88 branched peptides with AMP “arms”,89 narrow spectra pH activated peptides,90 shorter specifically targeted AMPs,91 use of non-peptide backbones as peptidomimetics92,93 and release from liposomes.94
3 Synthetic antimicrobial oligomers and macromolecules: current design and production strategies
Although natural AMPs are highly effective and generally have low toxicity, there are several shortcomings associated with AMPs which limit their effective usage. Firstly, natural AMPs are often present at low concentrations, especially when extracted from natural sources. Production by chemical synthesis or recombinant technology typically produces AMPs at the milligram scale, which renders them expensive to manufacture at larger scales.112 Secondly, natural AMPs are also susceptible to proteolysis and are salt-sensitive, making them rather unstable when used in a physiological environment.112 To overcome these limitations, efforts have been undertaken to develop synthetic antimicrobial materials that are just as effective at targeting microbes but without the disadvantages associated with natural AMPs.113,114 Synthetic antimicrobial materials have been designed to mimic AMP properties (i.e. having both cationicity and hydrophobicity), where these properties can be easily modulated by changing the material's functionalization chemistry.115 Recent progress in the design and production strategies of synthetic antimicrobial oligomers, macromolecules and hybrid materials are presented and discussed in the following sections.3.1 Synthetic AMP mimetic foldamers/oligomers with well-defined secondary structures or conformations
To closely mimic natural AMPs, oligomers with short specific sequences which fold into well-defined secondary structures or conformations (so-called foldamers), have been synthesized using various monomers and methods.116,117 Such oligomers are sequentially synthesized by coupling individual monomeric units, where these oligomers are generally more stable and less expensive to produce than natural AMPs.118 In the design of synthetic AMPs, the choices of cationic and hydrophobic amino acids are constrained by the total number of amino acids available, while the use of oligomers allows a broader choice of backbone and functional groups. The structures of oligomers are also easier to modify for optimisation and modulation of antimicrobial activity and toxicity.119In earlier studies, efforts were mainly directed towards designing the oligomers to resemble the α-helix structure of natural AMPs. Oligomers such as oligoamides, oligoureas, and N-alkyl-glycines (peptoids) have been found to be able to fold into well-defined and controlled α-helix secondary structures (Fig. 1a).120–126 It has been generally accepted that such foldamers could be adopted as scaffolds to distribute cationic and hydrophobic groups in a predictable manner, to mimic the amphiphilic structure of host-defense peptides. Eight-residue enantiopure N, N'-linked oligoureas synthesized by fluorenylmethyloxycarbonyl (Fmoc) chemistry displayed mimetic α-helical structures as determined by CD spectroscopy.124 These oligourea foldamers showed broad-spectrum activity against both Gram-negative and Gram-positive bacteria, and showed higher HC50 values (concentration that yields 50% hemolysis of red blood cells) than MICs.124 Another class of α-helical macromolecules is the poly-N-substituted glycine peptoid which has a conformation that is controlled by its backbone chirality and hydrogen bonding.125 These ‘solid phase chemistry'-synthesized peptoids showed broad-spectrum antimicrobial activity against six clinically relevant pathogens, and MICs which were comparable with natural AMPs like pexiganan. They exhibited an AMP-like membrane disrupting mechanism, which was characterized by X-ray reflectivity measurements, and were also found to be stable against proteases due to the lack of peptide bonds.
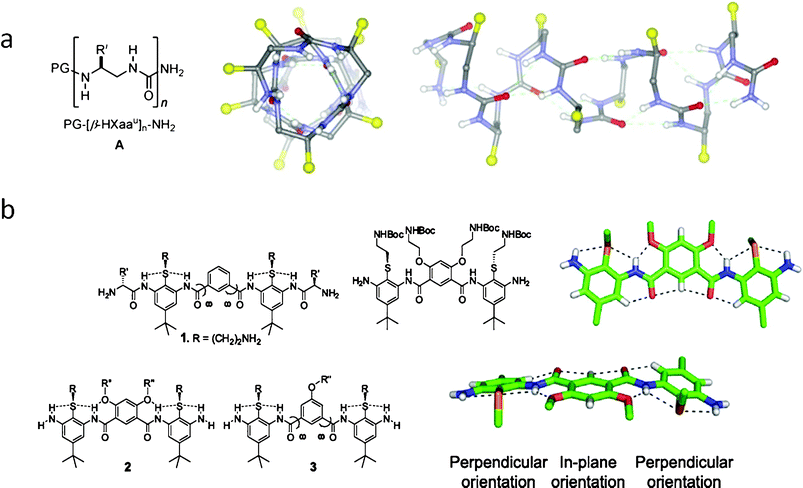 | ||
| Fig. 1 (a) Designed oligoureas fold into stable α-helix by H–bonded rings;124 (b) Non-helical, conformationally restrained, and facially amphiphilic antimicrobial aromatic acrylamide oligomers.127 | ||
Although early studies of synthetic AMP mimetic oligomers were initially focused on investigating the relationship between the folded α-helix structures and antimicrobial activity, the outcome of those studies showed that the well folded α-helical structure did not always correlate with antimicrobial activity, suggesting that the formation of an α-helical derived distinct amphiphilic face was not a pre-requisite for antimicrobial activity. In a recent study on α-helix oligoamide foldamers, the best helical folding isomer showed poor antimicrobial potency coupled with high haemolytic activity, while the scrambled isomer which did not fold well, showed lower MICs and haemolytic activity.123 Following the conclusion that there was little direct correlation between secondary structure and antimicrobial activity in synthetic oligomers, efforts were then gradually directed towards designing oligomers with amphipathic property, rather than α-helicity. DeGrado and coworkers studied the design of much simpler aromatic acrylamide oligomers which did not have a helix fold but contained facial amphipathic segments (Fig. 1b).118,127–129 Tew and co-workers also developed numerous antimicrobial oligomers by aromatic acrylurea and phenylene ethynylenes.117,130–136 The rigid aromatic backbone of these oligomers restrains the molecule's conformation, where hydrogen bonding between the inner molecule groups further stabilizes their facial amphipathic conformation. Many such non-helical oligomers have demonstrated low MICs.127,135 Recent results strongly suggest amphiphilicity rather than helicity to be the pre-requisite for antimicrobial activity of these oligomers.117,118,137
3.2 Synthetic AMP mimetic polymers without well-defined secondary structures
Compared to the synthetic peptides and oligomers discussed in Section 2.1 and 3.1, polymers are generally easier to design and synthesize. The sequence or structure of peptides or oligomers requires rigorous design, where the step-by-step synthesis method can be extremely tedious, even at small scale.138 Polymers, on the other hand, can be synthesized with significantly fewer reaction steps, and hence can be produced on the large scale relatively easily and cheaply. Antimicrobial polymers such as polynorbornenes,139 polymethacrylates,140 polypryidines,141 polyvinylether,142etc are designed to mimic the cationic and amphiphilic structure of natural AMPs (Table 7). An amphiphilic polymer is commonly designed either to be copolymerized by cationic and hydrophobic monomers or homopolymerized by amphiphilic monomers. Regardless of the monomer structure, cationic groups (such as protonated amine groups) and hydrophobic groups (such as alkyl chain) are usually present on the polymer chain, to confer the polymers cationic and hydrophobic properties, which are crucial for antimicrobial activity demonstration.143–149| Chemical structure | Name/description | MICa(μg ml−1) | HC50 (μg ml−1) | Selectivity | ||
|---|---|---|---|---|---|---|
| a (E): E. coli; (S): S. aureus. ND: not detected. | ||||||
| Homopolymer |
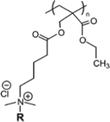
|
Polymethacrylate with pendant quaternary ammonium groups150 | 64 (E) | ND | ND | |
| 128 (S) | ||||||
(R![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C12H25) C12H25) |
||||||
|
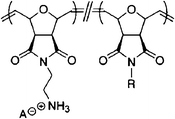
Homopolymer of facial amphiphilic oxanorbornene derived monomers139 | <3.8 (S) | >2000 | >533 | ||
|
Amphiphilic polynorbornene derivatives143 | 40 (E) | >4000 | >100 | ||
| 40 (S) | ||||||
| Copolymer | Random copolymer |
|
Random coplynorbornenes155 | 75 (S) | 1500 | 20 |
|
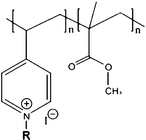
Random copolypyridinium methacrylate B4146 | 15 (E) | 0.23 | 0.02 | ||
|
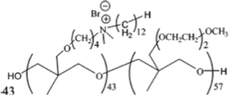
Poly[(3,3-quaternary/PEG)-copolyoxetanes]161 | 6 (E) | >60 | >10 | ||
| 5 (S) | ||||||
| Block copolymer |
|
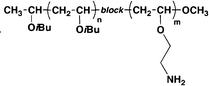
Poly(vinyl ether)142 | 62.5 (E) | >1000 | >16 | |
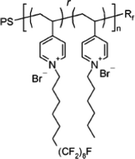
|
Fluorinated pyridinium block copolymers167 | almost 100% bactericidal effect of its coating | ND | ND | ||
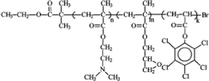
|
Self-quaternized P(DMAEMA-c-GMA)-b-PPCPA165 | >99% kill after 1 h contact with bacteria (E) of its nanofiber | ND | ND | ||
Sen and co-workers have developed three series copolymers of pyridinium and methacrylate.146 These pyridinium–methacrylate copolymers were synthesized by free radical polymerization followed by quarternization of the pyridine fragments, where the pendant alkyl tail was introduced into the polymer chain either on the quaternized pyridine site or the methacrylate site. It was reported that placing the cationic charge and hydrophobic alkyl tail on spatially separated centers amplified mammalian cytotoxicity, where the spatially separated hydrophobic alkyl tails interacted with RBCs more strongly. If the cationic charge and hydrophobic alkyl chain were placed on the same center, improved selectivity was observed despite lowered MIC values. The polymer with the highest selectivity in these series contained a four-carbon alkyl side chain and demonstrated a MIC of 30 μg ml−1 and HC50 of 1709 μg ml−1, leading to an HC50/MIC of 56.146 Gellman and co-workers also developed antimicrobial random copolymer based on nylon-3 derivatives.148,162 The copolymer with 60% lactam units was reported to show reduced hemolytic activity with increasing fraction of cationic monomers, leading to impressive selectivities above 200.148 Systematic studies on copolymers of cationic and hydrophobic methacrylate derivatives were also recently reported by Kuroda and co-workers.140,147,149,157,168 The amphiphilic polymethacrylate copolymers showed good antimicrobial activity (i.e. MIC of 16 μg ml−1 (E. coli)), but possessed HC50 values that were lower than MIC.140 The selectivity bottleneck was addressed by the systematic variation of the composition of the copolymers to fine-tune the cationic and hydrophobic balance, where the MIC values further decreased to 8 μg ml−1 for E. coli while the HC50 increase to 100 μg ml−1.147
Block copolymers need to be synthesized by living polymerizations which require more rigorous conditions, thus not many studies were reported for their antimicrobial applications.142,164,165,167,169 In an early study of block and random copolymers of styrene and quaternized pyridine, block copolymers (prepared as particles) were found to be more potent than random copolymers for both P. aeruginosa and S. aureus, although their hemolytic activities were unreported in this work.169 It was ascribed that preparation of the particles resulted in the concentration of positive charge of the pyridine block at the interface facing the hydrophilic solvent, which induced a higher surface charge to kill bacteria more effectively.169 In a more recent study, block and random polyvinylether copolymers were characterized with respect to both their antimicrobial and hemolytic activity.142 Block polyvinylether copolymers did not show improved antimicrobial activity compared to random ones, where the MIC of the block copolymer was higher than random ones at several copolymerization ratios. Interestingly, the block copolymer showed lower cytotoxicity behaviour than random ones, due to the conformational difference between the two copolymers. Random copolymers have a random coil structure, while block copolymers can fold into a hydrophobic/hydrophilic core/shell structure, where the hydrophobic segment of the block copolymer is shielded by the hydrophilic shell when making contact with the mammalian cell membrane.142 It has been demonstrated that the global hydrophobicity of these molecules is the dominant factor for hemolytic activity,115,147 thus the core/shell structure of block copolymer induced less hemolysis of RBCs.
3.3 Hybrid antimicrobial materials
Hybrid antimicrobial materials are compounds which contain two or more functional fragments combined. This synergistic merging of different functions is aimed at promoting desired properties such as biocompatibility, enhanced antimicrobial activity, and ease of applications etc.170 Antimicrobial macromolecules which are hybrids of different functional segments are setting new trends in the development of efficient, multi-functional and easily usable materials.171–175 Several classes of hybrid antimicrobial materials which have been reported to be effective in antimicrobial applications are presented in Fig. 2, and their design strategy and synergistic effects are discussed in this section.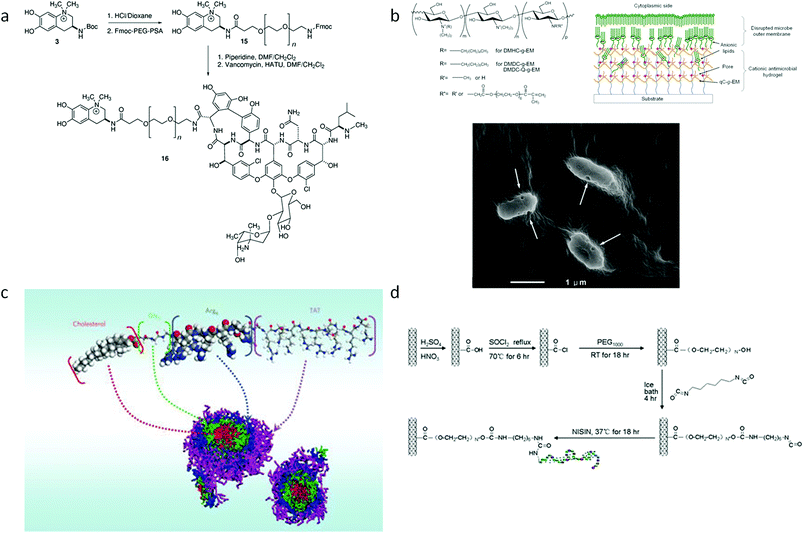 | ||
| Fig. 2 (a) Hybrids of anachelin with natural glycopeptide vancomycin.172 (b) Quaternized chitosan functionalized with acrylate PEG side-chains which forms a nanoporous hydrogel.182 (c) Self-assembly of peptide and cholesterol hybrids;173 (c) Conjugation of Amphotericin B to oxidized natural polysaccharides (dextran);171 (d) Conjugation of antimicrobial peptide with carbon nanotubes.175 | ||
Multi-functional hybrid materials can be obtained by linking natural products together. For example, the natural glycopeptide vancomycin was conjugated with the cyanobacterial-derived siderophore anachelin by a PEG linker using Fmoc chemistry.172 This hybrid material has three components, each with a different function: (i) vancomycin incurs antimicrobial activity, (ii) anachelin renders tethering functions to the system, while (iii) PEG chains with protein-resistant activity eliminates the adhesion of dead cells which will impede antimicrobial activity.172 The synergistic combination of these three parts renders an antimicrobial and antifouling surface with superior properties compared to the single use of these three molecules.172 In another study reported by Langer and co-workers, an antifungal agent, Amphotericin B (AmB), was chemically incorporated with a dextran chain. Dextran is a natural polysaccharide made of branched glucose chains, commonly used as scaffolds due to its biocompatible and protein-repellent properties,176,177 while AmB is a FDA-approved broad-spectrum antifungal agent widely used in clinical practice (Fig. 2a).171,177 By conjugation with the hydrophilic dextran–aldehyde polymer, the hydrophobic AmB was rendered water soluble, and the antifungal efficacy was retained.171 The AmB–Dextran conjugation is also cross-linkable and injectable for local antifungal therapy.171
Hybrid materials can also be obtained by introducing synthetic parts onto natural products by semi-synthesis or various modifications. Chitosan is a polysaccharide derived from chitin, which is a natural product rich in the exoskeleton of crustaceans, insects and the cell wall of fungi.178,179 Chitosan is a well-known biocompatible and non-toxic material which is widely used in various biomedical applications.180 In addition, chitosan also has inherent antimicrobial properties conferred by the cationic charge resulting from the protonation of its amine groups at the C2 position.178 However, its low pKa gives it a low net cationic charge, thus rendering somewhat poor antimicrobial activity to pristine chitosan.181 The low pKa also indicates that it is insoluble at neutral pH, which limits the use of chitosan. Therefore, many semi-synthetic methods were developed to introduce various groups or side-chains to chitosan to improve its charge and solubility properties.180 In our recent work, chitosan was modified by (i) alkyl side chain addition and quaternization of the amino group, (ii) hydrophilic polyethylene glycol with six ethylene glycol repeats (PEG6), and (iii) methacrylate functionality (Fig. 2b).182 Quaternization of the amine group and the introduction of pendent alkyl chains improved the positive charge and hydrophobicity of the polymer, respectively. PEGylation of polymers has been shown to decrease its hemolysis or cytotoxicity properties while the methacrylate functionalization makes the hybrid system cross-linkable for gelation. The final product takes the form of a nano-porous hydrogel system that acts like an “anion sponge” to effectively disrupt the anionic microbial cell wall or membrane.182,183 In another study, chitosan was hybridized with synthetic peptides to enhance its antimicrobial activity and biocompatibility.184 Peptidoglycan which consists of linear polysaccharide (glycan) chains and peptide side-chains, is a common component which exists in both Gram-negative and Gram-positive bacteria cell walls, but is absent in animal cells. The Gram-negative bacteria cell wall has two concentric lipid bilayer membranes (i.e. the cytoplasmic (inner) membrane and the outer membrane) and a thin peptidoglycan layer between them.185 The Gram-positive bacteria possesses a single anionic membrane (the cytoplasmic membrane), and a thick exposed peptidoglycan layer which is rich in anionic teichoic acids.186 Peptidopolysaccharide was designed to mimic the structure of peptidoglycan by using polysaccharide (chitosan) and polypeptide side-chains. The optimum peptidopolysaccharide comprised a copolymer of chitosan and polylysine (CS-g-K16), which was effective against both Gram-negative and Gram-positive bacteria, and fungi at low MICs (i.e. 5–20 μg ml−1). This peptidopolysaccharide also showed high selectivity (> 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) and low toxicity toward mammalian cells. The affinity of such peptidopolysaccharides with bacterial cell wall constituents, such as lipopolysaccharides and peptidoglycan, promotes its antimicrobial activity and selectivity. This new class of antimicrobial peptidopolysaccharides reveals a new direction for the design of antimicrobial molecules.
000) and low toxicity toward mammalian cells. The affinity of such peptidopolysaccharides with bacterial cell wall constituents, such as lipopolysaccharides and peptidoglycan, promotes its antimicrobial activity and selectivity. This new class of antimicrobial peptidopolysaccharides reveals a new direction for the design of antimicrobial molecules.
Self-assembly hybrid copolymers were recently designed by Yang and co-workers.173,174,187 Cationic cell-penetrating peptides were grafted with block cholesterol segments (assembly-driving component) to promote the ability to disintegrate microbial cell walls (Fig. 2c).173,187 This hybrid copolymer assembles into a nanoparticle with a core-shell structure, where cholesterol forms a hydrophobic core and the cationic peptide forms a hydrophilic shell which face the exterior. This nanoparticle efficiently inhibited the growth of drug-resistant Gram-positive bacteria and fungi at low MICs while inducing relatively low hemolytic activity. The hydrophobic core and cationic shell nanoparticle structure increases the local density of positive charge to render higher wall- and membrane-permeabilising ability, thereby improving antimicrobial potency.173,174,187,188 The design of this hybrid copolymer nanoparticle was further improved by adding degradable linkages between the cationic block and self-assembly driving block, to render it biocidal and biodegradable.174
Nanomaterials (i.e. carbon nanotubes, graphene and graphene oxides) have also been reported to be able to incur physical and microchemical damage to microbial cells.189–192 A natural AMP, Nisin, was covalently linked with multi-walled carbon nanotubes (MWNTs), which yielded higher antimicrobial activity (7-fold increase) and dramatically improved anti-biofilm activity than pristine MWNTs (Fig. 2d).175 In our recent work, the grafting of quaternized chitosan (QC) with graphene oxides (GO) showed that the GO–QC hybrids had superior bacterial cell wall damage property compared to GO alone (unpublished work).
3.4 Applications of synthetic antimicrobial oligomers and macromolecules
Armed with flexible short chains, non-peptide backbones, and higher in vivo efficacy and stability, antimicrobial polymers offer vast application opportunities in health care and biomedical implant coatings which include medical devices such as catheters, contact lenses, and stents.193–195 The development of biomimetic polymer–peptide hybrids allows the controlled release of AMPs which has applications in systemic administration for improved antimicrobial efficiency. Release of dermaceptin and Ponericin G1 from polymer thin films, for example, have shown to effectively inhibit S. aureus biofilms.196,197 Similar studies with gentamicin-loaded polymer beads and LL-37 from porous solid support systems have also shown effective killing of bacteria, indicating a promising system for the treatment of local infections.198 One of the bottlenecks to effective usage of coated AMP–polymer hybrids, however, is the instability or reduced efficacy of tethered AMPs. Extensive studies on immobilisation strategies of AMPs,197 factors influencing the activity of immobilised AMPs199 and tethering of AMPs on flexible polymer brushes200,201 are active areas of research and will continue to open new and improved avenues for application of antimicrobial polymers in healthcare.4 Concluding remarks
Since their discovery, AMPs are viewed as new generation antibiotics which can effectively treat infections without easily incurring pathogen resistance. However, production complexity and cost, coupled with peptide stability are major challenges in scaling up AMP production for therapeutic applications. Efforts to address these shortcomings which include peptide engineering efforts and the optimisation of synthesis protocols are actively underway. The use of ‘synthetic polymer’-based antimicrobial materials, on the other hand, does not pose any scale-up cost problems. Although the mechanism of polymer-based antimicrobial activity is less understood than AMPs, the outcomes from recent characterisation studies of some polymeric antimicrobial materials indicate that the overall polymer amphiphilicity is more important than secondary structure enhancement for antimicrobial activity, while the amphiphilicity balance appeared to be important to reduce cytotoxicity. The generally higher cytotoxicity demonstrated by antimicrobial polymeric materials compared to natural AMPs indicate the need for more fundamental research at the molecular level to fully elucidate the interaction between antimicrobial polymers and microbial/mammalian cells. As single functional materials cannot effectively confer antimicrobial properties on polymers, more multifunctional antimicrobial polymers including AMP–polymer hybrids are being designed and tested to further improve antimicrobial properties. Despite their exciting prospects, the use of these new antimicrobial macromolecules must still be administered with great care, as the development of pathogen resistance against these macromolecules over time cannot be completely ruled out. With improved understanding of the antimicrobial mechanism incurred, the design of these macromolecules will have to continue evolving over time to combat resistance developmentAcknowledgements
This work was funded and supported by a Biomedical Engineering Program, A*Star grant (Project SERC1031490001).References
- H. Breithaupt, Nat. Biotechnol., 1999, 17, 1165–1169 CrossRef CAS.
- B. Spellberg, R. Guidos, D. Gilbert, J. Bradley, H. W. Boucher, W. M. Scheld, J. G. Bartlett and J. Edwards Jr., Clin. Infect. Dis., 2008, 46, 155–164 CrossRef.
- T. van der Poll and S. M. Opal, Lancet Infect. Dis., 2008, 8, 32–43 CrossRef.
- G. Taubes, Science, 2008, 321, 356–361 CrossRef CAS.
- F. W. Goldstein, Clin. Microbiol. Infect., 2007, 13, 2–6 CrossRef CAS.
- R. E. Hancock, Lancet Infect. Dis., 2001, 1, 156–164 CrossRef CAS.
- L. Steinstraesser, U. Kraneburg, F. Jacobsen and S. Al-Benna, Immunobiology, 2011, 216, 322–333 CrossRef CAS.
- D. M. Bowdish and R. E. Hancock, J Endotoxin Res, 2005, 11, 230–236 Search PubMed.
- H. G. Boman, Immunol. Rev., 2000, 173, 5–16 CAS.
- R. E. Hancock and G. Diamond, Trends Microbiol., 2000, 8, 402–410 CrossRef CAS.
- P. Pristovsek and J. Kidric, Curr. Top. Med. Chem., 2004, 4, 1185–1201 CrossRef CAS.
- S. Bhattacharjya, Curr. Med. Chem., 2010, 17, 3080–3093 CrossRef CAS.
- E. Gazit, I. R. Miller, P. C. Biggin, M. S. Sansom and Y. Shai, J. Mol. Biol., 1996, 258, 860–870 CrossRef CAS.
- K. Matsuzaki, Biochim. Biophys. Acta, 1998, 1376, 391–400 CAS.
- W. T. Heller, A. J. Waring, R. I. Lehrer and H. W. Huang, Biochem., 1998, 37, 17331–17338 CrossRef CAS.
- H. G. Boman, B. Agerberth and A. Boman, Infect. Immun., 1993, 61, 2978–2984 CAS.
- B. Yasin, M. Pang, J. S. Turner, Y. Cho, N. N. Dinh, A. J. Waring, R. I. Lehrer and E. A. Wagar, Eur. J. Clin. Microbiol. Infect. Dis., 2000, 19, 187–194 CrossRef CAS.
- M. Wachinger, A. Kleinschmidt, D. Winder, N. von Pechmann, A. Ludvigsen, M. Neumann, R. Holle, B. Salmons, V. Erfle and R. Brack-Werner, J. Gen. Virol., 1998, 79(Pt 4), 731–740 CAS.
- C. Lorin, H. Saidi, A. Belaid, A. Zairi, F. Baleux, H. Hocini, L. Belec, K. Hani and F. Tangy, Virology, 2005, 334, 264–275 CrossRef CAS.
- A. Belaid, M. Aouni, R. Khelifa, A. Trabelsi, M. Jemmali and K. Hani, J. Med. Virol., 2002, 66, 229–234 CrossRef CAS.
- H. Jenssen, P. Hamill and R. E. Hancock, Clin. Microbiol. Rev., 2006, 19, 491–511 CrossRef CAS.
- M. Zasloff, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 5449–5453 CrossRef CAS.
- D. G. Lee, P. I. Kim, Y. Park, E. R. Woo, J. S. Choi, C. H. Choi and K. S. Hahm, Biochem. Biophys. Res. Commun., 2002, 293, 231–238 CrossRef CAS.
- D. G. Lee, D. H. Kim, Y. Park, H. K. Kim, H. N. Kim, Y. K. Shin, C. H. Choi and K. S. Hahm, Biochem. Biophys. Res. Commun., 2001, 282, 570–574 CrossRef CAS.
- D. G. Lee, H. K. Kim, S. A. Kim, Y. Park, S. C. Park, S. H. Jang and K. S. Hahm, Biochem. Biophys. Res. Commun., 2003, 305, 305-310 Search PubMed.
- A. J. de Lucca, J. M. Bland, T. J. Jacks, C. Grimm and T. J. Walsh, Med. Mycol., 1998, 36, 291–298 CAS.
- X. Li and S. S. Leong, J. Chromatogr., A, 2011, 1218, 3654–3659 CrossRef CAS.
- J. Harder, J. Bartels, E. Christophers and J. M. Schroder, Nature, 1997, 387, 861 CrossRef CAS.
- J. Harder, J. Bartels, E. Christophers and J. M. Schroder, J. Biol. Chem., 2001, 276, 5707–5713 CrossRef CAS.
- Y. Li, Biotechnol. Appl. Biochem., 2009, 54, 1–9 CrossRef CAS.
- R. B. Merrifield, Adv Enzymol Relat Areas Mol Biol, 1969, 32, 221–296 CAS.
- M. Pazgier, X. Li, W. Lu and J. Lubkowski, Curr. Pharm. Des., 2007, 13, 3096–3118 CrossRef CAS.
- K. Hamamoto, Y. Kida, Y. Zhang, T. Shimizu and K. Kuwano, Microbiol. Immunol., 2002, 46, 741–749 CAS.
- E. M. Molhoek, A. van Dijk, E. J. A. Veldhuizen, H. P. Haagsman and F. J. Bikker, Peptides, 2011, 32, 875–880 CrossRef CAS.
- L. J. M. Cross, M. Ennis, E. Krause, M. Dathe, D. Lorenz, G. Krause, M. Beyermann and M. Bienert, Eur. J. Pharmacol., Mol. Pharmacol. Sect., 1995, 291, 291–300 CrossRef CAS.
- D. L. Lee, J. P. S. Powers, K. Pflegerl, M. L. Vasil, R. E. W. Hancock and R. S. Hodges, J. Pept. Res., 2004, 63, 69–84 CrossRef CAS.
- K. Matsuzaki, Biochim. Biophys. Acta, Biomembr., 2009, 1788, 1687–1692 CrossRef CAS.
- S. Y. Hong, J. E. Oh and K. H. Lee, Biochem. Pharmacol., 1999, 58, 1775–1780 CrossRef CAS.
- S. M. Owen, D. Rudolph, W. Wang, A. M. Cole, M. A. Sherman, A. J. Waring, R. I. Lehrer and R. B. Lal, J. Pept. Res., 2004, 63, 469–476 CrossRef CAS.
- I. Guell, E. Badosa, M. Talleda, R. Ferre, J. Cabrefiga, E. Montesinos, E. Bardaji, L. Feliu and M. Planas, J. Pept. Sci., 2010, 16, 139–139 Search PubMed.
- E. A. Papanastasiou, Q. Hua, A. Sandouk, U. H. Son, A. J. Christenson, M. L. Van Hoek and B. M. Bishop, APMIS, 2009, 117, 492–499 CrossRef CAS.
- S. R. Dennison, F. Harris, T. Bhatt, J. Singh and D. A. Phoenix, Mol. Cell. Biochem., 2009, 332, 43–50 CrossRef CAS.
- S. R. Dennison and D. A. Phoenix, Biochemistry, 2011, 50, 1514–1523 CrossRef CAS.
- L. T. Nguyen, J. K. Chau, N. A. Perry, L. de Boer, S. A. J. Zaat and H. J. Vogel, PLoS One, 2010, 5 Search PubMed.
- S. P. Liu, L. Zhou, R. Lakshminarayanan and R. W. Beuerman, Int. J. Pept. Res. Ther., 2010, 16, 199–213 CrossRef CAS.
- Z. B. Wu, D. M. Hoover, D. Yang, C. Boulegue, F. Santamaria, J. J. Oppenheim, J. Lubkowski and W. Y. Lu, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 8880–8885 CrossRef CAS.
- M. Pazgier, D. M. Hoover, D. Yang, W. Lu and J. Lubkowski, Cell. Mol. Life Sci., 2006, 63, 1294–1313 CrossRef CAS.
- D. K. Tay, G. Rajagopalan, X. Li, Y. Chen, L. H. Lua and S. S. Leong, Biotechnol. Bioeng., 2011, 108, 572–581 CrossRef CAS.
- A. B. Ingham and R. J. Moore, Biotechnol. Appl. Biochem., 2007, 47, 1–9 CrossRef CAS.
- L. D. Cabrita and S. P. Bottomley, Biotechnol. Annu. Rev., 2004, 10, 31–50 CrossRef CAS.
- E. V. Valore and T. Ganz, Methods Mol Biol, 1997, 78, 115–131 CAS.
- J. M. Kim, S. A. Jang, B. J. Yu, B. H. Sung, J. H. Cho and S. C. Kim, Appl. Microbiol. Biotechnol., 2008, 78, 123–130 CrossRef CAS.
- H. Q. Chen, Z. N. Xu, L. Peng, X. M. Fang, X. F. Yin, N. Z. Xu and P. L. Cen, Peptides, 2006, 27, 931–940 CrossRef CAS.
- X. C. Rao, S. Li, J. C. Hu, X. L. Jin, X. M. Hu, J. J. Huang, Z. J. Chen, J. M. Zhu and F. Q. Hu, Protein Expression Purif., 2004, 36, 11–18 CrossRef CAS.
- M. Satakarni and R. Curtis, Protein Expression Purif., 2011, 78, 113–119 CrossRef CAS.
- C. Richard, D. Drider, K. Elmorjani, D. Marion and H. Prevost, J. Bacteriol., 2004, 186, 4276–4284 CrossRef CAS.
- L. Beaulieu, D. Tolkatchev, J. F. Jette, D. Groleau and M. Subirade, Can. J. Microbiol., 2007, 53, 1246–1258 CrossRef CAS.
- S. Kanai, H. Toh, T. Hayano and M. Kikuchi, J. Mol. Evol., 1998, 47, 200–210 CrossRef CAS.
- L. Huang, S. S. J. Leong and R. R. Jiang, Appl. Microbiol. Biotechnol., 2009, 84, 301–308 CrossRef CAS.
- L. Huang, C. B. Ching, R. Jiang and S. S. Leong, Protein Expression Purif., 2008, 61, 168–174 CrossRef CAS.
- X. J. Feng, C. L. Liu, J. Y. Guo, C. P. Bi, B. J. Cheng, Z. Y. Li, A. S. Shan and Z. Q. Li, Curr. Microbiol., 2010, 60, 179–184 CrossRef CAS.
- H. X. Luo, S. W. Chen, F. Z. Ren, H. Y. Guo, S. H. Lin and W. T. Xu, J. Dairy Res., 2007, 74, 233–238 CrossRef CAS.
- C. Ma, M. Gao, W. Liu, J. Zhu, H. Tian, X. Gao and W. Yao, Protein Pept. Lett., 2010, 17, 1245–1250 CrossRef CAS.
- C. Morassutti, F. De Amicis, A. Bandiera and S. Marchetti, Protein Expression Purif., 2005, 39, 160–168 CrossRef CAS.
- Y. F. Li, Biotechnol. Lett., 2011, 33, 869–881 CrossRef CAS.
- J. H. Lee, J. H. Kim, S. W. Hwang, W. J. Lee, H. K. Yoon, H. S. Lee and S. S. Hong, Biochem. Biophys. Res. Commun., 2000, 277, 575–580 CrossRef CAS.
- Y. L. Sun, T. C. Kuan, Y. J. Lin, Y. C. Chou and C. S. Lin, Ann. Microbiol., 2010, 60, 329–334 CrossRef CAS.
- A. B. Ingham and R. J. Moore, Biotechnol. Appl. Biochem., 2007, 47, 1–9 CrossRef CAS.
- G. Morreale, E. G. Lee, D. B. Jones and A. P. J. Middelberg, Biotechnol. Bioeng., 2004, 87, 912–923 CrossRef CAS.
- S. H. Pyo, J. H. Lee, H. B. Park, J. S. Cho, H. R. Kim, B. H. Han and Y. S. Park, Process Biochem., 2004, 39, 1731–1736 CrossRef CAS.
- R. C. Stevens, Structure, 2000, 8, R177–185 CrossRef CAS.
- W. Kaar, B. M. Hartmann, Y. Fan, B. Zeng, L. H. L. Lua, A. F. Dexter, R. J. Falconer and A. P. J. Middelberg, Biotechnol. Bioeng., 2009, 102, 176–187 CrossRef CAS.
- S. E. Blondelle and K. Lohner, Curr. Pharm. Des., 2010, 16, 3204–3211 CrossRef CAS.
- G. M. Nava, M. Escorcia and M. P. Castaneda, Compar. Funct. Genom., 2009 Search PubMed.
- B. O. Schroeder, Z. Wu, S. Nuding, S. Groscurth, M. Marcinowski, J. Beisner, J. Buchner, M. Schaller, E. F. Stange and J. Wehkamp, Nature, 2011, 469, 419–423 CrossRef CAS.
- D. M. Hoover, Z. Wu, K. Tucker, W. Lu and J. Lubkowski, Antimicrob. Agents Chemother., 2003, 47, 2804–2809 CrossRef CAS.
- Z. Wu, D. M. Hoover, D. Yang, C. Boulegue, F. Santamaria, J. J. Oppenheim, J. Lubkowski and W. Lu, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 8880–8885 CrossRef CAS.
- S. Liu, L. Zhou, J. Li, A. Suresh, C. Verma, Y. H. Foo, E. P. Yap, D. T. Tan and R. W. Beuerman, ChemBioChem, 2008, 9, 964–973 CrossRef CAS.
- K. Taylor, D. J. Clarke, B. McCullough, W. Chin, E. Seo, D. Yang, J. Oppenheim, D. Uhrin, J. R. Govan, D. J. Campopiano, D. MacMillan, P. Barran and J. R. Dorin, J. Biol. Chem., 2008, 283, 6631–6639 CrossRef CAS.
- K. B. Chandrababu, B. Ho and D. Yang, Biochemistry, 2009, 48, 6052–6061 CrossRef CAS.
- Y. Bai, S. Liu, P. Jiang, L. Zhou, J. Li, C. Tang, C. Verma, Y. Mu, R. W. Beuerman and K. Pervushin, Biochemistry, 2009, 48, 7229–7239 CrossRef CAS.
- O. Scudiero, S. Galdiero, M. Cantisani, R. Di Noto, M. Vitiello, M. Galdiero, G. Naclerio, J. J. Cassiman, C. Pedone, G. Castaldo and F. Salvatore, Antimicrob. Agents Chemother., 2010, 54, 2312–2322 CrossRef CAS.
- A. K. Marr, W. J. Gooderham and R. E. Hancock, Curr. Opin. Pharmacol., 2006, 6, 468–472 CrossRef CAS.
- Y. Bae, N. Nishiyama, S. Fukushima, H. Koyama, M. Yasuhiro and K. Kataoka, Bioconjugate Chem., 2005, 16, 122–130 CrossRef CAS.
- R. Eckert, Future Microbiol., 2011, 6, 635–651 CrossRef CAS.
- R. P. Hicks, J. B. Bhonsle, D. Venugopal, B. W. Koser and A. J. Magill, J. Med. Chem., 2007, 50, 3026–3036 CrossRef CAS.
- S. R. Dennison, F. Harris, T. Bhatt, J. Singh and D. A. Phoenix, Mol. Cell. Biochem., 2009, 332, 43–50 CrossRef CAS.
- A. Makovitzki, J. Baram and Y. Shai, Biochemistry, 2008, 47, 10630–10636 CrossRef CAS.
- J. He, M. H. Anderson, W. Shi and R. Eckert, Int. J. Antimicrob. Agents, 2009, 33, 532–537 CrossRef CAS.
- L. Li, J. He, R. Eckert, D. Yarbrough, R. Lux, M. Anderson and W. Shi, Chem. Biol. Drug Des., 2010, 75, 127–132 CAS.
- J. He, D. K. Yarbrough, J. Kreth, M. H. Anderson, W. Shi and R. Eckert, Antimicrob. Agents Chemother., 2010, 54, 2143–2151 CrossRef CAS.
- S. Rotem and A. Mor, Biochim. Biophys. Acta, Biomembr., 2009, 1788, 1582–1592 CrossRef CAS.
- N. Srinivas, P. Jetter, B. J. Ueberbacher, M. Werneburg, K. Zerbe, J. Steinmann, B. Van der Meijden, F. Bernardini, A. Lederer, R. L. Dias, P. E. Misson, H. Henze, J. Zumbrunn, F. O. Gombert, D. Obrecht, P. Hunziker, S. Schauer, U. Ziegler, A. Kach, L. Eberl, K. Riedel, S. J. DeMarco and J. A. Robinson, Science, 2010, 327, 1010–1013 CrossRef CAS.
- C. F. Lange, R. E. Hancock, J. Samuel and W. H. Finlay, J. Pharm. Sci., 2001, 90, 1647–1657 CrossRef CAS.
- R. Higgs, D. J. Lynn, S. Cahalane, I. Alana, C. M. Hewage, T. James, A. T. Lloyd and C. O'Farrelly, Immunogenetics, 2007, 59, 573–580 CrossRef CAS.
- A. Munoz, B. Lopez-Garcia, E. Perez-Paya and J. F. Marcos, Biochem. Biophys. Res. Commun., 2007, 354, 172–177 CrossRef CAS.
- M. Tamaki, Y. Imazeki, A. Shirane, K. Fujinuma, M. Shindo, M. Kimura and Y. Uchida, Bioorg. Med. Chem. Lett., 2011, 21, 440–443 CrossRef CAS.
- B. O. Schroeder, Z. H. Wu, S. Nuding, S. Groscurth, M. Marcinowski, J. Beisner, J. Buchner, M. Schaller, E. F. Stange and J. Wehkamp, Nature, 2011, 469, 419 CrossRef CAS.
- K. B. Chandrababu, B. Ho and D. W. Yang, Biochemistry, 2009, 48, 6052–6061 CrossRef CAS.
- Y. Orikasa, K. Ichinohe, J. Saito, S. Hashimoto, K. Matsumoto, T. Ooi and S. Taguchi, Biosci., Biotechnol., Biochem., 2009, 73, 1683–1684 CrossRef CAS.
- E. K. Lee, Y. C. Kim, Y. H. Nan and S. Y. Shin, Peptides, 2011, 32, 1123–1130 CrossRef CAS.
- N. Tsuda, Y. Koba, T. Hatakeyama, S. Ando, M. Shindo, Y. Uchida and H. Aoyagi, Bull. Chem. Soc. Jpn., 2008, 81, 1299–1303 CrossRef CAS.
- R. Saravanan, A. Bhunia and S. Bhattacharjya, Biochim. Biophys. Acta, Biomembr., 2010, 1798, 128–139 CrossRef CAS.
- N. L. Reynolds, M. De Cecco, K. Taylor, C. Stanton, F. Kilanowski, J. Kalapothakis, E. Seo, D. Uhrin, D. Campopiano, J. Govan, D. Macmillan, P. Barran and J. R. Dorin, Antimicrob. Agents Chemother., 2010, 54, 1922–1929 CrossRef CAS.
- L. Zhou, S. P. Liu, L. Y. Chen, J. Li, L. B. Ong, L. Guo, T. Wohland, C. C. Tang, R. Lakshminarayanan, J. Mavinahalli, C. Verma and R. W. Beuerman, Amino Acids, 2011, 40, 123–133 CrossRef CAS.
- S. Jung, J. Mysliwy, B. Spudy, I. Lorenzen, K. Reiss, C. Gelhaus, R. Podschun, M. Leippe and J. Grotzinger, Antimicrob. Agents Chemother., 2011, 55, 954–960 CrossRef CAS.
- J. G. M. Bolscher, R. Adao, K. Nazmi, P. A. M. van den Keybus, W. van't Hof, A. V. N. Amerongen, M. Bastos and E. C. I. Veerman, Biochimie, 2009, 91, 123–132 CrossRef CAS.
- I. Guell, J. Cabrefiga, E. Badosa, R. Ferre, M. Talleda, E. Bardaji, M. Planas, L. Feliu and E. Montesinos, Appl. Environ. Microbiol., 2011, 77, 2667–2675 CrossRef CAS.
- M. J. Oudhoff, K. L. Kroeze, K. Nazmi, P. A. M. van den Keijbus, W. van't Hof, M. Fernandez-Borja, P. L. Hordijk, S. Gibbs, J. G. M. Bolscher and E. C. I. Veerman, FASEB J., 2009, 23, 3928–3935 CrossRef CAS.
- C. T. T. Wong, M. Taichi, H. Nishio, Y. Nishiuchi and J. P. Tam, Biochemistry, 2011, 50, 7275–7283 CrossRef CAS.
- K. T. N. Giang, S. Zhang, T. K. N. Ngan, Q. T. N. Phuong, M. S. Chiu, A. Hardjojo and J. P. Tam, J. Biol. Chem., 2011, 286, 24275–24287 CrossRef.
- A. K. Marr, W. J. Gooderham and R. E. W. Hancock, Curr. Opin. Pharmacol., 2006, 6, 468–472 CrossRef CAS.
- E. R. Kenawy, S. D. Worley and R. Broughton, Biomacromolecules, 2007, 8, 1359–1384 CrossRef CAS.
- G. J. Gabriel, A. Som, A. E. Madkour, T. Eren and G. N. Tew, Mater. Sci. Eng., R, 2007, 57, 28–64 CrossRef.
- E. F. Palermo and K. Kuroda, Appl. Microbiol. Biotechnol., 2010, 87, 1605–1615 CrossRef CAS.
- A. Som, S. Vemparala, I. Ivanov and G. N. Tew, Biopolymers, 2008, 90, 83–93 CrossRef CAS.
- G. J. Gabriel and G. N. Tew, Org. Biomol. Chem., 2008, 6, 417–423 CAS.
- G. N. Tew, R. W. Scott, M. L. Klein and W. F. Degrado, Acc. Chem. Res., 2010, 43, 30–39 CrossRef CAS.
- C. M. Goodman, S. Choi, S. Shandler and W. F. DeGrado, Nat. Chem. Biol., 2007, 3, 252–262 CrossRef CAS.
- L. Arnt and G. N. Tew, J. Am. Chem. Soc., 2002, 124, 7664–7665 CrossRef CAS.
- P. Claudon, A. Violette, K. Lamour, M. Decossas, S. Fournel, B. Heurtault, J. Godet, Y. Mely, B. Jamart-Gregoire, M. C. Averlant-Petit, J. P. Briand, G. Duportail, H. Monteil and G. Guichard, Angewandte Chemie-International Edition, 2010, 49, 333–336 CAS.
- W. S. Horne and S. H. Gellman, Acc. Chem. Res., 2008, 41, 1399–1408 CrossRef CAS.
- M. A. Schmitt, B. Weisblum and S. H. Gellman, J. Am. Chem. Soc., 2004, 126, 6848–6849 CrossRef CAS.
- A. Violette, S. Fournel, K. Lamour, O. Chaloin, B. Frisch, J. P. Briand, H. Monteil and G. Guichard, Chem. Biol., 2006, 13, 531–538 CrossRef CAS.
- N. P. Chongsiriwatana, J. A. Patch, A. M. Czyzewski, M. T. Dohm, A. Ivankin, D. Gidalevitz, R. N. Zuckermann and A. E. Barron, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 2794–2799 CrossRef CAS.
- J. A. Patch and A. E. Barron, J. Am. Chem. Soc., 2003, 125, 12092–12093 CrossRef CAS.
- S. Choi, A. Isaacs, D. Clements, D. H. Liu, H. Kim, R. W. Scott, J. D. Winkler and W. F. DeGrado, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 6968–6973 CrossRef CAS.
- D. H. Liu, S. Choi, B. Chen, R. J. Doerksen, D. J. Clements, J. D. Winkler, M. L. Klein and W. F. DeGrado, Angew. Chem., Int. Ed., 2004, 43, 1158–1162 CrossRef CAS.
- B. Mensa, Y. H. Kim, S. Choi, R. Scott, G. A. Caputo and W. F. DeGrado, Antimicrob. Agents Chemother., 2011, 55, 5043–5053 CrossRef CAS.
- H. Tang, R. J. Doerksen, T. V. Jones, M. L. Klein and G. N. Tew, Chem. Biol., 2006, 13, 427–435 CrossRef CAS.
- G. N. Tew, D. Clements, H. Z. Tang, L. Arnt and R. W. Scott, Biochim. Biophys. Acta, Biomembr., 2006, 1758, 1387–1392 CrossRef CAS.
- H. Z. Tang, R. J. Doerksen and G. N. Tew, Chem. Commun., 2005, 1537–1539 RSC.
- Y. Ishitsuka, L. Arnt, J. Majewski, S. Frey, M. Ratajczek, K. Kjaer, G. N. Tew and K. Y. C. Lee, J. Am. Chem. Soc., 2006, 128, 13123–13129 CrossRef CAS.
- L. H. Yang, V. D. Gordon, A. Mishra, A. Sorn, K. R. Purdy, M. A. Davis, G. N. Tew and G. C. L. Wong, J. Am. Chem. Soc., 2007, 129, 12141–12147 CrossRef CAS.
- M. M. Slutsky, J. S. Phillip and G. N. Tew, New J. Chem., 2008, 32, 670–675 RSC.
- A. Som and G. N. Tew, J. Phys. Chem. B, 2008, 112, 3495–3502 CrossRef CAS.
- R. W. Scott, W. F. DeGrado and G. N. Tew, Curr. Opin. Biotechnol., 2008, 19, 620–627 CrossRef CAS.
- K. Lienkamp, A. Madkour and G. Tew, Springer Berlin/Heidelberg, Editon edn, 2011, 1–32 Search PubMed.
- K. Lienkamp, A. E. Madkour, A. Musante, C. F. Nelson, K. Nusslein and G. N. Tew, J. Am. Chem. Soc., 2008, 130, 9836–9843 CrossRef CAS.
- K. Kuroda and W. F. DeGrado, J. Am. Chem. Soc., 2005, 127, 4128–4129 CrossRef CAS.
- B. C. Allison, B. M. Applegate and J. P. Youngblood, Biomacromolecules, 2007, 8, 2995–2999 CrossRef CAS.
- Y. Oda, S. Kanaoka, T. Sato, S. Aoshima and K. Kuroda, Biomacromolecules, 2011, 12, 3581–3591 CrossRef CAS.
- M. F. Ilker, K. Nusslein, G. N. Tew and E. B. Coughlin, J. Am. Chem. Soc., 2004, 126, 15870–15875 CrossRef CAS.
- Z. M. Al-Badri, A. Som, S. Lyon, C. F. Nelson, K. Nusslein and G. N. Tew, Biomacromolecules, 2008, 9, 2805–2810 CrossRef CAS.
- E. Glukhov, L. L. Burrows and C. M. Deber, Biopolymers, 2008, 89, 360–371 CrossRef CAS.
- V. Sambhy, B. R. Peterson and A. Sen, Angew. Chem., Int. Ed., 2008, 47, 1250–1254 CrossRef CAS.
- K. Kuroda, G. A. Caputo and W. F. DeGrado, Chem.–Eur. J., 2009, 15, 1123–1133 CrossRef CAS.
- B. P. Mowery, A. H. Lindner, B. Weisblum, S. S. Stahl and S. H. Gellman, J. Am. Chem. Soc., 2009, 131, 9735–9745 CrossRef CAS.
- E. F. Palermo, I. Sovadinova and K. Kuroda, Biomacromolecules, 2009, 10, 3098–3107 CrossRef CAS.
- B. Dizman, M. O. Elasri and L. J. Mathias, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 5965–5973 CrossRef CAS.
- K. Lienkamp and G. N. Tew, Chem.–Eur. J., 2009, 15, 11784–11800 CrossRef CAS.
- S. Colak, C. F. Nelson, K. Nusslein and G. N. Tew, Biomacromolecules, 2009, 10, 353–359 CrossRef CAS.
- K. Lienkamp, K. N. Kumar, A. Som, K. Nusslein and G. N. Tew, Chem.–Eur. J., 2009, 15, 11710–11714 CrossRef CAS.
- K. Lienkamp, A. E. Madkour, K. N. Kumar, K. Nusslein and G. N. Tew, Chem.–Eur. J., 2009, 15, 11715–11722 CrossRef CAS.
- G. J. Gabriel, J. A. Maegerlein, C. E. Nelson, J. M. Dabkowski, T. Eren, K. Nusslein and G. N. Tew, Chem.–Eur. J., 2009, 15, 433–439 CrossRef CAS.
- S. Venkataraman, Y. Zhang, L. H. Liu and Y. Y. Yang, Biomaterials, 2010, 31, 1751–1756 CrossRef CAS.
- E. F. Palermo and K. Kuroda, Biomacromolecules, 2009, 10, 1416–1428 CrossRef CAS.
- T. R. Stratton, J. L. Rickus and J. P. Youngblood, Biomacromolecules, 2009, 10, 2550–2555 CrossRef CAS.
- T. R. Stratton, J. A. Howarter, B. C. Allison, B. M. Applegate and J. P. Youngblood, Biomacromolecules, 2010, 11, 1286–1290 CrossRef CAS.
- T. R. Stratton, B. M. Applegate and J. P. Youngblood, Biomacromolecules, 2011, 12, 50–56 CrossRef CAS.
- S. Chakrabarty, A. King, P. Kurt, W. Zhang, D. E. Ohman, L. F. Wood, C. Lovelace, R. Rao and K. J. Wynne, Biomacromolecules, 2011, 12, 757–769 CrossRef CAS.
- B. P. Mowery, S. E. Lee, D. A. Kissounko, R. F. Epand, R. M. Epand, B. Weisblum, S. S. Stahl and S. H. Gellman, J. Am. Chem. Soc., 2007, 129, 15474 CrossRef CAS.
- R. F. Epand, B. P. Mowery, S. E. Lee, S. S. Stahl, R. I. Lehrer, S. H. Gellman and R. M. Epand, J. Mol. Biol., 2008, 379, 38–50 CrossRef CAS.
- D. Park, J. A. Finlay, R. J. Ward, C. J. Weinman, S. Krishnan, M. Paik, K. E. Sohn, M. E. Callow, J. A. Callow, D. L. Handlin, C. L. Willis, D. A. Fischer, E. R. Angert, E. J. Kramer and C. K. Ober, ACS Appl. Mater. Interfaces, 2010, 2, 703–711 CAS.
- G. D. Fu, F. Yao, Z. G. Li and X. S. Li, J. Mater. Chem., 2008, 18, 859–867 RSC.
- P. H. Sellenet, B. Allison, B. M. Applegate and J. P. Youngblood, Biomacromolecules, 2007, 8, 19–23 CrossRef CAS.
- S. Krishnan, R. J. Ward, A. Hexemer, K. E. Sohn, K. L. Lee, E. R. Angert, D. A. Fischer, E. J. Kramer and C. K. Ober, Langmuir, 2006, 22, 11255–11266 CrossRef CAS.
- E. F. Palermo, D. K. Lee, A. Ramamoorthy and K. Kuroda, J. Phys. Chem. B, 2011, 115, 366–375 CrossRef CAS.
- E. S. Park, H. S. Kim, M. N. Kim and J. S. Yoon, Eur. Polym. J., 2004, 40, 2819–2822 CrossRef CAS.
- K. Gademann, Chimia, 2006, 60, 841–845 CrossRef CAS.
- S. P. Hudson, R. Langer, G. R. Fink and D. S. Kohane, Biomaterials, 2010, 31, 1444–1452 CrossRef CAS.
- J. Y. Wach, S. Bonazzi and K. Gademann, Angew. Chem., Int. Ed., 2008, 47, 7123–7126 CrossRef CAS.
- L. H. Liu, K. J. Xu, H. Y. Wang, P. K. J. Tan, W. M. Fan, S. S. Venkatraman, L. J. Li and Y. Y. Yang, Nat. Nanotechnol., 2009, 4, 457–463 CrossRef CAS.
- F. Nederberg, Y. Zhang, J. P. K. Tan, K. J. Xu, H. Y. Wang, C. Yang, S. J. Gao, X. D. Guo, K. Fukushima, L. J. Li, J. L. Hedrick and Y. Y. Yang, Nat. Chem., 2011, 3, 409–414 CrossRef CAS.
- X. B. Qi, G. Poernomo, K. A. Wang, Y. A. Chen, M. B. Chan-Park, R. Xu and M. W. Chang, Nanoscale, 2011, 3, 1874–1880 RSC.
- Y. X. Liu and M. B. Chan-Park, Biomaterials, 2009, 30, 196–207 CrossRef.
- A. Zumbuehl, L. Ferreira, D. Kuhn, A. Astashkina, L. Long, Y. Yeo, T. Iaconis, M. Ghannoum, G. R. Fink, R. Langer and D. S. Kohane, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 12994–12998 CrossRef CAS.
- M. Rinaudo, Prog. Polym. Sci., 2006, 31, 603–632 CrossRef CAS.
- J. P. Latge, Mol. Microbiol., 2007, 66, 279–290 CrossRef CAS.
- M. Dash, F. Chiellini, R. M. Ottenbrite and E. Chiellini, Prog. Polym. Sci., 2011, 36, 981–1014 CrossRef CAS.
- D. Raafat, K. von Bargen, A. Haas and H. G. Sahl, Appl. Environ. Microbiol., 2008, 74, 3764–3773 CrossRef CAS.
- P. Li, Y. F. Poon, W. F. Li, H. Y. Zhu, S. H. Yeap, Y. Cao, X. B. Qi, C. C. Zhou, M. Lamrani, R. W. Beuerman, E. T. Kang, Y. G. Mu, C. M. Li, M. W. Chang, S. S. J. Leong and M. B. Chan-Park, Nat. Mater., 2011, 10, 149–156 CrossRef CAS.
- C. C. Zhou, P. Li, X. B. Qi, A. R. M. Sharif, Y. F. Poon, Y. Cao, M. W. Chang, S. S. J. Leong and M. B. Chan-Park, Biomaterials, 2011, 32, 2704–2712 CrossRef CAS.
- P. Li, C. C. Zhou, S. Rayatpisheh, K. Ye, Y. F. Poon, P. T. Hammond, H. Duan and M. B. Chan-Park, Adv. Mater.., 2012 DOI:10.1002/adma. 201104186.
- T. J. Beveridge, Journal of Bacteriology, 1999, 181, 4725–4733 CAS.
- W. Fischer, Med. Microbiol. Immunol., 1994, 183, 61–76 CrossRef CAS.
- H. Y. Wang, K. J. Xu, L. H. Liu, J. P. K. Tan, Y. B. Chen, Y. T. Li, W. M. Fan, Z. Q. Wei, J. F. Sheng, Y. Y. Yang and L. J. Li, Biomaterials, 2010, 31, 2874–2881 CrossRef CAS.
- S. B. Darling, Prog. Polym. Sci., 2007, 32, 1152–1204 CrossRef CAS.
- S. B. Liu, L. Wei, L. Hao, N. Fang, M. W. Chang, R. Xu, Y. H. Yang and Y. Chen, ACS Nano, 2009, 3, 3891–3902 CrossRef CAS.
- S. B. Liu, T. H. Zeng, M. Hofmann, E. Burcombe, J. Wei, R. R. Jiang, J. Kong and Y. Chen, ACS Nano, 2011, 5, 6971–6980 CrossRef CAS.
- O. Akhavan and E. Ghaderi, ACS Nano, 2010, 4, 5731–5736 CrossRef CAS.
- W. B. Hu, C. Peng, W. J. Luo, M. Lv, X. M. Li, D. Li, Q. Huang and C. H. Fan, ACS Nano, 2010, 4, 4317–4323 CrossRef CAS.
- V. Acharya, C. R. Prabha and C. Narayanamurthy, Biomaterials, 2004, 25, 4555–4562 CrossRef CAS.
- E. B. Hume, J. Baveja, B. Muir, T. L. Schubert, N. Kumar, S. Kjelleberg, H. J. Griesser, H. Thissen, R. Read, L. A. Poole-Warren, K. Schindhelm and M. D. Willcox, Biomaterials, 2004, 25, 5023–5030 CrossRef CAS.
- R. Kenawy el, S. D. Worley and R. Broughton, Biomacromolecules, 2007, 8, 1359–1384 CrossRef.
- M. F. Zampa, I. M. Araujo, V. Costa, C. H. Nery Costa, J. R. Santos Jr., V. Zucolotto, C. Eiras and J. R. Leite, Nanomed.: Nanotechnol., Biol. Med., 2009, 5, 352–358 CrossRef CAS.
- A. Shukla, K. E. Fleming, H. F. Chuang, T. M. Chau, C. R. Loose, G. N. Stephanopoulos and P. T. Hammond, Biomaterials, 2010, 31, 2348–2357 CrossRef CAS.
- I. Izquierdo-Barba, M. Vallet-Regi, N. Kupferschmidt, O. Terasaki, A. Schmidtchen and M. Malmsten, Biomaterials, 2009, 30, 5729–5736 CrossRef CAS.
- K. Hilpert, M. Elliott, H. Jenssen, J. Kindrachuk, C. D. Fjell, J. Korner, D. F. Winkler, L. L. Weaver, P. Henklein, A. S. Ulrich, S. H. Chiang, S. W. Farmer, N. Pante, R. Volkmer and R. E. Hancock, Chem. Biol., 2009, 16, 58–69 CrossRef CAS.
- M. Bagheri, M. Beyermann and M. Dathe, Antimicrob. Agents Chemother., 2009, 53, 1132–1141 CrossRef CAS.
- S. A. Onaizi and S. S. Leong, Biotechnol. Adv., 2011, 29, 67–74 CrossRef CAS.
Footnote |
| † These two authors contributed equally to the paper. |
| This journal is © The Royal Society of Chemistry 2012 |