Diastereoselective addition to N-acyliminium ions with aryl- and alkenyl boronic acids via a Petasis-type reaction†
Satoshi
Mizuta
and
Osamu
Onomura
*
Graduate School of Biomedical Sciences, Nagasaki University, 1-14 Bunkyo-machi, Nagasaki 852-8521, Japan. E-mail: onomura@nagasaki-u.ac.jp; Fax: +81 95 819 2476; Tel: +81 95 819 2429
First published on 9th February 2012
Abstract
A highly diastereoselective synthesis of 2,3-disubstituted piperidines has been accomplished through nucleophilic additions to N-acyliminium ions with aryl- and alkenyl boronic acids. A reversal of stereoselectivity depending on a β-substituent on the piperidine ring was observed in the alkenylation reactions with (E)-styrylboronic acid. Our strategy was applied in the key step for the synthesis of the neurokinin NK1 receptor antagonist (±)-L-733,060.
Introduction
Substituted piperidines are found in numerous alkaloids and biologically active compounds, of which 2-aryl-3-hydroxypiperidines have been prominent structural motifs. For example, (+)-L-733,060 (1)1 and its N-analogue (+)-CP-99,9942 are selective neurokinin-1 substance P receptor antagonists (Fig. 1).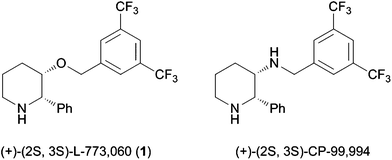 | ||
| Fig. 1 Biologically active 2,3-disubstituted piperidines. | ||
One of the most powerful methodologies to furnish substituted piperidines is nucleophilic addition to N-acyliminium ions.3 To date, a broad range of C-based nucleophiles have been known to react with cyclic N-acyliminium ions. We also have achieved asymmetric intermolecular reactions with a variety of enol derivatives.4
The Petasis reaction,5 which constitutes the efficient synthesis of allylamines and α-amino acids by the three-component coupling of an amine, an aldehyde and an organoboronic acid, is highly attractive because of the use of organoboronic acids as nucleophiles which are readily available and show little sensitivity toward air and water. Batey and co-workers have developed the procedure for the diastereoselective formation of functionalized N-heterocycles with alkenyl- and aryl boronates.6 Moreover asymmetric additions to chiral iminium ions derived from (S)-5-phenylmorpholin-2-one and aliphatic aldehydes with 2-furyl boronic acid have been reported by Harwood et al.7 In spite of some examples of arylation reactions of N-acyliminium ions,8 a synthetic protocol for the asymmetric arylation of piperidine rings with organoboronic acids as nucleophiles has not yet been reported. We herein disclose a novel approach to 2-aryl-3-hydroxypiperidines by the diastereocontrolled introduction of aromatic moieties in organoboronic acids to N-protected piperidinium ions mediated by a Lewis acid via a Petasis-type reaction (Scheme 1).
 | ||
| Scheme 1 Diastereoselective introduction of aryl moieties onto the 2-position of N-protected piperidinium ions. | ||
Results and discussion
To start with, the preparation of tetrahydropyridines from the respective N-protected piperidines was achieved according to our previously reported method, which consists of electrochemical methoxylation and elimination of methanol.9 The tetrahydropyridines were thus treated with oxone in methanol, yielding N,O-acetals 2–4 as N-acyliminium ion precursors (eqn (1)). | (1) |
As shown in Table 1, we demonstrated the Lewis acid-mediated reactions of 2–4 with phenylboronic acid. An initial survey using substrate 2 indicated that the reaction in the presence of BF3·Et2O did not proceed. The N,O-acetal 3, possessing an N-phenoxycarbonyl group, reacted slightly with phenylboronic acid, providing the desired product 6a. The corresponding boronate did not lead to any increase of yield.10 In contrast, the reaction of N-Cbz piperidine 4 proceeded with good diastereoselectivity (cis![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :trans = 8.3:1 by 1H NMR), and gave the cis-adduct 7a in 69% yield as a single isomer after purification by column chromatography. Otherwise, TiCl4 was found to be a poor mediator.
:trans = 8.3:1 by 1H NMR), and gave the cis-adduct 7a in 69% yield as a single isomer after purification by column chromatography. Otherwise, TiCl4 was found to be a poor mediator.
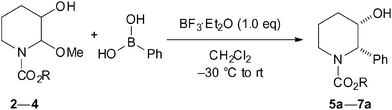
| Entry | R (Substrate) | Time/h | Product | dr (cis:trans) a |
Yield (%)b | |
|---|---|---|---|---|---|---|
| a The diastereomer ratio was determined by 1H NMR spectroscopy of the crude mixture. b Yield of isolated product after purification by column chromatography. c The reaction was performed with 1.0 equivalent of TiCl4 instead of BF3·Et2O. | ||||||
| 1 | Me | (2) | 18 | 5a | — | Trace |
| 2 | Ph | (3) | 18 | 6a | 7.2:1 |
14 |
| 3 | Bn | (4) | 24 | 7a | 8.3:1 |
69 |
| 4c | Bn | (4) | 24 | 7a | — | 0 |
By using substrate 4, we next examined the scope and limitation of this process with respect to substitution on the benzene ring of aryl boronic acids (Table 2, Entries 1–13). Overall adducts 7 were cis-formed as single diastereomers (>98:2 cis:trans by 1H NMR) in moderate to high yields. When a nucleophile with a methoxy substituent was used, the 2-arylated piperidine 7b was obtained in 86% yield. Moreover, nucleophiles bearing a methylthio, a methyl and a phenyl group provided the corresponding desired products 7c–g in moderate yields. Both 1- and 2-naphthylboronic acids also were well-tolerated in this process. It should be noted that acceptable substitutions on the phenyl group are characterized by electron-neutral or -donating groups. Substrates with electron-withdrawing groups are challenging for this reaction. For instance, the reactions of 4-fluoro and 4-chlorophenyl boronic acid gave adducts (7j and 7k) in less than 40% yield. Additionally, (4-cyanophenyl)boronic acid was ineffective. An attempt to use disubstituted phenyl boronic acids was also successful; arylated piperidines (7m and 7n) were obtained in 64% and 62% yields, respectively. The additions of other boronic acids involving an alkenyl and heteroaryl substitutes were explored (Entries 14–17). The reaction of (E)-2-styrylboronic acid gave cis-adduct 7o in 81% yield. The electron-rich boronic acids, 3-thienylboronic acid, 2-benzothienylboronic acid and 2-benzofuranylboronic acid, were effective, providing the diastereomerically pure 2-heteroarylated piperidines (7p–r) in good to high yield.

| Entry | Ar | Time/h | Product | Yield (%)a |
|---|---|---|---|---|
| a Yield of isolated product after purification by column chromatography. | ||||
| 1 | 4-MeOC6H4 | 5 | 7b | 86 |
| 2 | 4-MeSC6H4 | 5 | 7c | 71 |
| 3 | 4-MeC6H4 | 19 | 7d | 67 |
| 4 | 3-MeC6H4 | 24 | 7e | 66 |
| 5 | 2-MeC6H4 | 24 | 7f | 49 |
| 6 | 3-Biphenyl | 24 | 7g | 43 |
| 7 | 1-Naphthyl | 24 | 7h | 71 |
| 8 | 2-Naphthyl | 24 | 7i | 71 |
| 9 | 4-FC6H4 | 24 | 7j | 38 |
| 10 | 4-ClC6H4 | 24 | 7k | 33 |
| 11 | 4-NCC6H4 | 24 | 7l | 0 |
| 12 | 3,4-(MeO)2C6H3 | 5 | 7m | 64 |
| 13 |

|
14 | 7n | 62 |
| 14 | (E)-PhCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH CH |
24 | 7o | 81 |
| 15 | 3-Thienyl | 5 | 7p | 57 |
| 16 | 2-Benzothienyl | 24 | 7q | 60 |
| 17 | 2-Benzofuranyl | 24 | 7r | 90 |
In the case of 1-(N-methoxycarbonyl)indole-2-boronic acid, the reaction proceeded successfully, providing the cyclic carbamate compound 8 in 61% yield (Scheme 2).
 | ||
| Scheme 2 Addition of 1-(N-methoxycarbonyl)indole-2-boronic acid to N,O-acetal 4. | ||
Although we examined the temperature and solvent effect on the diastereoselectivity of 7a, and whether the epimerization of cis-7a caused by BF3·Et2O was present or not in order to explain the source of lower diastereoselectivity of 7a, we did not obtain any significant results.†
The synthesis of the cis-isomers (7a, 7b, 7d, and 7j) has been reported11 and the 1H NMR data of compounds obtained by us are consistent with literature examples. The coupling constant J2,3 values for the cis-isomers were 5.6–5.8 Hz, compared with <1.0 Hz for the corresponding trans-isomers.8,12 The stereochemistry of 7c, 7e, 7f–i, 7k, 7m–r and 8 was readily determined by the examination of the coupling constants J2,3, which were 5.2–6.1 Hz, consistent with J2,3 values for cis-isomers.
Next, we explored the scope of β-substitution with respect to the N-acyliminium ion partner. Indeed, neither the nucleophilic substitutions of N-benzyloxycarbonyl-2-methoxypiperidine nor β-halo substituted piperidine N,O-acetals 9a–c with 4-methoxyphenyl boronic acid provided the desired products. The requirement for a β-hydroxyl group adjacent to the α-carbon implied the coordination to organoboronic acid. The reaction intermediates I and II, which would be responsible for the reactivity and selectivity, are proposed (Scheme 3). First, the N-acyliminium ion tetracoordinate boronic acid as an intermediate I would be formed by Lewis acid activation of the N,O-acetal. Next, successive intramolecular attack of boronate I on the N-acyliminium ion by the same face bearing the β-oxygen results in the diastereospecific C–C bond formation to provide the cyclic intermediate II. Consequently, II undergoes the elimination of boronic acid leading to a cis-2,3-disubstituted piperidine derivative with a high level of diastereoselectivity.
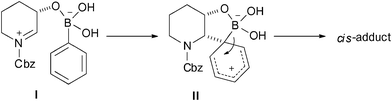 | ||
| Scheme 3 Plausible mechanism for the diastereoselective arylation of N-acyliminium ions. | ||
On the other hand, in our research, we found that (E)-2-styrylboronic acid as a nucleophile was suitable for nucleophilic additions to N-acyliminium ions derived from N,O-acetals 9a–c (Scheme 4). The reaction of 9a in the presence of BF3·Et2O gave the desired product 10a with good diastereoselectivity (dr = 12:1) in 62% yield. Interestingly, the reactions of 3-chloro- and 4-bromo-piperidine N,O-acetals 9b and 9c provided exclusively a single isomer in good yield, respectively. The major isomers 10a–c were confirmed to be trans-formed by the examination of their coupling constants J2,3 < 1.0 Hz, consistent with literature examples for trans-2-aryl-3-hydroxypiperidines.
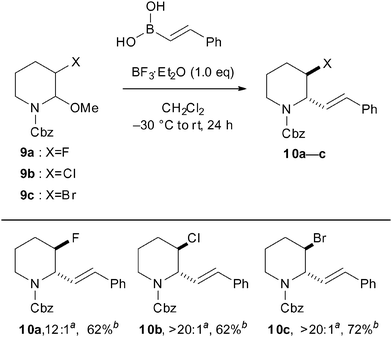 | ||
| Scheme 4 Scope of β-position substituents of N,O-acetals in the diastereoselective reaction with (E)-2-styrylboronic acid. a The diastereomeric ratio was determined by 1H NMR spectroscopy of the crude mixture. b The yield of the isolated product after purification by column chromatography. | ||
Further investigation will be required to elucidate the mechanism of the alkenylation reaction. We envisage that transient iminium ions (III–V) would be formed due to the high conformational control (Fig. 2). Organofluorine compounds tend to stabilize the conformations by hyperconjugative and electrostatic interactions.13 For instance, the [NH–FC] dipole effect in 3-fluoropiperidine derivatives has been observed by Snyder and Lankin et al.14 In addition, Gilmour and co-workers developed a glycosylation stereocontrolled by the polarized C–F bond, which would orient towards the electropositive center in a gluco-configured 2-fluoro-oxonium ion.15 We postulated that an electrostatic interaction between the partially negatively charged C–F bond and the N-acyliminium cation would lead to conformational rigidification of 3-fluoropiperidinium ion III because they are positioned closer together in a pseudoaxial conformer than in a pseudoequatorial conformer. On the other hand, 3-chloro and 3-bromopiperidinium ion intermediates should be indicated by the two proposed conformational states (IV and V). The alkenylations of 9b and 9c with (E)-2-styrylboronic acid would proceed via a pseudoaxial conformer V providing trans-products, and not via the pseudoequatorial conformer IV which might cause a steric repulsion between a halogen substitute and a nucleophile.
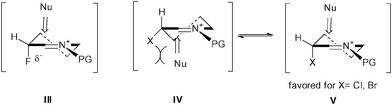 | ||
| Fig. 2 Conformational control in 3-halopiperidinium ions. | ||
Finally, we aimed at the synthesis of (±)-L-773,060 (1).16 With diastereomerically pure 7a in hand, the treatment with NaH for the deprotonation of the β-hydroxyl group to form the alkoxyl anion which reacted with 3,5-bis(trifluoromethyl)benzyl bromide afforded the O-benzyl ether product. The subsequent hydrogenolysis of N-Cbz with ammonium acetate and Pd/C according to Sajiki's conditions17 afforded the desired product (±)-L-773,060 (1) in excellent yield (Scheme 5).
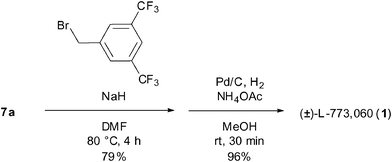 | ||
| Scheme 5 Preparation of 1 from 7a. | ||
Conclusions
In conclusion, we have disclosed a versatile approach to cis-2-aryl-3-hydroxypiperidine derivatives through highly diastereoselective α-arylations of piperidinium ions with a broad range of readily available arylboronic acids involving heteroaryl- and alkenyl boronic acids. In the alkenylation of N,O-acetals with (E)-styrylboronic acid, a reversal of stereoselectivity was observed depending on whether a hydroxyl group or a halogen was present as β-substituent. A concise synthesis of (±)-L-773,060 was realised using our method. Further investigation of the scope of the substrates and the detailed mechanism is currently ongoing.Acknowledgements
This research was supported by a Grant-in-Aid for Scientific Research on Innovative Areas “Molecular Activation Directed toward Straightforward Synthesis” (23105539) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.References
- (a) R. Baker, T. Harrison, G. J. Hollingworth, C. J. Swain and B. J. Williams, EP 0 528, 495A1, 1993 Search PubMed; (b) T. Harrison, B. J. Williams, C. J. Swain and R. G. Ball, Bioorg. Med. Chem. Lett., 1994, 4, 2545 CrossRef CAS.
- M. C. Desai, S. L. Lefkowitz, P. F. Thadeio, K. P. Longo and R. M. Snider, J. Med. Chem., 1992, 35, 4911 CrossRef CAS.
- For reviews of N-acyliminium ion chemistry: (a) W. N. Speckamp and M. J. Moolenaar, Tetrahedron, 2000, 56, 3817 CrossRef CAS; (b) A. Yazici and S. G. Pyne, Synthesis, 2009, 339 CAS; (c) A. Yazici and S. G. Pyne, Synthesis, 2009, 513 CAS; (d) H. de Koning and W. N. Speckamp, in Houben-Weyl, Stereoselective Synthesis, ed. G. Helmchen, R. W. Hoffmann, J. Mulzer and E. Schaumann, 1995, vol. E21, pp. 1953–2009 Search PubMed; (e) H. Hiemstra and W. N. Speckamp, in Comprehensive Organic Synthesis, ed. B. M. Trost, I. Fleming and C. H. Heathcock, Pergamon, Oxford, 1991, vol. 2, pp. 1047–1082 Search PubMed; (f) R. A. Volkmann, in Comprehensive Organic Synthesis, ed. B. M. Trost, I. Fleming and S. L. Schreiber, Pergamon, Oxford, 1991, vol. 1, pp. 335–396 Search PubMed.
- (a) Y. Matsumura, D. Minato and O. Onomura, J. Organomet. Chem., 2007, 692, 654 CrossRef CAS; (b) O. Onomura, Y. Kanda, Y. Nakamura, T. Maki and Y. Matsumura, Tetrahedron Lett., 2002, 43, 3229 CrossRef CAS; (c) Y. Matsumura, Y. Kanda, K. Shirai, O. Onomura and T. Maki, Tetrahedron, 2000, 56, 7411 CrossRef CAS.
- (a) N. A. Petasis and I. A. Zavialov, J. Am. Chem. Soc., 1998, 120, 11798 CrossRef CAS; (b) N. A. Petasis and I. A. Zavialov, J. Am. Chem. Soc., 1997, 119, 445 CrossRef CAS.
- (a) R. A. Batey, D. B. MacKay and V. Santhakumar, J. Am. Chem. Soc., 1999, 121, 5075 CrossRef CAS; (b) R. A. Batey and D. B. MacKay, Tetrahedron Lett., 2000, 41, 9935 CrossRef CAS.
- L. H. Harwood, G. S. Currie, M. G. B. Drew and R. W. A. Luke, Chem. Commun., 1996, 1953 RSC.
- (a) I. R. Morgan, A. Yazici and S. G. Pyne, Tetrahedron, 2008, 64, 1409 CrossRef CAS; (b) S. Suga, T. Nishida, D. Yamada, A. Nagaki and J. Yoshida, J. Am. Chem. Soc., 2004, 126, 14338 CrossRef CAS. Intramolecular reaction, see: (c) K. Tomooka, A. Nakazaki and Y. Nakai, J. Am. Chem. Soc., 2000, 122, 408 CrossRef CAS.
- (a) T. Shono, Y. Matsumura, O. Onomura and Y. Yamada, Tetrahedron Lett., 1987, 28, 4073 CrossRef CAS; (b) T. Shono, H. Hamaguchi and Y. Matsumura, J. Am. Chem. Soc., 1975, 97, 4264 CrossRef CAS.
- The use of ethylene glycol boronate did not afford practically the desired product 6a. For details, see ESI†.
- The relative configuration of the major isomer 7a was established to be cis by 1H-NMR spectroscopy and a precedent reference, see: M. I. Monterde, R. Brieva and V. Gotor, Tetrahedron: Asymmetry, 2001, 12, 525 CrossRef CAS.
- M. Atobe, N. Yamazaki and C. Kibayashi, J. Org. Chem., 2004, 69, 5595 CrossRef CAS.
- (a) J. J. Irwin, T.-K. Ha and J. D. Dunitz, Helv. Chim. Acta, 1990, 73, 1805 CrossRef CAS; (b) D. O'Hagan, C. Bilton, J. A. K. Howard, L. Knight and D. J. Tozer, J. Chem. Soc., Perkin Trans. 2, 2000, 605 RSC; (c) C. R. S. Briggs, D. O' Hagan, J. A. K. Howard and D. S. Yufit, J. Fluorine Chem., 2003, 119, 9 CrossRef CAS; (d) C. R. S. Briggs, M. J. Allen, D. O'Hagan, D. J. Tozer, A. M. Z. Slawin, A. E. Goeta and J. A. K. Howard, Org. Biomol. Chem., 2004, 2, 732 RSC; (e) C. Sparr, W. B. Schweizer, H. M. Senn and R. Gilmour, Angew. Chem., 2009, 121, 3111 CrossRef; (f) Angew. Chem. Int. Ed., 2009, 48, 3065 Search PubMed; (g) C. Bucher, C. Sparr, W. B. Schweizer and R. Gilmour, Chem.–Eur. J., 2009, 15, 7637 CrossRef CAS.
- (a) J. P. Snyder, N. S. Chandrakumar, H. Sato and D. C. Lankin, J. Am. Chem. Soc., 2000, 122, 544 CrossRef CAS; (b) D. C. Lankin, N. S. Chandrakumar, S. N. Rao, D. P. Spangler and J. P. Snyder, J. Am. Chem. Soc., 1993, 115, 3356 CrossRef CAS.
- (a) C. Bucher and R. Gilmour, Angew. Chem., 2010, 122, 8906 CrossRef; (b) Angew. Chem. Int. Ed., 2010, 49, 8724 Search PubMed.
- For the synthesis of chiral 1, see: (a) G. Kumaraswamy and A. Pitchaiah, Tetrahedron, 2011, 67, 2536 CrossRef CAS; (b) N. M. Garrido, M. García, M. R. Sánchez, D. Díez and J. G. Urones, Synlett, 2010, 387 CrossRef CAS; (c) S. Prévost, P. Phansavath and M. Haddad, Tetrahedron: Asymmetry, 2010, 21, 16 CrossRef; (d) J. L. Bilke, S. P. Moore, P. O'Brien and J. Gilday, Org. Lett., 2009, 11, 1935 CrossRef CAS; (e) F. A. Davis and T. Ramachandar, Tetrahedron Lett., 2008, 49, 870 CrossRef CAS; (f) S. K. Cherian and P. Kumar, Tetrahedron: Asymmetry, 2007, 18, 982 CrossRef CAS; (g) T. Oshitari and T. Mandai, Synlett, 2006, 3395 CAS; (h) Y.-J. Yoon, J.-E. Joo, K.-Y. Lee, Y.-H. Kim, C.-Y. Oh and W.-H. Ham, Tetrahedron Lett., 2005, 46, 739 CrossRef CAS; (i) S. Rao, V. Kandula and P. Kumar, Tetrahedron: Asymmetry, 2005, 16, 3579 CrossRef; (j) G. Bhaskar and B. V. Rao, Tetrahedron Lett., 2003, 44, 915 CrossRef CAS; (k) P.-Q. Huang, L.-X. Liu, B.-G. Wei and Y.-P. Ruan, Org. Lett., 2003, 5, 1927 CrossRef CAS. Also see: ref. 13; For the synthesis of precursor of 1, see: (l) M.-R. Tsai, B.-F. Chen, C.-C. Cheng and N.-C. Chang, J. Org. Chem., 2005, 70, 1780 CrossRef CAS; (m) N. Kise, K. Ohya, K. Arimoto, Y. Yamashita, Y. Hirano, T. Ono and N. Ueda, J. Org. Chem., 2004, 69, 7710 CrossRef CAS; (n) O. Calves and N. Langlois, Tetrahedron Lett., 1999, 40, 7099 CrossRef.
- H. Sajiki, Tetrahedron Lett., 1995, 36, 3465 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data. NMR spectra for all novel compounds. See DOI: 10.1039/c2ra01254e |
| This journal is © The Royal Society of Chemistry 2012 |