Controlled multistep oxidation of alcohols and aldehydes to carboxylic acids using air, sunlight and a robust metalloporphyrin sensitizer with a pH-switchable photoreactivity†
Mahdi
Hajimohammadi
ac,
Clemens
Schwarzinger
b and
Günther
Knör
*c
aDepartment of Chemistry, Shahid Beheshti University, G.C., Evin, 19839-63113, Teheran, Iran
bInstitut für Chemische Technologie Organischer Stoffe, Johannes Kepler Universität (JKU), Linz, 4040, Linz, Austria
cInstitut für Anorganische Chemie, Johannes Kepler Universität (JKU), Linz, 4040, Linz, Austria. E-mail: guenther.knoer@jku.at; Fax: (+43)-732-2468-9681
First published on 2nd March 2012
Abstract
A homogeneous photocatalytic reaction cascade for the stepwise biomimetic oxidation of alcohols to form either aldehydes or alternatively carboxylic acids is presented. The system is based on robust and abundant materials and efficiently utilizes dioxygen as a clean and sustainable oxidant. Photoassisted substrate transformation occurs upon exposure to visible light (<620 nm) or sunlight. The pH-dependent photoredox mechanism of the sensitizer can be reversibly switched between two different selectivity patterns to control the desired product formation.
Visible-light driven redox reactions display attractive features for the development of energy conserving and environmentally benign synthetic processes.1,2 For example, the solar chemical activation of dioxygen based on earth-abundant photosensitizer materials can be coupled to the selective catalytic transformation of organic compounds under very mild reaction conditions, thus avoiding undesired waste products and utilizing ambient air as a readily available chemical feedstock.3,4 The absorption of light can also be applied to trigger or regulate the catalytic performance of such systems, which has already been demonstrated to enable a very convenient on-off control of reactivity in several cases.2,5,6 A much less explored feature in this context is the systematic coupling of consecutive catalytic reactions to multistep cascade processes without intermediate recovery steps to carry out sustainable organic transformations in the natural way, as takes place in living cells.7,8 As part of our ongoing efforts to demonstrate the versatility of biomimetic and bio-inspired photocatalysis,2,6 we therefore started to investigate such kinds of “artificial metabolic pathway” sequences based on light-driven substrate transformations. Here we report our first results on a homogeneous photocatalytic reaction cascade for the stepwise oxidation of alcohols to form either aldehydes or carboxylic acids with excellent yields and selectivities. The redox mechanism of the catalyst system can be reversibly switched in situ by a simple ligand protonation step in order to control the desired product formation, while all other parameters of the one-pot system remain unchanged.
We have chosen the high-valent antimony tetraphenylporphyrin cation [Sb(tpp)(OH)2]+, (1) as the photosensitizer (Scheme 1).‡
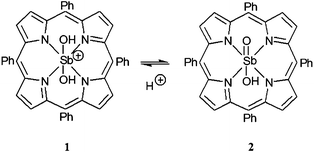 | ||
| Scheme 1 Structural representation of the protonated (1) and deprotonated (2) states of the photocatalyst (pKa = 9.7).4 | ||
Antimony porphyrin complexes such as 1 are powerful excited state oxidants with a broad potential for visible-light driven photocatalysis.9,10 In the deprotonated form SbO(tpp)(OH) (2), the tetraphenylporphyrin derivative has been shown4 to photo-oxidize primary alcohols such as methanol or ethanol with a quantum yield of Φ = 0.02 at 546 nm to the corresponding aldehydes with turnover numbers (TON) exceeding 4000 and a typical turnover frequency (TOF) of 180 h−1. It should be noted that a similar reaction has been reported using blue-light responsive flavin derivatives reaching a TON of up to 68.11 The oxidation catalyzed by 2 proceeds in aqueous solution exposed to sunlight under ambient conditions. Furthermore, no significant product formation occurs in the dark or when the protonated form of the sensitizer 1 is photoexcited under otherwise identical conditions. A monooxygenase type substrate conversion process according to eqn (1) has been proposed,4 where dioxygen is photochemically activated to form hydrogen peroxide.
| RCH2OH + 3O2 → RCHO + H2O2 | (1) |
For the present study, we have changed the reaction conditions to adopt this system for an unprecedented one-pot cascade transformation of alcohols to obtain either the two- or the four-electron oxidized carbonyl compounds using antimony porphyrin sensitizers 1 and 2 under simulated solar light irradiation.
In a typical experiment, 12 ml of a H2O–acetonitrile mixture (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 v/v) containing 6.5 × 10−5 M photocatalyst and 8.3 × 10−2 M benzyl alcohol substrate were irradiated under aerobic conditions with a solar simulator adjusted to an output of approximately 35000 lx at the sample position. When the hexafluorophosphate salt of cation 1 was used as a photosensitizer, no conversion of the alcohol substrate was observed even after 48 h of light exposure. Upon the formation of compound 2 by deprotonation with a stoichiometric amount of NaOH, the photogeneration of benzaldehyde and hydrogen peroxide took place as expected (eqn (1)). The primary aldehyde photoproduct on the other hand could be readily further photooxidized with the protonated form 1 of the catalyst under the same reaction conditions as described above. Thus, for example, a 63% conversion of benzaldehyde to benzoic acid was observed in 12 ml of a H2O–acetonitrile mixture (1:3 v/v) containing 6.5 × 10−5 M photocatalyst 1 and 8.3 × 10−2 M substrate when irradiated for 24 h under aerobic conditions (solar simulator, 35000 lx). Small amounts of benzene were detected in the product mixture under these conditions, which indicates the presence of a decarboxylation side reaction upon prolonged irradiation times. No other reaction products were formed. The consecutive one-pot conversion of the benzyl alcohol substrate is summarized in Scheme 2.
:3 v/v) containing 6.5 × 10−5 M photocatalyst and 8.3 × 10−2 M benzyl alcohol substrate were irradiated under aerobic conditions with a solar simulator adjusted to an output of approximately 35000 lx at the sample position. When the hexafluorophosphate salt of cation 1 was used as a photosensitizer, no conversion of the alcohol substrate was observed even after 48 h of light exposure. Upon the formation of compound 2 by deprotonation with a stoichiometric amount of NaOH, the photogeneration of benzaldehyde and hydrogen peroxide took place as expected (eqn (1)). The primary aldehyde photoproduct on the other hand could be readily further photooxidized with the protonated form 1 of the catalyst under the same reaction conditions as described above. Thus, for example, a 63% conversion of benzaldehyde to benzoic acid was observed in 12 ml of a H2O–acetonitrile mixture (1:3 v/v) containing 6.5 × 10−5 M photocatalyst 1 and 8.3 × 10−2 M substrate when irradiated for 24 h under aerobic conditions (solar simulator, 35000 lx). Small amounts of benzene were detected in the product mixture under these conditions, which indicates the presence of a decarboxylation side reaction upon prolonged irradiation times. No other reaction products were formed. The consecutive one-pot conversion of the benzyl alcohol substrate is summarized in Scheme 2.
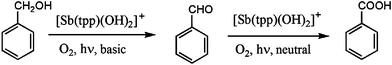 | ||
| Scheme 2 Consecutive photoreaction steps controlled by pH. | ||
In the course of the photolysis, which was also studied in modified solvent mixtures with variable water to acetonitrile ratios and for the transformation of several other aldehyde substrates, a gradual fading of the pink colour of the sensitizer 1 occurs (Fig. 1).
 | ||
| Fig. 1 Photocatalytic conversion of 4-chlorobenzaldehyde (a) to the corresponding carboxylic acid, which crystallizes from the irradiated reaction mixture (b). Reaction conditions: air saturated H2O–CH3CN solvent (1:1 v/v) containing 1 × 10−3 M of substrate and 7.8 × 10−7 M of catalyst 1 1 under simulated sunlight exposure (AM 1.5). | ||
This catalyst bleaching process is thought to be responsible for an incomplete substrate conversion even under extended irradiation times due to a decreasing absorption of actinic light (Fig. 2). Typical degradation quantum yields of the quite robust sensitizer 2 determined for monochromatic excitation with visible light are in the range of Φ = 0.003%.4 In the oxygenation reactions studied here with typical values of TON ≤ 600 and TOF ≤ 14 h−1 at λ ≤ 620 nm, the more rapid destruction of the photocatalyst suggests the operation of a different reaction mechanism for the light-dependent transformation of aldehydes mediated by the protonated complex 1. In the presence of sodium azide, which is acting as a scavenger of singlet oxygen, only traces of reaction products were formed. Furthermore, the efficiency of the process correlates with the reported decay rates of singlet oxygen in different solvents.13 Therefore, we propose that a 1O2-dependent reaction pathway according to eqn (2) is operative in the photocatalytic formation of carboxylic acids mediated by the [Sb(tpp)(OH)2]+ cation 1, which can formally be considered as a dioxygenase type substrate transformation, where both oxygen atoms of O2 are incorporated into the product molecule.
| 2 RCHO + 1O2 → 2 RCOOH | (2) |
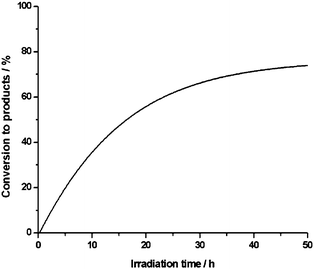 | ||
| Fig. 2 Irradiation time profile for the photochemical oxidation of benzaldehyde to benzoic acid with catalyst 1 in air saturated H2O–CH3CN (1:1 v/v). | ||
The quantum yield for 1O2 production using the [Sb(tpp)(OH)2]+ cation 1 as a sensitizer was determined as ΦΔ = 0.53 for monochromatic irradiation in the Q-band region (546 nm) using 1,3-diphenyl-isobenzofuran (DPBF) as a probe (Fig. 3).
![Degradation of the singlet oxygen scavenger DPBF sensitized by the protonated photocatalyst [Sb(tpp)(OH)2]+ under 546 nm irradiation recorded at t = 0 (a), 30, 60, 90, 120, 150, and 180 s (g). Inset: plot of DPBF absorbance versus time.](/image/article/2012/RA/c2ra01076c/c2ra01076c-f3.gif) | ||
| Fig. 3 Degradation of the singlet oxygen scavenger DPBF sensitized by the protonated photocatalyst [Sb(tpp)(OH)2]+ under 546 nm irradiation recorded at t = 0 (a), 30, 60, 90, 120, 150, and 180 s (g). Inset: plot of DPBF absorbance versus time. | ||
Generation of singlet oxygen in the system is also thought to be involved in the gradual degradation process of the antimony porphyrin catalyst 1 under turnover conditions. Compared to other well-established 1O2-sensitizers such as rose bengal or methylene blue, which were also tested for the transformation of aldehydes to carboxylic acids according to eqn (2), the [Sb(tpp)(OH)2]+ cation however turned out to be more robust and efficient under identical experimental conditions.
A first systematic study of the novel pH- and light-dependent oxidation process summarized in Scheme 2 was carried out including a variation of solvent mixtures, substrates and other relevant parameters in order to improve the conditions and yields for the photocatalytic conversion of aldehydes to carboxylic acids. In Table 1, some of the results obtained in air saturated H2O–CH3CN solvent (1:1 v/v) are summarized. Further details of these investigations can be found in the supplementary information† of the present work.
| Aldehyde | Acid | Time (h) | Conversion (%)b | TONc | TOFd |
|---|---|---|---|---|---|
|
a 6.5 × 10−5 M catalyst (1) and 8.3 × 10−2 M aldehydes as substrates in 12 ml of H2O–acetonitrile (1:1 v/v) in the presence of air and light (35000 lx, solar simulator).
b Conversion of aldehyde to acid.
c Turnover number of catalyst (calculated for two product molecules cycle−1 according to eqn (2)).
d Turnover frequency (h−1).
|
|||||
|
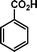
|
72 | 84 | 538 | 7.5 |
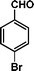
|
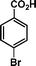
|
32 | 71 | 455 | 14.2 |
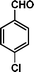
|
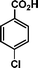
|
72 | 73 | 467 | 6.5 |
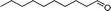
|
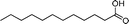
|
80 | 69 | 440 | 5.5 |
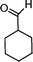
|
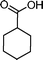
|
72 | 92 | 605 | 8.4 |
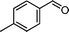
|
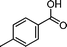
|
48 | 45 | 288 | 6.0 |
In summary, we have presented a novel type of bio-inspired photocatalytic process for the solar chemical conversion of aromatic and aliphatic alcohols to the corresponding aldehydes and carboxylic acids based on abundant and environmentally benign materials. The photosensitized consecutive reaction proceeds in aqueous solvent mixtures with excellent yields and selectivities under ambient conditions, requiring only air supply and visible light or sunlight exposure with a long-wavelength threshold limit of 620 nm. By means of a simple pH-dependent switch, the oxidation of substrates can be readily driven as a one-pot process in a controlled and stepwise fashion to determine the desired product distribution in situ. This unprecedented feature allows an isolation of either the two-electron oxidized compounds (aldehydes) or alternatively the four-electron oxidized products (carboxylic acids). The first mechanistic studies indicate that the photochemical dioxygen activation pathways involved in these reactions drastically differ in the protonated and deprotonated forms of the photosensitizer. While an SbO-based substrate transformation with monooxygenase characteristics occurs in alkaline solution,2,12 a singlet oxygen dependent dioxygenase reactivity with different TON and TOF parameters is observed at lower pH. Further investigations to exploit the synthetic potential of these sunlight-driven redox enzyme models for other applications in green chemical photocatalysis are currently underway.
Acknowledgements
Financial support of this work by the Austrian Science Fund (FWF project P21045: “Bio-inspired Multielectron Transfer Photosensitizers”) is gratefully acknowledged.References
- D. Ravelli, D. Dondi, M. Fagnoni and A. Albini, Chem. Soc. Rev., 2009, 38, 1999 RSC.
- G. Knör and U. Monkowius, Adv. Inorg. Chem., 2011, 63, 235 CrossRef.
- M. Hajimohammadi, N. Safari, H. Mofakham and F. Deyhimi, Green Chem., 2011, 13, 991 RSC.
- G. Knör, ChemBioChem, 2001, 2, 593 CrossRef.
- R. S. Stoll and S. Hecht, Angew. Chem., 2010, 122, 5176 CrossRef ; Angew. Chem. Int. Ed., 2010, 49, 5054.
- G. Knör, Chem.–Eur. J., 2009, 15, 568 CrossRef.
- T. Kieboom, in Catalysis for renewables—from feedstock to energy production, ed. G. Centi, R. A. van Santen, Wiley-VCH, Weinheim, 2007, ch. 13, p. 273 Search PubMed.
- A. Bruggink, R. Schoevaart and T. Kieboom, Org. Process Res. Dev., 2003, 7, 622 CrossRef CAS.
- T. Shiragami, J. Matsumoto, H. Inoue and M. Yasuda, J. Photochem. Photobiol., C, 2005, 6, 227 CrossRef CAS.
- (a) G. Knör, Coord. Chem. Rev., 1998, 171, 61 CrossRef; (b) G. Knör, Chem. Phys. Lett., 2000, 330, 383 CrossRef.
- H. Schmaderer, P. Hilgers, R. Lechner and B. König, Adv. Synth. Catal., 2009, 351, 163 CrossRef CAS.
- G. Stochel, M. Brindell, W. Macyk, Z. Stasicka, K. Szaciłiowski, Bioinorganic Photochemistry, Wiley, Chichester, 2009, ch. 12, p. 198 Search PubMed.
- F. Wilkinson, W. P. Helman and A. B. Ross, J. Phys. Chem. Ref. Data, 1995, 24, 663 CrossRef CAS.
- J. G. Calvert, J. N. Pitts, Photochemistry, Wiley, New York, 1966, p. 748 Search PubMed.
- T. L. C. Figueredo, R. A. W. Johnstone, A. M. P. SantAna Sørensen, D. Burget and P. Jaques, Photochem. Photobiol., 1999, 69, 517 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2ra01076c |
| ‡ Preparation of the photosensitizers: antimony tetraphenylporphyrins Sb(tpp)(OH)2]+ (1) and SbO(tpp)OH (2) were synthesized and purified according to the reported procedure.4 General method for the synthesis of carboxylic acids: the aldehyde (1 × 10−3 mol) and [Sb(tpp)(OH)2]PF6 (7.8 × 10−7 mol) were dissolved in 12 ml of H2O–acetonitrile (1:1 v/v) in a pyrex glass tube filtering off short wavelength UV-light.14 A stream of air at 1 atm pressure was gently bubbled through the solution and the sample was irradiated using simulated AM 1.5 solar light generated with the 300 W xenon lamp of a Luzchem LZC-SSR SPR-01 photoreactor equipped with a SolSim filter combination. The light intensity at the sample position was adjusted to 35000 lx using a Reliability Direct AR823 model digital lux meter. All quantum yields were determined at 546 nm using a Hanovia Xe–Hg 977 B-1 (1 kW) lamp equipped with a Schoeffel GM 250-1 monochromator. DPBF degradation in air-saturated acetone solution was used to determine the singlet oxygen quantum yield of 1 relative to free base tetraphenylporphyrin H2(tpp) as a standard (ΦΔ = 0.67).15 The consumption of the starting aldehydes and the formation of the corresponding carboxylic acids were monitored using a Thermo Finnigan Trace GC with a Fisons MD 800 mass spectrometer. The products were separated on a Restek RTX5 MS column (30 m × 0.32 mm × 0.25 μm) using helium as carrier gas with the following oven program: 40 °C (1 min) to 280 °C (1 min) with 12 °C min−1. Spectral changes were measured with a Varian Cary 300 Bio spectrophotometer. |
| This journal is © The Royal Society of Chemistry 2012 |