H-bonding promotion of peptide solubility and cyclization by fluorinated alcohols†
Hiroshi
Hinou
*ab,
Kei
Hyugaji
a,
Fayna
Garcia-Martin
a,
Shin-Ichiro
Nishimura
a and
Fernando
Albericio
*bc
aGraduate School of Life Science, and Frontier Research Center for Post-Genome Science and Technology, N21W11, Kita-ku, Sapporo 0010021, Japan. E-mail: hinou@sci.hokudai.ac.jp; Fax: 81 11 706 9042; Tel: 81 11 706 9040
bInstitute for Research in Biomedicine (IRB), Barcelona Science Park (PCB) and CIBER-BBN, Networking Centre on Bioengineering, Biomaterials and Nanomedicine, PCB Baldiri Reixac 10, Barcelona 08028, Spain
cDepartment of Organic Chemistry, University of Barcelona, Martii Franques 1–11, Barcelona 08028, Spain
First published on 27th February 2012
Abstract
A promotion effect for peptide cyclization by strong H-bonding of fluorinated alcohols was revealed via a synthetic study of a cyclic AFGP skeleton. Combination of fluorinated alcohol-DCM solvent system and DIC-additive system afforded the cyclic hexapeptide skeleton in more than 80% yield. The ratio of intra- vs. inter-peptide condensation depended upon the H-bonding donor strength. This effect was quenched by H-bond acceptor solvents.
Cyclic peptides and peptide mimetics are often involved in potent and selective molecular recognition because of the combination of its stabilized peptide skeleton and variety of side chains.1 However, the syntheses of cyclic peptides, especially in the case of all-L-cyclic peptide, often encountered low efficiency at the cyclization step because of the poor solubility, steric hindrance, and/or conformational mismatch of the corresponding linear peptide. As is known, the presence of N-alkylation in, for example, Pro and/or D-amino acids favor the cis-peptide bond and therefore the cyclization kinetics.2,3 Thus, in addition to the resin cyclization, which could also favor the intrachain reaction,4 the use of N-backbone protecting groups2 and of pseudoproline moieties3 have been proposed to force the cis form and to overcome these problems. Otherwise highly diluted conditions were adopted to avoid the problem of oligomerization and low solubility.2 However, these techniques were costly, time-consuming and/or difficult to scale up.
Bearing in mind the idea of forcing the cis configuration, we recall the use of fluorinated alcohols. These alcohols, such as 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) and 2,2,2-trifluoroethanol (TFE), are well known solvents to enhance the solubility of the peptides by denaturing the peptide bond structure through their strong hydrogen-bond donor capacity.5 However, studies utilizing them for peptide condensation are quite limited6 because of the inactivation property of intermolecular coupling by the H-bonding;6b to the best of our knowledge, fluorinated alcohols have not been used for peptide cyclization.
In the case of cyclic peptides, the promotion of a strong hydrogen bond between the alcohol and the O of the peptide bond can favor the cis configuration because the existence of the alcohol makes it more stable. Herein, we describe an approach to improve simultaneously solubility and efficiency of cyclization for the practical synthesis of the cyclic hexapeptide 1, a skeleton of cyclic antifreeze glycopeptide 27 (cyclic AFGP;8Fig. 1), from the corresponding linear hexapeptide by using fluorinated alcohol as a co-solvent.
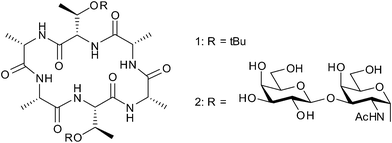 | ||
| Fig. 1 Smallest cyclic AFGP 2 and its cyclic peptide skeleton. | ||
The three possible precursor hexapeptides for the cyclic skeleton (1), H–Thr(tBu)–Ala–Ala–Thr(tBu)–Ala–Ala–OH (3), H–Ala–Thr(tBu)–Ala–Ala–Thr(tBu)–Ala–OH (4), and H–Ala–Ala–Thr(tBu)–Ala–Ala–Thr(tBu)–OH (5), were prepared respectively by solid phase method using 2-chlorotrityl (2-ClTr) resin.9 However, linear hexapeptides 3–5 showed poor solubility in common solvents, such as N,N-dimethylformamide (DMF) and dichloromethane (DCM), for peptide synthesis. To overcome this problem, we focused on the HFIP-DCM mixture, which is well known as a potential solvent system for protected peptides and was often used as cleavage solvent system for the 2-ClTr resin.10 To avoid too much dilution during the scale-up process, 10 mM peptide solution was set as the basic concentration for cyclization tests. All attempts of cyclization of 3 in HFIP-DCM (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4) using stand-alone coupling reagents11 such as 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo-[4,5-b]pyridinium hexafluorophosphate 3-oxide (HATU), 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo-[4,5-b]pyridinium hexafluorophosphate 3-oxide (HBTU), 1-[(1-(Cyano-2-ethoxy-2-oxoethylideneaminooxy)-dimethylamino-morpholinomethylene)]methanaminium Hexafluorophosphate (COMU), azabenzotriazol-1-yl-N-oxy-tris(pyrrolidino)phosphoniumhexafluorophosphate (PyAOP), benzotriazol-1-yl-N-oxy-tris(pyrrolidino)phosphonium hexafluorophosphate (PyBOP), 1-(Cyano-2-ethoxy-2-oxoethylideneaminooxy) tris(pyrrolidino)phosphoniumhexafluorophosphate (PyOxP),11c and diphenyl phosphorazidate (DPPA) in the presence of base failed and unreacted linear peptide was recovered. Only traces of the cyclic product 1 and HFIP ester were detected by LC-ESI/MS when the most potent COMU or HATU were used. Then, we tried 1:1 mixture of N,N′-diisopropylcarbodiimide (DIC) and 7-aza-1-hydroxybenzotriazole(3-hydroxy-3H-1,2,3-triazolo-[4,5-b]pyridine) (HOAt) (DIC-HOAt) as a non-base activation system in HFIP-DCM (1:3) solvent system because of gel formation of 5 in HFIP-DCM (1:4). Interestingly, 3 afforded cyclic hexapeptide 1 in 81.2% in 1 h with a little cyclic dodecapeptide 6 (5.8%), HFIP ester 7 (11.3%), and a trace of the N-DIC adduct of 7. (Fig. 2)
:4) using stand-alone coupling reagents11 such as 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo-[4,5-b]pyridinium hexafluorophosphate 3-oxide (HATU), 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo-[4,5-b]pyridinium hexafluorophosphate 3-oxide (HBTU), 1-[(1-(Cyano-2-ethoxy-2-oxoethylideneaminooxy)-dimethylamino-morpholinomethylene)]methanaminium Hexafluorophosphate (COMU), azabenzotriazol-1-yl-N-oxy-tris(pyrrolidino)phosphoniumhexafluorophosphate (PyAOP), benzotriazol-1-yl-N-oxy-tris(pyrrolidino)phosphonium hexafluorophosphate (PyBOP), 1-(Cyano-2-ethoxy-2-oxoethylideneaminooxy) tris(pyrrolidino)phosphoniumhexafluorophosphate (PyOxP),11c and diphenyl phosphorazidate (DPPA) in the presence of base failed and unreacted linear peptide was recovered. Only traces of the cyclic product 1 and HFIP ester were detected by LC-ESI/MS when the most potent COMU or HATU were used. Then, we tried 1:1 mixture of N,N′-diisopropylcarbodiimide (DIC) and 7-aza-1-hydroxybenzotriazole(3-hydroxy-3H-1,2,3-triazolo-[4,5-b]pyridine) (HOAt) (DIC-HOAt) as a non-base activation system in HFIP-DCM (1:3) solvent system because of gel formation of 5 in HFIP-DCM (1:4). Interestingly, 3 afforded cyclic hexapeptide 1 in 81.2% in 1 h with a little cyclic dodecapeptide 6 (5.8%), HFIP ester 7 (11.3%), and a trace of the N-DIC adduct of 7. (Fig. 2)
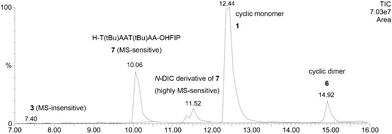 | ||
| Fig. 2 Total ion chromatogram (0.5–2.0 kDa) of LC-ESI/MS for reaction mixture for 3 activated with DIC-HOAt in HFIP-DCM (1:3). | ||
On the other hand, hexapeptides 4 and 5 also afforded 1 in 13.5 and 24.5% yield respectively with the corresponding HFIP ester (75.4% and 50.2%) as the main product. Replacement of the C-terminal amino acid with the D-isomer (compound 8) selectively promoted cyclization as reported2 to yield cyclic hexapeptide 9 in 80.6% yield without detectable cyclic dimer, and the corresponding HFIP ester was found only in 3.9% (Table 1).
:3) for 1 h
| Linear peptides | Cyclic monomera | Cyclic dimera | HFIP estera |
|---|---|---|---|
| a Yields (%) were estimated from HPLC peak area (220 nm). b Detected by LC-ESI/MS. c ND = not detected. | |||
| T(tBu)AAT(tBu)AA (3) | 81.2 (1) | 5.8 (6) | 11.3 (7) |
| AT(tBu)AAT(tBu)A (4) | 13.5 | 3.8 | 75.4 |
| AAT(tBu)AAT(tBu) (5) | 24.9 | Traceb | 50.2 |
| T(tBu)AAT(tBu)AdA (8) | 80.6 (9) | NDc | 3.9 |
Next, we focused on the effect of the DIC additive as well as the solvent system for peptide 3 (Table 2). Without additives, HFIP ester was formed as the major product (# 1), showing the power of the O-acylisourea as the active species. Replacement of HOAt by 1-hydroxybenzotriazole (HOBt) or ethyl cyanoglyoxylate-2-oxime (Oxyma)11d decreased the yield of 1 to 20.6% and 14.2% respectively in HFIP-DCM (1:4). However, Oxyma seemed to suppress the formation of cyclic dimer (# 2–4). Using a non-fluorinated alcohol-DCM system, such as MeOH, EtOH, 2-PrOH, and tBuOH, and DMF decreased both solubility of 3 and reaction rate with the DIC-HOAt system (# 5–9). Nishino et al. reported that HFIP itself is just a barren medium for peptide coupling but this character can be changed by mixing it with a proton accepting solvent such as DMF.6b In contrast to this, HFIP is well known as a potent promoting solvent of double bond oxidation by hydrogen peroxide,5 but this property is quenched by mixing it with a H-bond acceptor such as dioxane.5a To elucidate how the H-bonding property of HFIP influenced the peptide cyclization and solvation, these two solvent systems, HFIP-DMF (1:1) and HFIP-dioxane (1:1), were applied to the cyclization reaction with DIC-HOAt (# 10, 11). As a result, both solvent systems decrease not only the reactivity but solubility of 3. Peptide 3 (10 mM) seemed to dissolve in the HFIP-DMF system but formed clear gel and afforded only 5.1% of 1 after 1 h reaction (# 10). The HFIP-dioxane system forms a suspension mixture with 3 (10 mM) and afforded 7.1% of 1 after 1 h reaction (# 11). In contrast to these results, the same reaction in the 1:1 mixture of HFIP-DCM afforded 66.6% of 1 and decreased cyclic dimer 6 to only 1.2% (# 12). Although formation of HFIP ester 7 was increased, the more the HFIP in the HFIP-DCM type solvent system that was used, the greater was the ratio of cyclic monomer 1versus dimer 2 that was afforded (# 2, 12). These results indicate that the H-bonding property of HFIP facilitates the peptide cyclization by DIC-HOAt system as well as increasing the solubility of the peptide.
| # | Solvent system | Additive | 1 a | 6 a | Estera |
|---|---|---|---|---|---|
| a Yields (%) were estimated from HPLC peak area (220 nm). b Detected by LC-ESI/MS. c Suspended. d ND = not detected. | |||||
| 1 | HFIP-DCM (1:4) |
— | 2.2 | traceb | 93.5 |
| 2 | HFIP-DCM (1:4) |
HOAt | 83.0 | 5.2 | 8.4 |
| 3 | HFIP-DCM (1:4) |
HOBt | 20.6 | 2.5 | 52.3 |
| 4 | HFIP-DCM (1:4) |
Oxyma | 14.2 | traceb | 26.9 |
| 5 | MeOH-DCM (1:1)c |
HOAt | 4.6 | traceb | 2.8 |
| 6 | EtOH-DCM (1:1)c |
HOAt | 0.7 | traceb | 1.1 |
| 7 | 2-PrOH-DCM (1:1)c |
HOAt | 0.3 | traceb | 0.8 |
| 8 |
tBuOH-DCM (1:1)c |
HOAt | 0.2 | traceb | NDd |
| 9 | DMFc | HOAt | 4.8 | 0.4 | — |
| 10 | HFIP-DMF (1:1) |
HOAt | 5.1 | 0.4 | 5.7 |
| 11 | HFIP-dioxane (1:1)c |
HOAt | 7.1 | 0.4 | 8.2 |
| 12 | HFIP-DCM (1:1) |
HOAt | 66.6 | 1.2 | 16.9 |
Next, we focused on TFE which has weaker H-bonding properties (pKa = 12.9) than HFIP (pKa = 9.3). The reaction was tested in a 1:1 mixture with DCM because 10 mM of 3 formed a clear gel in TFE-DCM (1:4). All results using this solvent system are shown in Table 3. As in the HFIP system, a large formation of ester product took place without additive (# 1). In the presence of HOAt, cyclic monomer 1 was afforded in excellent yield (82.1%) and formation of ester by-product was decreased (2.5%) compared to HFIP-DCM system, but the formation of the cyclic dimer 6 was increased (11.2%) (# 2). These results also suggested that stronger H-bonding or larger steric hindrance of the HFIP contribute to the higher selectivity for cyclic monomer 1 than dimer 6. However, the larger pKa of the TFE contributes to suppressing ester formation. These results indicate that the stronger hydrogen bonding suppresses the inter-peptide reaction and favors the intra reaction. These differences between HFIP and TFE were reproduced after changing the peptide sequence from 3 to 4, 5, or 8 (# 3–5). In all cases ester formation was suppressed. Alanine terminated sequence 4 was preferred to inter-peptide reaction. This property of 4 might contribute to the condensation polymerization to afford the linear AFGP oligomer in non H-bonding solvents such as DMF, as shown in our previous study (# 3).8a The peptide 5 which has a threonine residue on the C-terminal preferred intra-peptide coupling to afford 1 as the major product (# 4). The peptide 8, the C-terminal D-isomer of 3, afforded the cyclic monomer 9 almost quantitatively (# 5). In contrast to the HFIP system, replacement of HOAt with HOBt or Oxyma also afforded 1 in high yield and Oxyma showed a suppression effect of dimerization which supposedly came from the lower steric hindrance of Oxyma11 than HOAt (# 3, 4). Dilution of the reaction condition might also contribute to the selectivity of 1 without increasing ester formation. In all cases, racemized product 9 (> 1%) could not be detected. Sakakibara also reported a reduced racemization during fragment coupling of long peptides in the presence of fluorinated alcohol.6c The strong H-bonding and weak H-withdrawing property of fluorinated alcohol might be attributed to the lowered racemization.
:1) for 1 h
| # | Linear peptides | Additive | Cyclic monomera | Cyclic dimera | TFE estera |
|---|---|---|---|---|---|
| a Yields (%) were estimated from HPLC peak area (220 nm). b ND = not detected. c Detected by LC-ESI/MS. | |||||
| 1 | 3 | — | 5.3 | 1.3 | 88.7 |
| 2 | 3 | HOAt | 82.1 | 11.2 | 2.5 |
| 3 | 4 | HOAt | 22.1 | 53.0 | 15.9 |
| 4 | 5 | HOAt | 72.1 | 11.5 | 12.6 |
| 5 | 8 | HOAt | 96.9 | NDb | Tracec |
| 6 | 3 | HOBt | 78.0 | 11.0 | 10.1 |
| 7 | 3 | Oxyma | 84.7 | 6.1 | 2.5 |
In summary, we have developed an efficient peptide cyclization method by the combination of a fluorinated alcohol containing solvent system and DIC-additive activation system. The strong H-bonding property not only enhanced the solubility and intra-peptide cyclization by both the denaturing effect for peptide structure and by favoring the cis configuration. Furthermore, it inhibited the inter-peptide coupling. Although the efficiency of cyclization still depends on the sequence of the linear peptide used, this study could afford a potential strategy for the synthesis of cyclic peptides and peptide mimetics1–4 without any modification or highly diluted conditions.
Acknowledgements
This work was supported by Excellent Young Researcher Overseas Visit Program (no. 21-4145) from JSPS, Grant-in-Aid for Scientific Research (B, no. 23350074) from MEXT Japan, and b CICYT (CTQ2009-07758), the Generalitat de Catalunya (2009SGR 1024), the Institute for Research in Biomedicine, and the Barcelona Science Park.References
- (a) Reviews of functions of cyclic peptides and peptide mimetics: D. P. Fairlie, G. Abbenante and D. R. March, Curr. Med. Chem., 1995, 2, 654–686 CAS; (b) J. D. Hartgerink, T. D. Clark and M. R. Ghadiri, Chem.–Eur. J., 1998, 4, 1367–1372 CrossRef CAS; (c) E. M. Nolan and C. T. Walsh, ChemBioChem, 2009, 10, 34–53 CrossRef CAS.
- A. Ehrlich, H.-U. Heyne, R. Winter, M. Beyermann, H. Haber, L. A. Carpino and M. Bienert, J. Org. Chem., 1996, 61, 8831–8838 CrossRef CAS.
- (a) D. S. Skropeta, K. A. Jolliffe and P. Turner, J. Org. Chem., 2004, 69, 8804–8809 CrossRef CAS; (b) N. Sayyadi, D. Skropeta and K. A. Jolliffe, Org. Lett., 2005, 7, 5497–5499 CrossRef CAS; (c) K. A. Fairweather, N. Sayyadi, I. J. Luck, J. K. Clegg and K. A. Jolliffe, Org. Lett., 2010, 12, 3136–3139 CrossRef CAS.
- (a) S. A. Kates, N. A. Solé, C. R. Johnson, D. Hudson, G. Barany and F. Albericio, Tetrahedron Lett., 1993, 34, 1549–1552 CrossRef CAS; (b) J. N. Lambert, J. P. Mitchell and K. D. Roberts, J. Chem. Soc., Perkin Trans. 1, 2001, 471–484 RSC; (c) J. M. Humphrey and A. R. Chamberlin, Chem. Rev., 1997, 97, 2243–2266 CrossRef CAS.
- (a) A. Berkessel, J. A. Adrio, D. Hüttenhain and J. M. Neudörfl, J. Am. Chem. Soc., 2006, 128, 8421–8426 CrossRef CAS; (b) N. H. Andersen, R. B. Dyer, R. M. Fesinmeyer, F. Gai, Z. H. Liu, J. W. Neidigh and H. Tong, J. Am. Chem. Soc., 1999, 121, 9879–9880 CrossRef CAS; (c) H. R. Mulla and A. Cammers-Goodwin, J. Am. Chem. Soc., 2000, 122, 738–739 CrossRef CAS.
- (a) S. C. F. Milton and R. C. de L. Milton, Int. J. Pept. Protein Res., 1990, 36, 193–196 CrossRef CAS; (b) N. Nishino, H. Mihara, Y. Makinose and T. Fujimoto, Tetrahedron Lett., 1992, 33, 7007–7010 CrossRef CAS; (c) S. Sakakibara, Biopolymers, 1999, 51, 279–296 CrossRef CAS.
- M. Hachisu, H. Hinou, M. Takamichi, S. Tsuda, S. Koshida and S.-I. Nishimura, Chem. Commun., 2009, 1641–1643 RSC.
- (a) Y. Tachibana, G. L. Fletcher, N. Fujitani, S. Tsuda, K. Monde and S.-I. Nishimura, Angew. Chem., Int. Ed., 2004, 43, 856–862 CrossRef CAS; (b) R. Peltier, M. A. Brimble, J. M. Wojnar, D. E. Williams, C. W. Evans and A. L. DeVries, Chem. Sci., 2010, 1, 538–551 RSC; (c) J. Garmer and M. M. Harding, ChemBioChem, 2010, 11, 2489–2498 CrossRef.
- K. Barlos, D. Gatos, J. Kallitsis, G. Papaphotiu, P. Sotiriu, Y. Wenqing and W. Schafer, Tetrahedron Lett., 1989, 30, 3943–3946 CrossRef CAS.
- R. Bollhagen, M. Schmiedberger, K. Barlos and E. Grell, J. Chem. Soc., Chem. Commun., 1994, 2559–2560 RSC.
- (a) Recent review of coupling reagents: E. Valeur and M. Bradley, Chem. Soc. Rev., 2009, 38, 606–631 RSC; (b) A. El-Faham, R. Subirós-Funosas, R. Prohens and F. Albericio, Chem.–Eur. J., 2009, 15, 9404–9416 CrossRef CAS; (c) R. Subirós-Funosas, A. El-Faham and F. Albericio, Org. Biomol. Chem., 2010, 8, 3665–3673 RSC; (d) R. Subirós-Funosas, R. Prohens, R. Barbas, A. El-Faham and F. Albericio, Chem.–Eur. J., 2009, 15, 9394–9403 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2ra01043g |
| This journal is © The Royal Society of Chemistry 2012 |