Ionic liquids designed for advanced applications in bioelectrochemistry
Kyoko
Fujita
,
Kenichi
Murata
,
Miyuki
Masuda
,
Nobuhumi
Nakamura
and
Hiroyuki
Ohno
*
Department of Biotechnology, Tokyo University of Agriculture and Technology, Koganei, Tokyo, 184-8588, Japan. E-mail: ohnoh@cc.tuat.ac.jp; Fax: +81 42 388 7024; Tel: +81 42 388 7024
First published on 6th February 2012
Abstract
Recent applications of ionic liquids (ILs) as sustainable media in biochemistry and bioelectrochemistry are reviewed. The use of ILs as solvents for biopolymers such as proteins is particularly important in bioelectrochemistry. Maintenance of the higher-ordered structure of proteins after dissolution in IL is essential for the applications envisaged. The affinity between ILs and proteins is discussed in relation to the design of new solvents. IL-modified electrodes are another important topic for bioelectrochemistry. Some bio-related facets of ILs are also reviewed for the understanding of the usefulness of ILs for this field. The use of ILs in bioscience has led to the development of new types of biodevices, and has also been mentioned.
 Kyoko Fujita (back, left), Miyuki Masuda (back, middle), Kenichi Murata (back, right) Nobuhumi Nakamura (front, left), Hiroyuki Ohno (front, right) | Kyoko Fujita received her PhD degree (2004) from Tokyo University of Agriculture and Technology (TUAT; Japan). During her PhD studies in Prof. Ohno’s group, she received a Research Fellowship for Young Scientists at the Japan Society for the Promotion of Science (JSPS). Before working at TUAT, she worked as a postdoctoral research fellow with Prof. D. R. MacFarlane and Prof. M. Forsyth at Monash University (Australia) until 2006. She is a postdoctoral research fellow in TUAT. Her research interests are bioelectrochemistry especially ionic liquids as protein solvents and the effective water state for bioscience. |
Kenichi Murata received his BEng (2006), MEng (2007), and PhD (2009) degrees in biotechnology all from TUAT. This was followed by a JSPS postdoctoral research fellow under the direction of Prof. N. Nakamura until 2010. His thesis was focused on enzymatic biofuel cells. In 2010, he started research at Sony Corporation (Tokyo, Japan). His scientific interests are focused on bioelectronic devices. |
Miyuki Masuda received her BEng (2008) and MEng (2010) degrees from TUAT. She is currently a PhD candidate in the Department of Biotechnology in TUAT under the direction of Prof. N. Nakamura. Her PhD research is focused on ethanol–O2 enzymatic biofuel cells. |
Nobuhumi Nakamura received his BSc (1988) and MSc (1990) degrees in Chemistry from Tokyo University of Science (Japan) and his PhD degree from Osaka University (Japan) in 1993. He spent 5 years as a postdoctoral research fellow (at Osaka University as a JSPS postdoctoral fellow, at Oregon Graduate Institute (USA), and at Kyushu University (Fukuoka, Japan)). In 1998 he moved to TUAT as a lecturer and then he was promoted to an Associate Professor in 2003. His research interests are focused on electron transfer of enzymes and biodevices. |
Hiroyuki Ohno received a PhD degree in 1981 from Waseda University, Tokyo, Japan. After working at Waseda University and Case Western Reserve University (USA), he moved to TUAT as an Associate Professor in 1988. He was promoted to professor in 1997. His recent research activities are concentrated on the science of ionic liquids, especially the design of functional ionic liquids. He has published more than 100 papers and reviews on ionic liquids in this decade. He served as director of the university library, Vice Dean and Dean of his university. FRSC since 2008. |
1. Introduction
Chemists are now familiar with the words “ionic liquid” as these compounds have totally different properties from molecular liquids. The key point of ILs is their non-volatile nature. Reports on ILs are still increasing. The research areas with ILs gradually shifted from electrochemistry to biochemistry and bioelectrochemistry. The performance of bioelectrochemical devices can be improved significantly by using ILs instead of volatile molecular liquids. We also envisage ILs as superior alternatives to volatile organic solvents. In the early stages, research on ILs focused on the two fields of organic synthesis and electrochemical technology. Many interesting findings have already been summarised in various books and reviews.1–6 In recent years, research on ILs has diversified into various areas, including biotechnology and nanotechnology. In the field of biotechnology, extensive studies have been made on biotransformation,7–13 two-phase extraction systems,14 and biomass treatment.15–19 Electrodeposition of metals20–23 and synthesis of metal nanoparticles24,25 are major topics in nanotechnology. As the boundary between electrochemistry and bioscience is another important field for ILs, this review concentrates on research related to both bioscience and electrochemistry.2. Advanced properties of ILs
2.1 ILs for electrochemical devices
ILs are melts of organic salts that exist in the liquid state over a wide temperature range, and sometimes below room temperature. These compounds are generally composed of an organic cation and an organic or inorganic anion. The presence of a bulky and structurally asymmetric cation gives rise to lower cohesive energies for crystalline forms than in inorganic salts such as NaCl, and guarantees the ionic state of the liquid phase. The interaction between the ions that make up the IL are strong, long-range Coulomb forces and are responsible for the remarkable physicochemical properties of ILs, such as low melting point, almost zero pressure of saturated vapour, non-combustibility, and a wide electrochemical potential window. These properties make it possible to improve the durability and safety of electrochemical devices, extending the range of operating temperature and enabling improvements in power and energy density.6 Therefore, ILs are superior alternatives for electrolyte solutions used in electrochemical devices such as electric double layer capacitors, fuel cells, lithium batteries and solar cells.ILs are generally redox-robust. They typically have a wide potential window, which is one of the most important advantages of ILs as electrolytes.26 Potential windows of 4.5–5.0 V have been reported for ILs, and even an electrochemical potential window of 7.0 V for 1-butyl-3-methylimidazolium tetrafluoroborate ([C4mim]BF4) and 1-butyl-3-methylimidazolium hexafluorophosphate ([C4mim]PF6).27 These values are not reached by ionic liquids that contain water, however. Addition of more than 3 wt% water to [C4mim]BF4 and [C4mim]PF6 decreased the electrochemical window by at least 2.0 V,28 although water reduced the viscosity and thereby increased the conductivity. In bioelectrochemistry, a small amount of water is often added to ILs. In particular, the electrodes described below in the sub-section “ILs for modified electrodes” operate in contact with aqueous solution.29 When using ILs with water, the resulting change in physicochemical properties should be taken into account.
2.2 ILs for biosciences
The use of ILs as a protein matrix is the main spur for the development of ILs for application in the biosciences. ILs are currently being studied as a novel matrix, so that proteins can be used in a wider range of conditions,8,30 to improve reaction efficiency,31 in new devices,32 and for the preservation of biomaterials.33,34 Proteins generally are handled in a buffer solution. Concentrated salt solutions are not used for dissolving proteins, because the concentration of ions leads to denaturation of the protein. In fact, proteins do not dissolve readily in general ILs.30,35,36 When proteins dissolve in ILs, structural changes have been observed.36–38 A homogeneous solution of proteins in ILs is not essential for the study of ILs as protein solvents, because it has been reported that enzymes dispersed in ILs exhibit much greater activity than in buffer.30,39 Protein dissolution is attractive from the viewpoint of reaction efficiency and application. It is important to understand the properties of ILs which make them capable of dissolving proteins in a homogeneous manner. This would further the capability and durability of bioelectrochemical devices in which ILs are used as an alternative electrolyte solution.| Cations | Anions | ||
|---|---|---|---|
| a Welton et al. described that the abbreviation of methyl groups on both imidazolium and pyrrolidinium cations should be “C1”, not “m”. We referred to a methyl group only here, so the methyl group was abbreviated to “m”. | |||
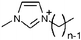
|
1-Alkyl-3-methylimidazoliuma | BF4− | Tetrafluoroborate |
| [Cnmim] (n = 2–12) | BF4 | ||
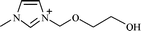
|
1-(2-Ethoxyethanol)-3-methylimidazoliuma | PF6− | Hexafluorophosphate |
| [et2mim] | PF6 | ||
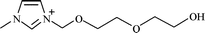
|
1-[2-(2-Ethoxy)-ethoxyethanol]-3-methylimidazoliuma |
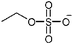
|
Ethylsulfate |
| [et3mim] | [C2SO4] | ||

|
N-Alkyl-N-methylpyrrolidiniuma | Br− | Bromide |
| [Cnmpyrr] (n = 3, 4) | Br | ||

|
Cholinium | Cl− | Chloride |
| [ch] | Cl | ||
|
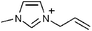
1-Allyl-3-methylimidazolium |

|
Dihydrogen phosphate |
| [amim] | [dhp] | ||
|
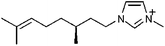
1-Methyl-3-(2,6-(S)-dimethylocten-2-yl)-imidazolium |

|
Bis(trifluoromethanesulfonyl)imide |
| [mdim] | [NTf2] | ||

|
1-Octylpyridinium | HCOO− | Formate |
| [C8pyr] | HCOO | ||

|
1-Allyl-3-ethylimidazolium |

|
Methylsulfate |
| [aeim] | [CH3OSO3] | ||

|
1-Ethylimidazolium | ||
| [C2im] | |||
This section looks at the design of ILs; chemical modification of proteins is introduced by focusing on the dissolving of proteins.
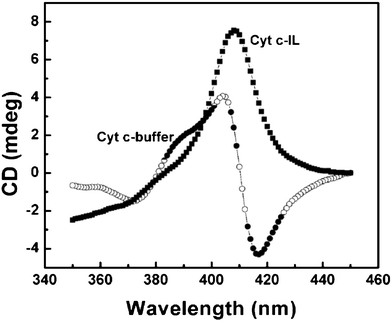 | ||
| Fig. 1 Soret region CD spectrum of cyt c in 0.01 M, pH 7 phosphate buffer, and IL.40 | ||
Polarity is an important factor in the dissolving of various materials in ILs. The polarity of an IL is generally estimated via solvatochromism of some organic dyes.41–43 The hydrogen bond donor ability (α value), hydrogen bond acceptor ability (β value) and the dipolarity–polarisability (π* value) were calculated from the affinity between an IL and several dyes, inferred from observations of the maximum absorption wavelength. Anions in ILs giving high β values are reported to facilitate the dissolution of cellulose; this would be an important practical breakthrough.44 ILs with a high β value have been recognised to break intra- and inter-molecular hydrogen bonds in cellulose fibres. Such highly polar ILs also dissolved silk and other biopolymers.45 The maintenance of protein structure after dissolution is important in retaining function, yet the change to the higher ordered structure generally causes loss of function when proteins dissolve in such highly polar solvents.
It has been reported that dried polar ILs having a chloride anion dissolve cyt c.46 Cyt c was mixed in several ILs and was stirred for 30 min at 80 °C. Cyt c was dissolved in 1-butyl-3-methylimidazolium chloride ([C4mim]Cl) at a final concentration of 10 mM, and in 1-butyl-3-methylimidazolium bromide ([C4mim]Br) at concentrations of 10 mM and 0.5 mM. The solubility of cyt c has been found to depend on the cation as well as the anion. To investigate the factors influencing solubility, the polarity of ILs was estimated in terms of the Kamlet–Taft parameters. Polar ILs having β > 0.7 proved effective in dissolving cyt c. (Fig. 2). No clear correlation between the value of α and the solubility of cyt c was found. Against this, correlation between the values of π* and β and the solubility of cyt c was observed. These results suggest that β and π* are significant parameters for the dissolution of cyt c. A Soret band was observed at 409 nm for cyt c in these ILs by UV-vis spectroscopy. The charge transfer (CT) band at 695 nm was not confirmed. This data suggests that the electronic state of the heme was not changed, but that there is a perturbation or loss of the axial sulphur ligand of the heme. Tuning of the polarity is a valuable procedure for selecting ILs to dissolve proteins.
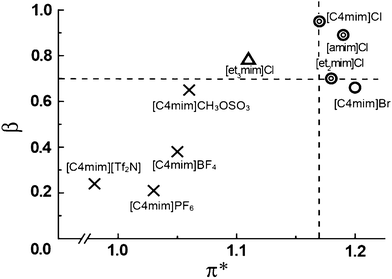 | ||
| Fig. 2 Solubility dependence of cyt c on the π* and β values of the ILs (X, insoluble; △, < 0.05 mM; ○, 0.5 mM; and ⊚, 10 mM).46 | ||
Improved stability of protein was observed in hydrated ILs; this is impossible in an aqueous phase. Long-term stability of cyt c stored in hydrated IL containing 20 wt% water (three water molecules per ion pair) has been confirmed. The IL was composed of cholinium ([ch]) cations and dihydrogen phosphate ([dhp]) anions. Upon keeping cyt c in hydrated form [ch][dhp] (Hy[ch][dhp]) for three weeks at room temperature, about 90% of redox activity for the freshly prepared buffer solution was observed.48 The secondary and tertiary structures of cyt c dissolved in several hydrated ILs were investigated using circular dichroism (CD) spectroscopy and Raman spectroscopy. The resulting spectra suggested that the vicinity of the active site was slightly changed in most hydrated ILs, such as N-butyl-N-methyl pyrrolidinium dihydrogen phosphate ([C4mpyrr][dhp]). Cyt c dissolved in Hy[ch][dhp] maintained a higher ordered structure, as in buffer (Fig. 3). Cyt c dissolved in Hy[ch][dhp] retained 70% of activity after storage at room temperature for 18 months.48 The thermal stability of cyt c was also improved, unlike in buffer solution.
![CD spectra of dissolved cyt c in (a) [C4mpyrr][dhp], (b) Hy[ch][dhp] and (c) buffer.51](/image/article/2012/RA/c2ra01045c/c2ra01045c-f3.gif) | ||
| Fig. 3 CD spectra of dissolved cyt c in (a) [C4mpyrr][dhp], (b) Hy[ch][dhp] and (c) buffer.51 | ||
The solubility in Hy[ch][dhp] of proteins other than cyt c has also been investigated. Horseradish peroxidase (HRP), hemoglobin (Hb), myoglobin (Mb), azurin, pseudoazurin and ascorbate oxidase were added to Hy[ch][dhp] containing 20 wt% water. Hemoglobin and myoglobin dissolved only slightly in Hy[ch][dhp]; the other proteins dissolved readily.50,51 Albumin, with a high helix content of about 70%, was almost completely insoluble. The reason is still unclear. Although it is difficult to dissolve any proteins in Hy[ch][dhp], dissolution of target proteins should be possible by suitable design of the ion structure and by controlling the water content.
3. Application of ILs in biodevices
3.1 ILs as a matrix for proteins
ILs should be capable of acting as a device matrix such as a battery electrolyte, since they are thermally and chemically stable compared with traditional matrices.1,3,60 In regard to biodevices,61,62 the availability of ILs should improve the conductivity and the thermal and long-term stability of proteins. In this section, recent studies of the electrochemistry of proteins in IL solvents will be reviewed.The redox reaction of PEO-modified cyt c dissolved in 1-butyl-3-methylimidazolium bis(trifluoromethanesulfonyl)imide ([C4mim][NTf2]) has been analysed by optoelectrochemistry.52 Optical waveguide spectroscopy is an advanced method for detecting and analysing redox species dissolved in ILs.2 Progress in the electron transfer reaction of PEO-modified cyt c dissolved in [C4mim][NTf2] was observed only after addition of KCl. The role of the chloride anion has been estimated as a charge compensator for heme.54
The redox reaction of cyt c dissolved in a pure polar IL was also analysed by optical waveguide spectroscopy.46 Cyt c was dissolved in 1-allyl-3-methylimidazolium chloride ([amim]Cl), which is a highly polar IL having low viscosity among the chloride salts. After dissolution of cyt c at 80 °C, the redox response was analysed at room temperature by optical waveguide spectroscopy. A spectral shift of the Soret band was observed with an applied potential. A similar spectral shift was observed even at 140 °C. This is a typical example of the fact that ILs provide a thermally stable matrix for proteins due to their relatively high viscosity; the thermal motion of molecules is less vigorous than in ordinary liquids with lower viscosity.
The redox reaction has been studied of protein dissolved in a mixture of IL and water using an electrochemical method, thin layer cyclic voltammetry.63 Direct and rapid redox reaction of Hb or Mb was observed in an aqueous solution containing 1 M [C4mim]BF4. The IL was observed to play a key role in the electron transfer. In general, no redox reaction of Hb was observed in aqueous KCl solution containing low concentrations of [C4mim]BF4. A higher [C4mim]BF4 concentration gives a better signal-to-noise ratio, up to 2 M. A good redox response was observed by using some IL, although it is difficult to use IL as a solvent.
Wang et al. reported the redox responses of heme proteins immobilised on the electrode with agarose hydrogel films in [C4mim]PF6 and [C4mim]BF4.65–67 Direct electron transfer (DET) reactions were analysed for Hb, Mb, peroxidase, cyt c and catalase.65 The electrocatalytic reaction of these heme proteins entrapped in agarose hydrogel on the electrode was also investigated using trichloroacetic acid and tert-butyl hydroperoxide in [C4mim]PF6. Electrocatalytic reduction of H2O2 by heme proteins entrapped in agarose hydrogel films was also observed. A small amount of water in [C4mim]BF4 is necessary to maintain the electrochemical activities of these heme proteins.66 The authors concluded that addition of water to the hydrophilic IL was necessary to realise the activity of proteins entrapped in agarose hydrogel. However, as mentioned by Gorton,68 it is important to check the redox potential and the state of proteins immobilised on the electrode in order to gain accurate insight into the redox activity. A desorption of heme from the protein might occur during immobilisation of proteins on the electrode.
DET reaction of heme proteins and catalytic activity has also been investigated using cetyltrimethylammonium bromide films containing single-wall carbon nanotubes (SWCNTs–CTAB).69 A pair of well defined and nearly reversible responses was observed in [C4mim]PF6. The electrode was prepared by casting the mixture of protein solution and SWCNTs–CTAB, followed by drying at room temperature. The resulting electrode was expected to retain the water molecules in the SWCNTs–CTAB film. As a result of the hydrophobicity of [C4mim]PF6, water molecules were believed to be trapped at the electrode surface, and contributed to the enzymatic reaction. When proteinase K–SWCNTs conjugates were introduced into ILs containing 5.0% (v/v) water, greater enzymatic activity was observed in hydrophobic ILs.70 The enzymatic activity in highly hydrophobic ILs was about 10 times greater than in hydrophilic ILs.
A similar effect of hydrophobicity of ILs on the electron transfer reaction of cyt c3 has been reported.71 The redox reaction of cyt c3 immobilised on pyrolytic graphite by casting was investigated in [C2mim]BF4, [C4mim]BF4, and [C2mim][NTf2]. A better response was observed in hydrophobic [C2mim][NTf2], and the current was almost 30 times greater than that in 4-(2-hydroxyethyl)piperazine-1-(ethanesulfonic acid) (HEPES) buffer, as seen in Fig. 4. This result suggests that a hydration layer of some sort was formed around proteins in hydrophobic ILs. No increase in the peak current and potential shift was observed when an electrode modified with cyt c3 was directly transferred from HEPES buffer to [C2mim][NTf2]. However, no such increase of redox current observed in [C2mim][NTf2] was observed in [C2mim]BF4 or [C4mim]BF4 even though the redox response was confirmed. The signal in [C4mim]BF4 was not stable with time, and it shifted gradually toward the positive potential side. Based on these results, hydrophilic ILs are considered to absorb water and destroy hydrate shells around proteins, as hydrophilic organic solvents did. This might explain the denaturation of proteins. These results are closely related to the kosmotropicity. The redox response of cyt c3 in [C4mim]BF4 was greater than that in [C2mim]BF4, because the [C4mim] cation was less chaotropic than [C2mim]. Although the direct electron transfer reaction of cyt c3 was confirmed in ILs, H2 oxidation of hydrogenase was not observed in the three ILs described above, [C2mim]BF4, [C4mim]BF4 and [C2mim][NTf2]. A small amount of water was believed to exist so as to realise the direct electron transfer reaction of proteins immobilised on the electrode. More hydrophobic ILs are suitable as the matrix for electron transfer reaction of proteins. Water saturated hydrophobic ILs have the capability to act as a matrix for protein electrochemistry.
![Cyclic voltammograms at 20 mV s−1 of cyt c3 adsorbed onto the PG electrode in RTILs: [C4mim]BF4 (black line), [C2mim][NTf2] (dark grey line), [C2mim]BF4 (fine grey line).71](/image/article/2012/RA/c2ra01045c/c2ra01045c-f4.gif) | ||
| Fig. 4 Cyclic voltammograms at 20 mV s−1 of cyt c3 adsorbed onto the PG electrode in RTILs: [C4mim]BF4 (black line), [C2mim][NTf2] (dark grey line), [C2mim]BF4 (fine grey line).71 | ||
In terms of water removal from proteins, hydrophobic ILs are preferable for electrochemistry. Proteins should be active when sufficient (but not excess) water molecules are maintained around them. ILs containing a certain amount of water, so-called hydrated ILs, have accordingly been analysed as solvents for proteins, as mentioned above. A well-defined redox response was observed in hydrated ILs. Hy[ch][dhp] exhibited good affinity with some proteins, and an improvement in thermal and long-term stability of the dissolved proteins was confirmed.48,51 In Hy[ch][dhp], a redox response of enzymes was also observed.72
A fructose dehydrogenase (FDH)-immobilised gold nanoparticle electrode was studied in Hy[ch][dhp]. The electron transfer reaction, based on the electrochemically catalysed oxidation of D-fructose via direct electron transfer between the active center of FDH and the electrodes, was clearly detected in Hy[ch][dhp] (Fig. 5 (a)). No redox response of FDH was observed in the absence of fructose (see Fig. 5 (b)). Similarly, a CDH-immobilised nanoparticle electrode gave rise to a catalytic current in the presence of cellobiose. CDH certainly catalysed the oxidation of cellobiose through a DET reaction between the active site of CDH domains and the electrode in Hy[ch][dhp]. Furthermore, substrate inhibition was observed, the same as that in the water phase. The long term stability of the bioelectroactivity of the CDH immobilised electrode in Hy[ch][dhp] was investigated. The activity of CDH immobilised on the electrode was maintained after three weeks at room temperature in Hy[ch][dhp], although no reaction was detected in the water phase. These results indicate that Hy[ch][dhp] is a candidate for the construction of durable biodevices.
![Cyclic voltammograms of FDH-modified MET–AuNP electrode in the presence of (89 mM) (a) and absence (b) of d-fructose in Hy[ch][dhp].72](/image/article/2012/RA/c2ra01045c/c2ra01045c-f5.gif) | ||
| Fig. 5 Cyclic voltammograms of FDH-modified MET–AuNP electrode in the presence of (89 mM) (a) and absence (b) of D-fructose in Hy[ch][dhp].72 | ||
In the protein-immobilised system, many parameters were not controlled well, such as the protein orientation on the electrode surface. Selection of the mediator for the DET reaction of proteins was a further issue. In the fixing system, however, the effect of diffusion was eliminated even in ILs having high viscosity. The construction of a stable device should prove possible
3.2 ILs for modified electrodes
ILs can be used not only as the supporting electrolyte for the electrochemical process, but also as the modifier for chemically modified electrodes. The high ionic conductivity of ILs makes them preferable for electrode materials in sensing devices. Microdroplets and thin film deposits of 1-methyl-3-(2,6-(S)-dimethyloctene-2-yl)-imidazolium tetrafluoroborate ([mdim]BF4) on a pyrolytic graphite electrode surface were first studied by Wadhawan et al.73 Subsequently, many papers have been published about IL-modified electrodes for sensing devices.![SEM images of (a) CPE and (b) IL-CPE 50 : 50 of graphite–[C8pyr]PF6 electrodes.76](/image/article/2012/RA/c2ra01045c/c2ra01045c-f6.gif) | ||
Fig. 6 SEM images of (a) CPE and (b) IL-CPE 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 of graphite–[C8pyr]PF6 electrodes.76 :50 of graphite–[C8pyr]PF6 electrodes.76 | ||
Both biocompatibility and a large surface area-to-volume ratio for high enzyme loading are necessary for the electrode material to immobilise enzymes. To realise the stable immobilisation of enzymes in biocompatible polymers, nanoparticles and/or other supporting materials have been added, although the immobilisation of enzymes without any modifier except for IL has also been reported.81,82 Many biocompatible materials immobilise enzymes when incorporated in IL-modified electrodes; for instance, polymers,83–91 polymerised ILs,92–95 sol–gel derived glass,96 sol–gel ILs,97 mesoporous molecular sieve MCM-41,98 CaCO3 nanoparticles,99 and clay.100–103 These films can retain the native structure of enzymes and enhance the electrochemical response at electrodes. The biocompatibility of these composite films is most commonly assessed by UV-vis absorption spectroscopy and FT-IR spectroscopy. Lu et al.83 showed that a chitosan–[C4mim]BF4–Hb composite is more thermally stable than a chitosan–Hb mixture, according to thermogravimetric analysis data. This is a good example of an IL which provides high ionic conductivity and further benefits to a composite film.
It is possible to realise a large surface area for high enzyme loading by using electro-conductive nanomaterials. Single-walled carbon nanotubes (SWCNTs) can be dispersed in imidazolium-based ILs by mechanical milling so as to form a thermally stable gel,104 although SWCNTs are easily entangled with each other to form agglomerates. The SWCNTs in the gel exist as a three-dimensional network of considerably untangled and much finer bundles that are physically cross-linked due to cation–π and π–π interactions between the imidazolium cations and CNTs.104,105 In bioelectrochemistry, SWCNT,106–108 MWCNT93,109–118 and graphene95,119–122 are commonly used to fabricate such composites. Furthermore, metal nanoparticles123–128 and several inorganic semiconductive nanomaterials, such as NiO,129 MnO2,130 CoO2,131 V2O5,132 Fe2O3,133 and CdS,134 have also been used to form IL-nanomaterial composites. The extremely low volatility of ILs makes it possible to study their morphology by SEM; this is not possible for other molecular liquid-modified electrodes. It has been reported that IL–carbon nanomaterial composites at electrodes show a unique structure and form a relatively uniform surface.115,135
IL-modified electrodes used in an aqueous solution have been considered. Since the solubility of hydrophobic ILs in water is much smaller than that of water in the IL, the IL-modified electrodes function stably. Hydrophilic ILs have also been used, and the resulting electrodes are stable, at least on a voltammetric timescale. This may be a result of the adsorption of ILs on the electrode. The stability of IL-modified electrodes should therefore be discussed carefully. To prevent leaking of ILs, polymerised IL-wrapped CNTs were designed and fabricated by direct polymerisation of IL monomers in the presence of CNTs.93 The resulting stable IL-functionalised composites are considered to be based on the strong π–π stacking interaction between the imidazolium ion moieties and the CNTs.136
Biodevices, such as biosensors and biofuel cells based on enzymatic reactions, consist of two components: a bioreceptor and a transducer. The bioreceptor is an enzyme that recognises the target analyte, whereas the transducer converts the recognition at the molecular level into a measurable signal. The transducer system should be selected and designed by taking into account aspects of the enzymatic reaction. In enzymatic biosensors based on an IL-modified electrode, the amperometric technique is commonly used to measure the signal, although a transistor-type sensing system has recently been reported.137
For some enzymes, such as hydrolase (including organophosphorus hydrolase,113 lipase,112 acetylcholine esterase116) and some oxidases (including laccase102,103,125,138 and polyphenol oxidase101), the products are redox active and can easily be detected at the substrate electrode. In particular, organophosphorus hydrolase catalyses hydrolysis of p-nitrophenolphosphate to p-nitrophenol, and the resulting p-nitrophenol, is electrochemically detectable.113
It has been reported that IL–nanomaterial composites electrochemically catalyse the electrochemical reaction of biomolecules with low overpotentials. The lowered overpotential is attributed to the properties of the nanoparticles, but the IL also has inherent catalytic activity.78 NADH and H2O2 can be detected with a low overpotential at IL-modified electrodes. Since NADH and H2O2 are involved in many enzymatic reactions, it is important to detect them with a lower overpotential. The combination of H2O2-producing oxidases and IL–nanomaterial composites provides a biosensor for the corresponding substrate of the oxidases. Many IL-modified electrodes based on materials that reduce H2O2 have been reported for the detection of glucose,92,93,95,107,110,119 cholesterol,126 and glutamate127 by applying the corresponding oxidases.
Many oxidoreductases can communicate electrochemically with the electrode via a reversible redox active molecule known as an “electron transfer mediator”. In this system, mediators and/or cofactors should be immobilised at the electrode surface with the enzyme. HRP was immobilised with ferrocene as a mediator in a IL-based sol–gel matrix.96 The efficient mediated bioelectrocatalysis of NADH-dependent dehydrogenase is difficult. This is because it is not easy to immobilise NADH on the electrodes together with enzymes, and NADH often has to be added to the analyte solution.115 Yu et al. successfully immobilised NADH by developing a NADH-based IL and forming a SWCNT–IL bucky gel as shown in Fig. 7.108
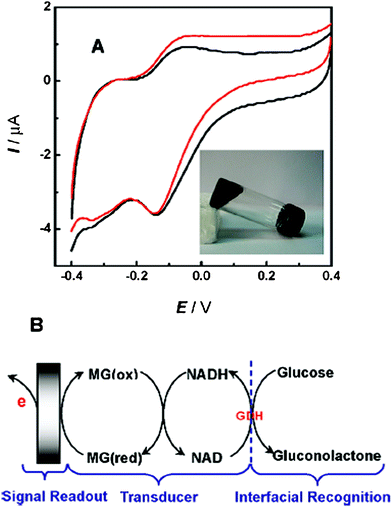 | ||
| Fig. 7 (A) Bioelectrocatalytic oxidation of glucose at the multifunctional gel based biosensor in 0.1 M phosphate buffer (pH 7.0) in the absence (black current) or presence (red curve) of 40 mM glucose. Scan rate, 1 mV s−1. Inset, digital picture of the prepared multifunctional gel. (B) Illustration of the reaction schemes involved in the bioelectrocatalytic oxidation of glucose at the multifunctional gel based biosensor.108 Methylene green (MG) was used together with NADH as transducers. | ||
DET reactions, in which enzymes directly exchange electrons with electrodes without a mediator, can be performed using some oxidoreductases and redox proteins. However, the DET reaction between the enzymes and the traditional bare working electrode is problematic, because of deeply buried electroactive prosthetic groups in the apoprotein domain. Modified electrodes have therefore been designed to provide a suitable microenvironment for enzymes and facilitate DET reactions. In the last few years, ILs have also been used to fabricate DET-type electrodes as components for modified electrodes. IL-modified electrodes for Hb,83,99,111 Mb,82,106,139 cyt c,106 and HRP109,132 have all been reported to improve the DET reaction toward biosensors. Multicopper oxidases such as laccase and bilirubin oxidase, which oxidise a wide variety of compounds with concomitant oxygen-reduction to H2O, have also been incorporated into IL-modified electrodes to facilitate the DET reaction.97,117,140 Electrodes with multicopper oxidase can be used both as a sensing component for their substrate, as phenols (electron donor) and oxygen (electron acceptor), and as a catalyst of oxygen reduction for biofuel cells. IL–MWCNT bucky-paper (BP) facilitates the diffusion of oxygen more readily than agglomerated MWCNT, and the composite facilitates the DET reaction of laccase as shown in Fig. 8.118
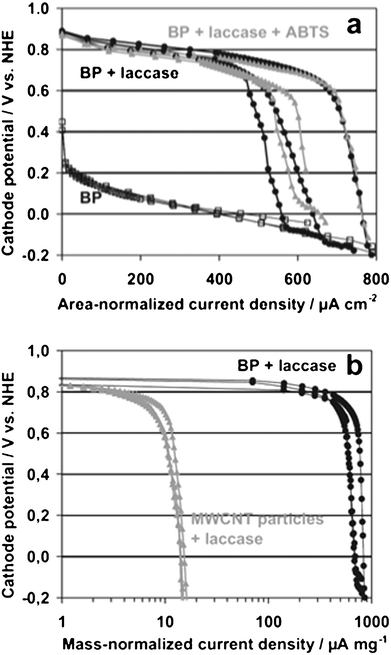 | ||
| Fig. 8 Galvanostatic polarisation curves of different electrodes in O2-saturated citrate buffer. (a) 30 μM thick bucky-paper cathodes without laccase (black squares), with adsorbed laccase (DET, black circles), and 2,2′-azinobis(3-ethylebenzthiazoline-6-sulfonic acid) diammonium salt (ABTS) (mediated electron transfer, gray triangles). (b) Comparison of different MWCNT-based electrodes with the current density normalised to the mass of carbon nanotubes (note the logarithmic scale). Electrodes fabricated as bucky-paper (black circles) and by the conventional technique (gray triangles) were investigated with adsorbed laccase in the absence of a mediator.118 | ||
The study of IL-modified electrodes for biofuel cells is still in its early stages.61,141 The design of ILs for biofuel cell is interesting from the viewpoint of their high ionic conductivity. ILs have tremendous potential as solvents for biofuel cells.61
As stated, IL-modified electrodes for biodevices have been developed by hybridising enzymes, ILs, and polymers or nanomaterials. Because of the synergetic effects of electro-conductive materials together with biocompatible polymers and ionic conductive ILs, biosensors developed with a composite of these materials have fast responses, high sensitivity, low detection limits, and high stability. The value of ILs for electrode modification has been recognised, and led to a rapid increase in the number of papers on this subject.29
4. Application of ILs in electrochemical devices using biopolymers
Biopolymers, such as enzymes and proteins, have been widely studied as transducers for amperometric biosensors and biofuel cells. Some biopolymers have also been regarded as functional materials. There are many reports on the preparation of composites of ILs and biopolymers. This section reviews recent reports of composites as polymer electrolytes for electrodevices.To prepare polymer electrolytes composed of ILs and biopolymers, many biopolymers have been utilised, including gelatin,142 DNA,143–148 and several polysaccharides such as cellulose,149–151 chitosan,152,153 chitin,154 and agarose.155,156 These biopolymer-based composites possess high mechanical strength, flexibility, and biodegradability. In addition, the added ILs gave high ionic conductivity to the composites. These properties are necessary for electrolyte-substituent membranes in electrodevices such as actuators, lithium ion batteries, and solar cells.157 Most of these polymer electrolytes were prepared by simple mixing of biopolymers with ILs. Another method includes preparing IL-like domains in biopolymers through the neutralisation of charged groups in the biopolymers.145,148 These ionic liquidised biopolymers provide novel applications as ion conducting materials.
4.1 Composites comprised of ILs and saccharides
In the field of electrodevices, cellulose has been regarded as a passive element and has been used as a separator in solid-state batteries.158 As cellulose does not dissolve in conventional molecular liquids, it is difficult to prepare cellulose-based composites. Against this, it is known that ILs having chloride anions are good solvents of cellulose.159 As mentioned above, some ILs are expected to dissolve cellulose, based on their high polarity. Recently, a few polar ILs have been designed to dissolve cellulose under mild conditions.44,160 These experiments are furthering the preparation of cellulose-based composites using ILs as solvents.161A supercapacitor has been prepared based on a polymer electrolyte composed of cellulose and MWCNTs, using [C4mim]Cl.149 This IL was used both as a solvent for cellulose and as an electrolyte in the resulting composite. The composite has high mechanical strength and flexibility. According to electrochemical measurement, the specific capacitance of this composite is 22 F g−1. The power density of this supercapacitor is 1.5 kW kg−1, which is comparable to commercially available ones (0.01–10 kW kg−1). This supercapacitor could be operated from −78 to +150 °C, a much wider range of temperatures than for existing capacitors (from −50 to +85 °C).
The incorporation of an organoboron unit improves the selectivity of target cation transport in polymer electrolytes, and/or enhances the ionic conductivity of the matrix.162 It is also known that polysaccharides, including cellulose, bind boric acid via the condensation reaction.163 Incorporation of a boric acid unit into cellulose using this reaction is a useful way to prepare highly ion conductive materials.150 As shown schematically in Fig. 9, an organoboron gel composed of 1-allyl-3-ethylimidazolium formate ([aeim]HCOO), boric acid, and lithium hydroxide had high ionic conductivity, of 1.98 × 10−3 S cm−1 at 30 °C, comparable to that of pure IL (1.74 × 10−3 S cm−1).150
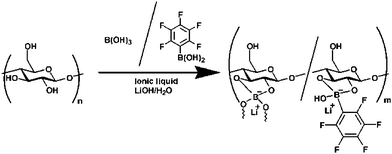 | ||
| Fig. 9 Synthesis of an organoboron ion gel.150 | ||
As another way to improve the lithium ion conduction of cellulose based-composites, cellulose triacetate (CTA) was used as a supporting polymer electrolyte.151 ILs composed of cations having an ester group have been reported to improve lithium ion conduction.164 The use of CTA as a supporting polymer for the preparation of polymer electrolyte affects the lithium ion conduction because CTA contains functional ester groups. The resulting polymer electrolyte composed of CTA, lithium bis(trifluoromethanesulfonyl)imide (Li[NTf2]), and N-methyl-N′-propylpyrrolidinium bis(trifluoromethanesulfonyl)imide ([C3mpyrr][NTf2]) exhibited high ionic conductivity (10−3–10−4 S cm−1). FT-IR and conductivity measurements confirm that the ester group of CTA is responsible for enhancing the dissociation of Li[NTf2].
Other biopolymers have been used as supporting scaffolds of polymer electrolytes containing ILs. ILs generally have high intrinsic ionic conductivity. An ion gel prepared with agarose was reported to have ionic conductivity almost as high as the neat ILs.155,156 This is because the agarose-based ion gel could be prepared by adding only a very small amount of agarose to ILs. The addition of a small amount of gelator facilitates the preparation of highly ion conductive gels with ILs.
4.2 ILs and DNA composites
There is interest in using DNA, which is a representative biopolymer, to create functional materials for electronics. Using metallointercalator-tethered DNA, rapid photo-induced electron transfer was demonstrated over a distance of at least 40 Å between a ruthenium complex as a donor, and a rhodium complex as an acceptor.165 This finding encouraged the development of DNA-based electron-conducting nanowires.166 Apart from its notable role in genetics, DNA is a useful biopolymer, like polysaccharides or other polypeptides. By combining DNA with ILs, novel functional materials have been prepared for electrochemical devices. The dissolution of DNA in ILs and/or the interaction between DNA and ILs has also been investigated.167–170 Since DNA has a large number of heteroaromatic bases, it should have a high affinity with ILs. Accordingly, DNA films containing ILs were prepared by mixing DNA with ILs.144 The ionic conductivity of a DNA film containing 93.9 wt% 1-ethylimidazolium tetrafluoroborate ([C2im]BF4) was 5.05 × 10−3 S cm−1 at 50 °C, and is comparable to that of bulk [C2im]BF4 (2.02 × 10−2 S cm−1). The [C2im]BF4 used here was easily prepared by the neutralisation of ethylimidazole with tetrafluoroboric acid, which was sometimes expected to show proton conduction due to relatively labile protons.Ohno et al. prepared ionic liquidised DNA and applied it to ion conductive materials.144,148 Ionic liquidised DNA was synthesised by neutralisation of DNA with tetrafluoroboric acid.148 The resulting ionic liquidised DNA has low ionic conductivity. Of the four nucleic acid bases, only adenine and cytosine formed salts with BF4−. For this reason, a continuous ionic conduction path could not be formed in the DNA chains. To prepare a film with higher ionic conductivity, ionic liquidised DNA was mixed with [C2im]BF4. The ionic conductivity of the resulting film was improved by increasing the concentration of the added [C2im]BF4. The hydrogen bonding between complementary base pairs was broken by the neutralisation, however, and the double stranded helix structure of DNA was not maintained in the film. An attempt to maintain the double stranded helix structure of DNA after ionic liquidisation was made by neutralising phosphate anion groups with imidazolium cations.145 Additionally, the hydrophobicity of ionic liquidised DNA can be controlled by changing the alkyl chain length of the imidazolium cation.
A serious problem with organic dye-based electrochromic systems is the irreversible dimerisation of organic dyes. It is well known that π-conjugated heteroaromatic compounds form a complex with DNA by a process called intercalation or groove-binding.171 To realise the reversible redox reaction of organic dyes without dimerisation, DNA was used as a host for the dyes.143 However, N,N′-diheptyl-4,4′-bipyridinium dibromide (HV), which is a kind of dye, is considered to be not only groove-bound but also freely dissolved, and free HVs formed dimers, lessening the reversibility. To improve the binding stability between HVs and DNA, ionic liquidised DNA was used as a host matrix.146,147 Since ionic liquidised DNA needs to retain the double stranded-helical structure for use as a host for dyes, ionic liquidised DNA was prepared by exchanging the Na+, counter cation of DNA, with a alkylimidazolium cation.145 DNA having 1-dodecyl-3-methylimidazolium cations ([C12mim]-DNA) is water-insoluble. HVs form a stable complex with [C12mim]-DNA through groove-binding. HVs were complexed with ionic liquidised DNA on an indium-tin oxide coated glass electrode. Upon combining N,N,N′,N′-tetramethyl-p-phenylenediamine as an active material at the counter electrode, and the IL copolymer as ion sources to the ionic liquidised DNA, an electrochromic cell was constructed based on DNA. Upon applying potentials of just ±1.0 V, reversible colour changes were observed accompanied with the redox reaction of the complexed organic dyes.146 There was little decrease in the performance on the colour change with time due to compartmented dyes in the DNA.
5. Conclusion and future aspects
This review has summarised applications of ILs in certain fields of bioscience. ILs are widely used as solvents in many fields. In some cases ILs have simply been used to replace organic solvents. In bioscience, IL studies are starting to dominate the classical research fields of reaction solvents and/or electrolyte materials. Water is believed to be the best solvent for biomolecules, but we are confident that water will eventually yield to ILs in many areas of bioscience as a result of their remarkable characteristics. Further research should be based on what ILs alone can do that water or other solvents cannot, as more is learned about them.Acknowledgements
The authors were supported partly by a Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (No. 21225007).References
- Ionic liquids in synthesis, ed. P. Wasserscheid, T. Welton, Wiley-VCH, Weinheim, Germany, 2007 Search PubMed.
- Electrochemical aspects of ionic liquids, 2nd edn, ed. H. Ohno, John Wiley & Sons, Hoboken, NJ, USA, 2011 Search PubMed.
- T. Welton, Chem. Rev., 1999, 99, 2071–2083 CrossRef CAS.
- N. Jain, A. Kumar, S. Chauhan and S. M. S. Chauhan, Tetrahedron, 2005, 61, 1015–1060 CrossRef CAS.
- D. R. Macfarlane, M. Forsyth, P. C. Howlett, J. M. Pringle, J. Sun, G. Annat, W. Neil and E. I. Izgorodina, Acc. Chem. Res., 2007, 40, 1165–1173 CrossRef CAS.
- P. Hapiot and C. Lagrost, Chem. Rev., 2008, 108, 2238–2264 CrossRef CAS.
- U. Kragl, M. Eckstein and N. Kaftzik, Curr. Opin. Biotechnol., 2002, 13, 565–571 CrossRef CAS.
- R. A. Sheldon, R. M. Lau, M. J. Sorgedrager, F. van Rantwijk and K. R. Seddon, Green Chem., 2002, 4, 147–151 RSC.
- F. van Rantwijk, R. M. Lau and R. A. Sheldon, Trends Biotechnol., 2003, 21, 131–138 CrossRef CAS.
- S. Park and R. J. Kazlauskas, Curr. Opin. Biotechnol., 2003, 14, 432–437 CrossRef CAS.
- P. C. A. G. Pinto, M. L. M. F. S. Saraiva and J. L. F. C. Lima, Anal. Sci., 2008, 24, 1231–1238 CrossRef CAS.
- C. Roosen, P. Mueller and L. Greiner, Appl. Microbiol. Biotechnol., 2008, 81, 607–614 CrossRef CAS.
- P. D. de Maria and Z. Maugeri, Curr. Opin. Chem. Biol., 2011, 15, 220–225 CrossRef.
- S. Oppermann, F. Stein and U. Kragl, Appl. Microbiol. Biotechnol., 2011, 89, 493–499 CrossRef CAS.
- S. D. Zhu, Y. X. Wu, Q. M. Chen, Z. N. Yu, C. W. Wang, S. W. Jin, Y. G. Ding and G. Wu, Green Chem., 2006, 8, 325–327 RSC.
- L. Feng and Z.-I. Chen, J. Mol. Liq., 2008, 142, 1–5 CrossRef.
- H. Ohno and Y. Fukaya, Chem. Lett., 2009, 38, 2–7 CrossRef CAS.
- A. Pinkert, K. N. Marsh, S. Pang and M. P. Staiger, Chem. Rev., 2009, 109, 6712–6728 CrossRef CAS.
- M. Mora-Pale, L. Meli, T. V. Doherty, R. J. Linhardt and J. S. Dordick, Biotechnol. Bioeng., 2011, 108, 1229–1245 CrossRef CAS.
- F. Endres, ChemPhysChem, 2002, 3, 144–154 CrossRef CAS.
- A. P. Abbott and K. J. McKenzie, Phys. Chem. Chem. Phys., 2006, 8, 4265–4279 RSC.
- S. Z. El Abedin, E. M. Moustafa, R. Hempelmann, H. Natter and F. Endres, ChemPhysChem, 2006, 7, 1535–1543 CrossRef.
- Y.-Z. Su, Y.-C. Fu, Y.-M. Wei, J.-W. Yan and B.-W. Mao, ChemPhysChem, 2010, 11, 2764–2778 CrossRef CAS.
- T. Torimoto, T. Tsuda, K.-I. Okazaki and S. Kuwabata, Adv. Mater., 2010, 22, 1196–1221 CrossRef CAS.
- C. Vollmer and C. Janiak, Coord. Chem. Rev., 2011, 255, 2039–2057 CrossRef CAS.
- M. C. Buzzeo, R. G. Evans and R. G. Compton, ChemPhysChem, 2004, 5, 1106–1120 CrossRef CAS.
- P. A. Z. Suarez, V. M. Selbach, J. E. L. Dullius, S. Einloft, C. M. S. Piatnicki, D. S. Azambuja, R. F. de Souza and J. Dupont, Electrochim. Acta, 1997, 42, 2533–2535 CrossRef CAS.
- U. Schroder, J. D. Wadhawan, R. G. Compton, F. Marken, P. A. Z. Suarez, C. S. Consorti, R. F. de Souza and J. Dupont, New J. Chem., 2000, 24, 1009–1015 RSC.
- M. Opallo and A. Lesniewski, J. Electroanal. Chem., 2011, 656, 2–16 CrossRef CAS.
- S. V. Muginova, A. Z. Galimova, A. E. Polyakov and T. N. Shekhovtsova, J. Anal. Chem., 2010, 65, 331–351 CrossRef CAS.
- H. Zhao, J. Chem. Technol. Biotechnol., 2010, 85, 891–907 CrossRef CAS.
- M. J. A. Shiddiky and A. A. J. Torriero, Biosens. Bioelectron., 2011, 26, 1775–1787 CrossRef CAS.
- P. Majewski, A. Pernak, M. Grzymislawski, K. Iwanik and J. Pernak, Acta Histochem., 2003, 105, 135–142 CrossRef CAS.
- K. Fujita, M. Forsyth, D. R. MacFarlane, R. W. Reid and G. D. Elliott, Biotechnol. Bioeng., 2006, 94, 1209–1213 CrossRef CAS.
- N. Kimizuka and T. Nakashima, Langmuir, 2001, 17, 6759–6761 CrossRef CAS.
- R. Madeira Lau, M. J. Sorgedrager, G. Carrea, F. van Rantwijk, F. Secundo and R. A. Sheldon, Green Chem., 2004, 6, 483–487 RSC.
- M. Erbeldinger, A. J. Mesiano and A. J. Russell, Biotechnol. Prog., 2000, 16, 1131–1133 CrossRef CAS.
- T. de Diego, P. Lozano, S. Gmouh, M. Vaultier and J. L. Iborra, Biomacromolecules, 2005, 6, 1457–1464 CrossRef CAS.
- T. Itoh, J. Synth. Org. Chem. Jpn., 2009, 67, 143–155 CrossRef CAS.
- M. Bihari, T. P. Russell and D. A. Hoagland, Biomacromolecules, 2010, 11, 2944–2948 CrossRef CAS.
- C. Reichardt, Green Chem., 2005, 7, 339–351 RSC.
- M. A. Ab Rani, A. Brant, L. Crowhurst, A. Dolan, M. Lui, N. H. Hassan, J. P. Hallett, P. A. Hunt, H. Niedermeyer, J. M. Perez-Arlandis, M. Schrems, T. Welton and R. Wilding, Phys. Chem. Chem. Phys., 2011, 13, 16831–16840 RSC.
- L. Crowhurst, P. R. Mawdsley, J. M. Perez-Arlandis, P. A. Salter and T. Welton, Phys. Chem. Chem. Phys., 2003, 5, 2790–2794 RSC.
- Y. Fukaya, K. Hayashi, M. Wada and H. Ohno, Green Chem., 2008, 10, 44–46 RSC.
- D. M. Phillips, L. F. Drummy, D. G. Conrady, D. M. Fox, R. R. Naik, M. O. Stone, P. C. Trulove, H. C. de Long and R. A. Mantz, J. Am. Chem. Soc., 2004, 126, 14350–14351 CrossRef CAS.
- K. Tamura, N. Nakamura and H. Ohno, Biotechnol. Bioeng., 2012, 109, 729–735 CrossRef CAS.
- K. Fujita, D. R. MacFarlane and M. Forsyth, Chem. Commun., 2005, 4804–4806 RSC.
- K. Fujita, D. R. MacFarlane, M. Forsyth, M. Yoshizawa-Fujita, K. Murata, N. Nakamura and H. Ohno, Biomacromolecules, 2007, 8, 2080–2086 CrossRef CAS.
- H. Zhao, J. Mol. Catal. B: Enzym., 2005, 37, 16–25 CrossRef CAS.
- K. Fujita, N. Nakamura, K. Igarashi, M. Samejima and H. Ohno, Green Chem., 2009, 11, 351–354 RSC.
- K. Fujita and H. Ohno, Biopolymers, 2010, 93, 1093–1099 CrossRef CAS.
- H. Ohno, C. Suzuki, K. Fukumoto, M. Yoshizawa and K. Fujita, Chem. Lett., 2003, 32, 450–451 CrossRef CAS.
- K. Nakashima, T. Maruyama, N. Kamiya and M. Goto, Chem. Commun., 2005, 4297–4299 RSC.
- H. Ohno, C. Suzuki and K. Fujita, Electrochim. Acta, 2006, 51, 3685–3691 CrossRef CAS.
- K. Shimojo, K. Nakashima, N. Kamiya and M. Goto, Biomacromolecules, 2006, 7, 2–5 CrossRef CAS.
- T. Itoh, Y. Matsushita, Y. Abe, S.-H. Han, S. Wada, S. Hayase, M. Kawatsura, S. Takai, M. Morimoto and Y. Hirose, Chem.–Eur. J., 2006, 12, 9228–9237 CrossRef CAS.
- T. Maruyama, S. Nagasawa and M. Goto, Biotechnol. Lett., 2002, 24, 1341–1345 CrossRef CAS.
- K. Nakashima, N. Kamiya, D. Koda, T. Maruyama and M. Goto, Org. Biomol. Chem., 2009, 7, 2353–2358 CAS.
- A. Tsurumaki, J. Kagimoto and H. Ohno, Polym. Adv. Technol., 2011, 22, 1223–1228 CrossRef CAS.
- K. R. Seddon, J. Chem. Technol. Biotechnol., 1997, 68, 351–356 CrossRef CAS.
- M. Armand, F. Endres, D. R. MacFarlane, H. Ohno and B. Scrosati, Nat. Mater., 2009, 8, 621–629 CrossRef CAS.
- Ionic liquids: the front and future of material development, ed. H. Ohno, CMC Press, Tokyo, 2003 Search PubMed.
- G. Loget, S. Chevance, C. Poriel, G. Simonneaux, C. Lagrost and J. Rault-Berthelot, ChemPhysChem, 2011, 12, 411–418 CrossRef CAS.
- D. L. Compton and J. A. Laszlo, J. Electroanal. Chem., 2003, 553, 187–190 CrossRef CAS.
- S. F. Wang, T. Chen, Z. L. Zhang, X. C. Shen, Z. X. Lu, D. W. Pang and K. Y. Wong, Langmuir, 2005, 21, 9260–9266 CrossRef CAS.
- S. F. Wang, T. Chen, Z. L. Zhang, D. W. Pang and K. Y. Wong, Electrochem. Commun., 2007, 9, 1709–1714 CrossRef CAS.
- S. Wang, Z. Guo and H. Zhang, Bioelectrochemistry, 2011, 82, 55–62 CrossRef CAS.
- Z. Brusova, L. Gorton and E. Magner, Langmuir, 2006, 22, 11453–11455 CrossRef CAS.
- H. Xu, H.-Y. Xiong, Q.-X. Zeng, L. Jia, Y. Wang and S.-F. Wang, Electrochem. Commun., 2009, 11, 286–289 CrossRef CAS.
- B. Eker, P. Asuri, S. Murugesan, R. J. Linhardt and J. S. Dordick, Appl. Biochem. Biotechnol., 2007, 143, 153–163 CrossRef CAS.
- A. Ciaccafava, M. Alberola, S. Hameury, P. Infossi, M. T. Giudici-Orticoni and E. Lojou, Electrochim. Acta, 2011, 56, 3359–3368 CrossRef CAS.
- K. Fujita, N. Nakamura, K. Murata, K. Igarashi, M. Samejima and H. Ohno, Electrochim. Acta, 2011, 56, 7224–7227 CrossRef CAS.
- J. D. Wadhawan, U. Schroder, A. Neudeck, S. J. Wilkins, R. G. Compton, F. Marken, C. S. Consorti, R. F. de Souza and J. Dupont, J. Electroanal. Chem., 2000, 493, 75–83 CrossRef CAS.
- H. T. Liu, P. He, Z. Y. Li, C. Y. Sun, L. H. Shi, Y. Liu, G. Y. Zhu and J. H. Li, Electrochem. Commun., 2005, 7, 1357–1363 CrossRef CAS.
- G. Shul, J. Sirieix-Plenet, L. Gaillon and M. Opallo, Electrochem. Commun., 2006, 8, 1111–1114 CrossRef CAS.
- N. Maleki, A. Safavi and F. Tajabadi, Anal. Chem., 2006, 78, 3820–3826 CrossRef CAS.
- I. Svancara, K. Vytras, K. Kalcher, A. Walcarius and J. Wang, Electroanalysis, 2009, 21, 7–28 CrossRef CAS.
- N. Maleki, A. Safavi and F. Tajabadi, Electroanalysis, 2007, 19, 2247–2250 CrossRef CAS.
- R. T. Kachoosangi, M. M. Musameh, I. Abu-Yousef, J. M. Yousef, S. M. Kanan, L. Xiao, S. G. Davies, A. Russell and R. G. Compton, Anal. Chem., 2009, 81, 435–442 CrossRef CAS.
- D. Wei and A. Ivaska, Anal. Chim. Acta, 2008, 607, 126–135 CrossRef CAS.
- P. Yu, Y. Q. Lin, L. Xiang, L. Su, J. Zhang and L. Q. Mao, Langmuir, 2005, 21, 9000–9006 CrossRef CAS.
- X. D. Shangguan and J. B. Zheng, Electroanalysis, 2009, 21, 881–886 CrossRef CAS.
- X. B. Lu, J. Q. Hu, X. Yao, Z. P. Wang and J. H. Li, Biomacromolecules, 2006, 7, 975–980 CrossRef CAS.
- Y. X. Xu, C. G. Hu and S. S. Hu, Anal. Chim. Acta, 2010, 663, 19–26 CrossRef CAS.
- A. Cao, H. Ai, Y. Ding, C. Dai and J. Fei, Sens. Actuators, B, 2011, 155, 632–638 CrossRef.
- X. D. Shangguan, J. B. Zheng and Q. L. Sheng, Electroanalysis, 2009, 21, 1469–1474 CrossRef CAS.
- R. Gao and J. Zheng, Electrochem. Commun., 2009, 11, 1527–1529 CrossRef CAS.
- Z. Zhu, Z. Sun, Y. Wang, Y. Zeng, W. Sun and X. Huang, J. Electroanal. Chem., 2010, 650, 31–35 CrossRef CAS.
- S. K. Moccelini, A. C. Franzoi, I. C. Vieira, J. Dupont and C. W. Scheeren, Biosens. Bioelectron., 2011, 26, 3549–3554 CrossRef CAS.
- R. Gao, J. Zheng and L. Qiao, Electroanalysis, 2010, 22, 1084–1089 CAS.
- A. Safavi and F. Farjami, Anal. Biochem., 2010, 402, 20–25 CrossRef CAS.
- M. S. P. Lopez, D. Mecerreyes, E. Lopez-Cabarcos and B. Lopez-Ruiz, Biosens. Bioelectron., 2006, 21, 2320–2328 CrossRef.
- C. Xiao, X. Chu, B. Wu, H. Pang, X. Zhang and J. Chen, Talanta, 2010, 80, 1719–1724 CrossRef CAS.
- Q. Zhang, X. Lv, Y. Qiao, L. Zhang, D. L. Liu, W. Zhang, G. X. Han and X. M. Song, Electroanalysis, 2010, 22, 1000–1004 CAS.
- Q. Zhang, S. Y. Wu, L. Zhang, J. Lu, F. Verproot, Y. Liu, Z. Q. Xing, J. H. Li and X. M. Song, Biosens. Bioelectron., 2011, 26, 2632–2637 CrossRef CAS.
- Y. Liu, L. H. Shi, M. J. Wang, Z. Y. Li, H. T. Liu and J. H. Li, Green Chem., 2005, 7, 655–658 RSC.
- K. Szot, R. P. Lynch, A. Lesniewski, E. Majewska, J. Sirieix-Plenet, L. Gaillon and M. Opallo, Electrochim. Acta, 2011, 56, 10306–10312 CrossRef CAS.
- Y. H. Li, X. D. Zeng, X. Y. Liu, X. S. Liu, W. Z. Wei and S. L. Luo, Colloids Surf., B, 2010, 79, 241–245 CrossRef CAS.
- W. Sun, R. F. Gao and K. Jiao, J. Phys. Chem. B, 2007, 111, 4560–4567 CrossRef CAS.
- Z. Dai, Y. Xiao, X. Yu, Z. Mai, X. Zhao and X. Zou, Biosens. Bioelectron., 2009, 24, 1629–1634 CrossRef CAS.
- E. Zapp, D. Brondani, I. C. Vieira, J. Dupont and C. W. Scheerenb, Electroanalysis, 2011, 23, 1124–1133 CrossRef CAS.
- E. Zapp, D. Brondani, I. C. Vieira, J. Dupont and C. W. Scheeren, Electroanalysis, 2011, 23, 764–776 CrossRef CAS.
- E. Zapp, D. Brondani, I. C. Vieira, C. W. Scheeren, J. Dupont, A. M. J. Barbosa and V. S. Ferreira, Sens. Actuators, B, 2011, 155, 331–339 CrossRef.
- T. Fukushima, A. Kosaka, Y. Ishimura, T. Yamamoto, T. Takigawa, N. Ishii and T. Aida, Science, 2003, 300, 2072–2074 CrossRef CAS.
- J. Wang, H. Chu and Y. Li, ACS Nano, 2008, 2, 2540–2546 CrossRef CAS.
- P. Du, S. N. Liu, P. Wu and C. X. Cai, Electrochim. Acta, 2007, 52, 6534–6547 CrossRef CAS.
- Y. Zhang, Y. Shen, D. Han, Z. Wang, J. Song, F. Li and L. Niu, Biosens. Bioelectron., 2007, 23, 438–443 CrossRef.
- P. Yu, H. Zhou, H. Cheng, Q. Qian and L. Mao, Anal. Chem., 2011, 83, 5715–5720 CrossRef CAS.
- F. Zhao, X. Wu, M. K. Wang, Y. Liu, L. X. Gao and S. J. Dong, Anal. Chem., 2004, 76, 4960–4967 CrossRef CAS.
- Y. Liu, X. Zou and S. Dong, Electrochem. Commun., 2006, 8, 1429–1434 CrossRef CAS.
- W. Sun, X. Q. Li, Y. Wang, R. J. Zhao and K. Jiao, Electrochim. Acta, 2009, 54, 4141–4148 CrossRef CAS.
- R. Pauliukaite, A. P. Doherty, K. D. Murnaghan and C. M. A. Brett, J. Electroanal. Chem., 2011, 656, 96–101 CrossRef CAS.
- B. G. Choi, H. Park, T. J. Park, D. H. Kim, S. Y. Lee and W. H. Hong, Electrochem. Commun., 2009, 11, 672–675 CrossRef CAS.
- Z. Zhu, L. Qu, X. Li, Y. Zeng, W. Sun and X. Huang, Electrochim. Acta, 2010, 55, 5959–5965 CrossRef CAS.
- A. Salimi, S. Lasghari and A. Noorbakhash, Electroanalysis, 2010, 22, 1707–1716 CrossRef CAS.
- L. G. Zamfir, L. Rotariu and C. Bala, Biosens. Bioelectron., 2011, 26, 3692–3695 CrossRef CAS.
- Y. Liu, L. Huang and S. Dong, Biosens. Bioelectron., 2007, 23, 35–41 CrossRef CAS.
- L. Hussein, S. Rubenwolf, F. von Stetten, G. Urban, R. Zengerle, M. Krueger and S. Kerzenmacher, Biosens. Bioelectron., 2011, 26, 4133–4138 CrossRef CAS.
- C. Shan, H. Yang, J. Song, D. Han, A. Ivaska and L. Niu, Anal. Chem., 2009, 81, 2378–2382 CrossRef CAS.
- H. G. Liao, H. Wu, J. Wang, J. Liu, Y. X. Jiang, S. G. Sun and Y. H. Lin, Electroanalysis, 2010, 22, 2297–2302 CrossRef CAS.
- L. Wang, X. H. Zhang, H. Y. Xiong and S. F. Wang, Biosens. Bioelectron., 2010, 26, 991–995 CrossRef CAS.
- Y. Qiu, X. Qu, J. Dong, S. Ai and R. Han, J. Hazard. Mater., 2011, 190, 480–485 CrossRef CAS.
- W.-L. Zhu, Y. Zhou and J.-R. Zhang, Talanta, 2009, 80, 224–230 CrossRef CAS.
- Y. Zhang, R. Yan, F. Zhao and B. Zeng, Colloids Surf., B, 2009, 71, 288–292 CrossRef CAS.
- A. C. Franzoi, I. C. Vieira, C. W. Scheeren and J. Dupont, Electroanalysis, 2010, 22, 1376–1385 CrossRef CAS.
- A. Safavi and F. Farjami, Biosens. Bioelectron., 2011, 26, 2547–2552 CrossRef CAS.
- Y. Yu, X. Liu, D. Jiang, Q. Sun, T. Zhou, M. Zhu, L. Jin and G. Shi, Biosens. Bioelectron., 2011, 26, 3227–3232 CrossRef CAS.
- J. Huang, J. Zheng and Q. Sheng, Microchim. Acta, 2011, 173, 157–163 CrossRef CAS.
- S. Dong, P. Zhang, H. Liu, N. Li and T. Huang, Biosens. Bioelectron., 2011, 26, 4082–4087 CrossRef CAS.
- Z. Zhu, L. Qu, Q. Niu, Y. Zeng, W. Sun and X. Huang, Biosens. Bioelectron., 2011, 26, 2119–2124 CrossRef CAS.
- Z. Zhu, X. Li, Y. Zeng, W. Sun, W. Zhu and X. Huang, J. Phys. Chem. C, 2011, 115, 12547–12553 CAS.
- Z. Zhu, X. Sun, Y. Wang, Y. Zeng, W. Sun and X. Huang, Mater. Chem. Phys., 2010, 124, 488–492 CrossRef CAS.
- Q. Shi, J. Zhang, C. Zhang, W. Nie, B. Zhang and H. Zhang, J. Colloid Interface Sci., 2010, 343, 188–193 CrossRef CAS.
- Z. Zhu, X. Li, Y. Wang, Y. Zeng, W. Sun and X. Huang, Anal. Chim. Acta, 2010, 670, 51–56 CrossRef CAS.
- Y. Xu, C. Hu and S. Hu, Anal. Chim. Acta, 2010, 663, 19–26 CrossRef CAS.
- T. Fukushima and T. Aida, Chem.–Eur. J., 2007, 13, 5048–5058 CrossRef CAS.
- S. Y. Yang, F. Cicoira, R. Byrne, F. Benito-Lopez, D. Diamond, R. M. Owens and G. G. Malliaras, Chem. Commun., 2010, 46, 7972–7974 RSC.
- S. K. Moccelini, A. C. Franzoi, I. C. Vieira, J. Dupont and C. W. Scheeren, Biosens. Bioelectron., 2011, 26, 3549–3554 CrossRef CAS.
- T. Zhan, M. Xi, Y. Wang, W. Sun and W. Hou, J. Colloid Interface Sci., 2010, 346, 188–193 CrossRef CAS.
- L. Hussein, S. Rubenwolf, F. von Stetten, G. Urban, R. Zengerle, M. Krueger and S. Kerzenmacher, Biosens. Bioelectron., 2011, 26, 4133–4138 CrossRef CAS.
- X. Wu, F. Zhao, J. R. Varcoe, A. E. Thumser, C. Avignone-Rossa and R. C. T. Slade, Biosens. Bioelectron., 2009, 25, 326–331 CrossRef CAS.
- P. Vidinha, N. M. T. Lourenco, C. Pinheiro, A. R. Bras, T. Carvalho, T. Santos-Silva, A. Mukhopadhyay, M. J. Romao, J. Parola, M. Dionisio, J. M. S. Cabral, C. A. M. Afonso and S. Barreiros, Chem. Commun., 2008, 5842–5844 RSC.
- T. Kakibe and H. Ohno, Chem. Commun., 2008, 377–379 RSC.
- H. Ohno and N. Nishimura, J. Electrochem. Soc., 2001, 148, E168–E170 CrossRef CAS.
- N. Nishimura, Y. Nomura, N. Nakamura and H. Ohno, Biomaterials, 2005, 26, 5558–5563 CrossRef CAS.
- T. Kakibe and H. Ohno, J. Mater. Chem., 2009, 19, 4960–4964 RSC.
- T. Kakibe, R. Kurihara and H. Ohno, Chem. Lett., 2009, 38, 494–495 CrossRef CAS.
- N. Nishimura and H. Ohno, J. Mater. Chem., 2002, 12, 2299–2304 RSC.
- V. L. Pushparaj, M. M. Shaijumon, A. Kumar, S. Murugesan, L. Ci, R. Vajtai, R. J. Linhardt, O. Nalamasu and P. M. Ajayan, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 13574–13577 CrossRef CAS.
- N. Matsumi, Y. Nakamura, K. Aoi, T. Watanabe, T. Mizumo and H. Ohno, Polym. J., 2009, 41, 437–441 CrossRef CAS.
- J. M. Lee, D. Q. Nguyen, S. B. Lee, H. Kim, B. S. Ahn, H. Lee and H. S. Kim, J. Appl. Polym. Sci., 2010, 115, 32–36 CrossRef CAS.
- P. K. Singh, B. Bhattacharya, R. K. Nagarale, K.-W. Kim and H.-W. Rhee, Synth. Met., 2010, 160, 139–142 CrossRef CAS.
- L. H. Lu and W. Chen, Adv. Mater., 2010, 22, 3745–3748 CrossRef CAS.
- S. Yamazaki, A. Takegawa, Y. Kaneko, J. Kadokawa, M. Yamagata and M. Ishikawa, Electrochem. Commun., 2009, 11, 68–70 CrossRef CAS.
- K. Suzuki, M. Yamaguchi, M. Kumagai, N. Tanabe and S. Yanagida, C. R. Chim., 2006, 9, 611–616 CrossRef CAS.
- T. Singh, T. J. Trivedi and A. Kumar, Green Chem., 2010, 12, 1029–1035 RSC.
- J. Le Bideau, L. Viau and A. Vioux, Chem. Soc. Rev., 2011, 40, 907–925 RSC.
- I. Ferreira, B. Bras, J. I. Martins, N. Correia, P. Barquinha, E. Fortunato and R. Martins, Electrochim. Acta, 2011, 56, 1099–1105 CrossRef CAS.
- R. P. Swatloski, S. K. Spear, J. D. Holbrey and R. D. Rogers, J. Am. Chem. Soc., 2002, 124, 4974–4975 CrossRef CAS.
- M. Abe, Y. Fukaya and H. Ohno, Green Chem., 2010, 12, 1274–1280 RSC.
- M. B. Turner, S. K. Spear, J. D. Holbrey and R. D. Rogers, Biomacromolecules, 2004, 5, 1379–1384 CrossRef CAS.
- M. A. Mehta and T. Fujinami, Chem. Lett., 1997, 915–916 CrossRef CAS.
- T. Sato, Y. Tsujii, T. Fukuda and T. Miyamoto, Macromolecules, 1992, 25, 3890–3895 CrossRef CAS.
- D. Q. Nguyen, J. Hwang, J. S. Lee, H. Kim, H. Lee, M. Cheong, B. Lee and H. S. Kim, Electrochem. Commun., 2007, 9, 109–114 CrossRef CAS.
- C. J. Murphy, M. R. Arkin, Y. Jenkins, N. D. Ghatlia, S. H. Bossmann, N. J. Turro and J. K. Barton, Science, 1993, 262, 1025–1029 CAS.
- V. D. Lakhno, Int. J. Quantum Chem., 2008, 108, 1970–1981 CrossRef CAS.
- J. H. Wang, D. H. Cheng, X. W. Chen, Z. Du and Z. L. Fang, Anal. Chem., 2007, 79, 620–625 CrossRef CAS.
- R. Vijayaraghavan, A. Izgorodin, V. Ganesh, M. Surianarayanan and D. R. MacFarlane, Angew. Chem., Int. Ed., 2010, 49, 1631–1633 CAS.
- Y. Ding, L. Zhang, J. Xie and R. Guo, J. Phys. Chem. B, 2010, 114, 2033–2043 CrossRef CAS.
- L. Cardoso and N. M. Micaelo, ChemPhysChem, 2011, 12, 275–277 CrossRef CAS.
- S. Takenaka, H. Sato, T. Ihara and M. Takagi, J. Heterocycl. Chem., 1997, 34, 123–127 CrossRef.
| This journal is © The Royal Society of Chemistry 2012 |