DOI:
10.1039/C2RA00981A
(Paper)
RSC Adv., 2012,
2, 3541-3547
Advanced electrochemical properties of Mo-doped Li4Ti5O12 anode material for power lithium ion battery
Received
29th October 2011
, Accepted 2nd February 2012
First published on 2nd March 2012
Abstract
Mo6+-doped Li4Ti5−xMoxO12 (0 ≤ x ≤ 0.2) samples have been synthesized via a simple solid-state reaction. The products were characterized by X-ray diffraction (XRD), Raman spectroscopy (RS), scanning electronic microscope (SEM), cyclic voltammetry (CV), electrochemical impedance spectroscopy (EIS), and galvanostatic charge-discharge testing. Li4Ti5−xMoxO12 (x = 0, 0.05) shows the pure phase structure, but several impurity peak can be detected when x ≥ 0.1. Mo-doping did not change the electrochemical reaction process and basic spinel structure of Li4Ti5O12. The particle size of the Li4Ti5−xMoxO12 (0≤ x ≤ 0.2) sample was about 2–3 μm and Li4Ti5O12 has less agglomeration. Electrochemical results show that the Mo6+-doped Li4Ti5O12 samples display a larger diffusion coefficient of lithium ions, lower charge transfer resistance, higher rate capability and excellent reversibility. The Li4Ti5−xMoxO12 (x = 0.1, 0.15) sample maintained considerable capacities until 6 C rates, whereas pristine Li4Ti5O12 shows a severe capacity decline at high rates. After 100 cycles, the specific reversible capacities of Li4Ti5O12 and Li4Ti4.9Mo0.1O12 are 195.8 and 210.8 mAh g−1, respectively. The superior cycling performance and wide discharge voltage range, as well as simple synthesis route and low synthesis cost of the Mo-doped Li4Ti5O12 are expected to show a potential commercial application.
1. Introduction
Rechargeable lithium-ion batteries have been considered as the most advanced energy storage systems for future electric vehicles (EVs) and hybrid electric vehicles (HEVs). However, safety remains the foremost factor delaying the rapid implementation of Li-ion technology into the field of automotive transportation. In order to enhance the safety of lithium ion batteries, flame retardants have been widely used to improve thermal stability.1,2 However, safety concerns related to the combination of high energy electrode materials and electrolytes are still a main obstacle to the commercialization and use of large scale lithium-ion batteries. Developing electrode materials with high safety and combination with electrolytes is one of the key challenges for lithium-ion batteries. One of the key safety issues in anodes for LIBs would be the dendritic lithium growth on the anode surface at high charging current.3 Although many attempts have been made and certain results have been achieved, the search for an ideal anode material is still necessary for the improvement of cell safety. Spinel Li4Ti5O12 (LTO) has been viewed as one promising alternative to graphite to be a safe anode material in lithium-ion batteries due to the high insertion potential of 1.55 V,4 which can avoid the reduction of electrolyte on the surface of electrode.5–7 In addition, Li4Ti5O12 has no solid–electrolyte interface, and this allows the electrode to meet the high power and abuse tolerance requirements needed for power sources.8 Considering the safety of lithium-ion batteries supplying the HEV, an attempt has been developed by combining the 1.5 V Li4Ti5O12 anodes with another cathode, and the safety of lithium-ion batteries can be obviously improved.9 There will be various possible candidates to replace the C/LiCoO2 cell, such as 2 V Li4Ti5O12/LiCoO210 and Li4Ti5O12/LiFePO4 (LFP),11 2.5 V Li4Ti5O12/LiMn2O4(LMO) 12,13 and the 3 V Li4Ti5O12/LiNi0.5Mn1.5O4 cells.14,15 Unfortunately, Li4Ti5O12 exhibits poor electronic and lithium ionic conductivities, and these limit its high rate-capability. Our group has reviewed the doping and surface modification of LTO to improve the electrochemical performance.16 It can be found that the conductivity and electrochemical performance of Li4Ti5O12 can be improved by doping some ions. Recently, Gao et al. reported that Mo doping can enhance the intrinsic conductivity and Li-ion diffusion coefficient, and then improve the electrochemical performance of LiFePO4.17 Zhong 18 reported Mo4+-doped Li4Ti5−xMoxO12 (0.5 ≤ x ≤ 1.5) synthesized by the Pechini method at low oxygen partial pressure (1.8 × 10−12 Pa), but the rate-capability performance is not reported. In addition, it has been reported that there is a possibility to increase the electronic conductivity of Li4Ti5O12 is to substitute multiply charged ions (i.e., M5+ or M6+) on a Ti4+ site, which should lead to an increase in electron concentration.19 According to previous reports, the electronic conductivity of Li4Ti5O12 can be increased by a substitution of Ti4+ site by high valence ions (such as V5+, Nb5+).20,21 As we know, the replacement of graphite by LTO in a lithium-ion battery will result in a reduction in the operating cell voltage, and then reduce the overall energy density at the cell level. Therefore, it is necessary to study the electrochemical behaviors of Li4Ti5O12 anode materials at lower voltage (down to 0 V) considering the safety and energy density. From a commercial viewpoint, the solid-state synthesis of LTO powders exhibit a potential commercial application due to the simple synthesis route and low synthesis cost. To our knowledge, no investigation has reported the rate cycling performance of Mo6+-doped Li4Ti5O12, discharged to 0 V, synthesized by a solid state method. In the present work, we report the rate cycling performance of Mo6+-doped Li4Ti5−xMoxO12 compound (0≤ x ≤ 0.2) prepared by a solid-state method at air. The electrochemical performances of Mo6+-doped Li4Ti5−xMoxO12, including cycling stability and rate capability, were extensively evaluated in half cells, and Mo doping achieved some encouraging results.
2. Experimental
Mo6+-doped Li4Ti5−xMoxO12 compounds (0≤ x ≤ 0.2) were prepared using a solid-state method from TiO2 (rutile structure, AR), LiOH (AR) and (NH4)2Mo2O7 (AR). The stoichiometric molar ratio of Li![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ti:Mo was maintained as 4.3:5−x:x. The starting materials were ball-milled in an ethanol liquid medium to form a slurry, and then dried in a vacuum chamber at 120 °C for 8 h. The dried and mixed reactant mixture was heated at 500 °C for 5 h in air. Then, the resulting precursor was ground into a fine powder and calcined at 830 °C in air for 18 h to obtain the final spinel product. Excessive Li was provided to compensate for the volatilization of Li during synthesis.
:Ti:Mo was maintained as 4.3:5−x:x. The starting materials were ball-milled in an ethanol liquid medium to form a slurry, and then dried in a vacuum chamber at 120 °C for 8 h. The dried and mixed reactant mixture was heated at 500 °C for 5 h in air. Then, the resulting precursor was ground into a fine powder and calcined at 830 °C in air for 18 h to obtain the final spinel product. Excessive Li was provided to compensate for the volatilization of Li during synthesis.
Powder X-ray diffraction (XRD) employing Cu-Kα1 (10° < 2θ < 80°) radiation was used to identify the crystalline phase of the prepared powders. A Hitachi S-4000 scanning electron microscope (SEM) was used for morphological studies. Raman measurements were performed on a SPEX-1403 Raman spectrometer. The laser light source was the 488 nm line of an Ar+ laser excited at 400 mW. All the powder samples were pressed into pellets before measurements. Cyclic voltammetry in two electrode coin-type cells, were carried out using a CHI 852C electrochemical working station between 0 and 3 V (vs. Li/Li+) at a scanning rate of 0.2 mV s−1. EIS measurements were carried out in two-electrode cells by using a Zahner Zennium IM6ex electrochemical workstation with a ±5 mV AC signal and a frequency range from 104 to 0.1 Hz. Galvanostatic charge–discharge cycling was carried out using a multi-channel battery tester from Land 2000 T (Wuhan, China) Battery Testing System, in two electrode coin-type cells (CR 2032). The cells were tested at 0.1 C (1 C = 175 mA g−1), 3.5 C and 6 C charge–discharge rate between 0.0 and 2.5 V (versus Li/Li+), respectively.
For the electrochemical measurements of the Li4Ti5−xMoxO12 samples, a stoichiometric ratio of 80:10:10 of active material (Li4Ti5−xMoxO12):conductive carbon (carbon black):polyvinylidene fluoride (PVDF) binder dissolved in N-methylpyrolline was used for electrode fabrication. The mixture was prepared as a slurry and spread onto copper foil using the doctor blade technique. Following evaporation of the solvent and drying, electrodes were incorporated into an electrochemical cell (coin cell CR 2032) in an Ar-filled glove box using a metal lithium foil as the counter electrode, 1M LiPF6 in a 1:1 (v/v) mixture of ethylene carbonate (EC) and dimethyl carbonate (DMC) as the electrolyte and Celgard 2300 polypropylene as the separator. After assembling, the cells were stored at room temperature for about 12 h to ensure complete impregnation of the electrodes and separators with the electrolyte solutions.
3. Results and discussion
The synthesis condition of Li4Ti5−xMoxO12 in the present paper is in air, and the molybdenum ion tends to be Mo6+ that may incorporate into targeted phases of Li4Ti5−xMoxO12. Fig. 1 shows the XRD patterns of Li4Ti5−xMoxO12 (0≤ x ≤ 0.2) synthesized by the solid state method. The pattern in Fig. 1b indicates the enlarged (111) peaks. The diffraction peaks of all the samples belong to an ordered spinel indexed in cubic Fd-3m. Li4Ti5−xMoxO12 (x = 0, 0.05) shows the pure phase structure, but several impurity peaks can be detected when x ≥ 0.1. It implies only a few substituted Mo6+ cations can enter into the lattices to form a solid solution instead of a mixture. As we know, Mo6+ (0.59 Å 22) ions have a smaller ionic radius than those of Li+ ions, Ti4+ ions (0.605 22), and Ti3+ ions (0.67 22) considering the coordination numbers. According to our previous work, the ionic radii of Li+ in the octahedron and tetrahedron site of Li4Ti5O12 are 0.76 and 0.64 Å, respectively.20Fig. 1b reveals that the peaks almost do not shift with the doping of Mo6+ when x ≥ 0.1, but there is a slight shift in the diffraction peaks to the lower 2θ values for the sample Li4Ti4.95Mo0.05O12 compared to LTO because the Ti4+ can be reduced to Ti3+. The valence change may influence the electronic configuration and generate some electron holes, which are helpful to enhance electron conductivity. Hence, it can be expected that Mo-doped Li4Ti5O12 materials have a higher electronic conductivity than that of Li4Ti5O12.
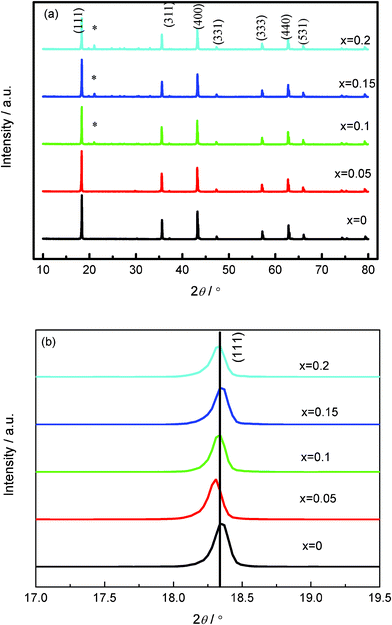 |
| | Fig. 1 XRD patterns for Mo6+-doped Li4Ti5−xMoxO12 (0 ≤ x ≤ 0.2). | |
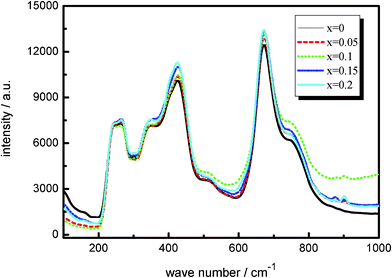
The typical Raman scattering features of the doped and non-doped Li4Ti5O12 are well-evidenced in all the samples as shown in Fig. 2. Each spectrum shows five main Raman bands at about 263, 342, 424, 671, and 745 cm−1 representing the features of the spinel structure (A1g + Eg + 3F2u).23,24 The only difference is that the intensities of the Raman signals for the doped samples are higher than those for the non-doped compound. This observation demonstrated that the doping of Mo6+ can not change the basic spinel structure of Li4Ti5O12.
Fig. 3 shows the scanning electron microscope (SEM) images of Li4Ti5−xMoxO12 (0 ≤ x ≤ 0.1). All materials show a particle size of about 2–3 μm. At the same time, Li4Ti5O12 is more agglomerated than the substituted materials. A good dispersion of Mo-doped Li4Ti5O12 may increase the electrode–electrolyte contact area and facilitate the Li+ and electron transportation, and so improve the high rate performance.
 |
| | Fig. 3 SEM images of Li4Ti5−xMoxO12 (a) x = 0, (b) x = 0.05 and (c) x = 0.1. | |
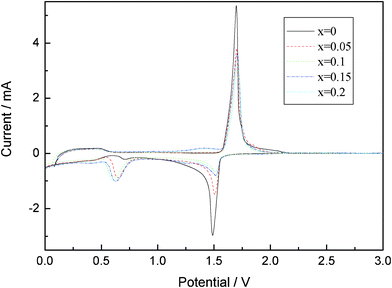 |
| | Fig. 4 Cyclic voltammetry (CV) between 0 V and 3 V with a scan rate of 0.2 mV s−1 of Li4Ti5−xMoxO12 (0 ≤ x ≤ 0.2). | |
Fig. 4 presents the typical steady-state cyclic voltammetry (CV) of Li4Ti5−xMoxO12 (0 ≤ x ≤ 0.2) obtained at a slow scan rate of 0.2 mV s−1. The oxidation and reduction peaks of Li4Ti5O12 are similar to those of Mo-doped Li4Ti5O12. This reveals that the basic spinel structure of Li4Ti5O12 is not changed by Mo6+ doping. The couple of peaks at about 1.7 V could be attributed to the redox of Ti4+/Ti3+,25 and the reduction and oxidation peaks below 0.6 V caused by a multi step restoration of Ti4+.26,27 Even with such a slow rate in the CV, the degree of the polarization was observed to be different among the samples. The potential difference between the anodic and cathodic peaks of Li4Ti5−xMoxO12 (x = 0, 0.05, 0.1, 0.15, 0.2) is about 210, 194, 188, 189 and 196 mV. This implies to some extent the enhanced electrochemical kinetics of Mo-doped Li4Ti5O12 electrode compared with pure Li4Ti5O12. This observation suggests that the Mo doping is beneficial to the reversible intercalation and de-intercalation of Li+, and then enhances the reversibility of the Li4Ti5O12.20,28Fig. 5 shows the corresponding charge–discharge curves at a 0.1 C rate. Fig. 6 shows the differential capacity discharge and charge curves for the first cycle obtained from Fig. 5. It can be seen that all the discharge curves present two flat plateaus near 1.55 and 0.7 V, and an inclined curve from 0.6 to 0.0 V. The charge and discharge flat plateaus near 1.5 V indicate the characteristic of a two phase reaction from Li4Ti5O12 to Li7Ti5O12, as the lithium ions occupy the vacant 8a and 16c sites of the host spinel. The incline in the voltage curves for the samples most probably indicates the existence of a single phase region. The appearance of the sloped regions indicates the new electrochemical insertion processes below 1.0 V. The first discharge capacities of Li4Ti5−xMoxO12 (x = 0, 0.05, 0.1, 0.15, 0.2) between 0 and 2.5 V at a 0.1 C charge–discharge rate are 253, 246, 276, 308 and 356 mAh g−1, respectively. It is obvious that an appropriate Mo doping improves the initial discharge capacity of Li4Ti5O12, and the initial discharge capacity of Li4Ti5−xMoxO12 (x = 0.15, 0.2) is higher than those of the theoretical capacities of 298 mAh g−1. As reported by our group,20 it is known that extra capacity is contributed to the electrolyte decomposition to form a surface film on the sample. The 1st, 10th, 20th and 30th discharge profiles of Li4Ti5O12 at 0.1 C and 3.5 C rate are plotted in Fig. 7. From Fig. 7, it can be seen that Li4Ti5O12 shows a gradual loss of the 1.55 V plateau with increasing the cycling number at low-rate charge–discharge (0.1 C), and this may be due to the low discharge voltage (discharged to 0 V). However, a rapid loss of the 1.55 V plateau with increasing the cycling number can be found, and this may be due to the high electrochemical polarization during high-rate charge–discharge (3.5 C).
 |
| | Fig. 5 Charge–discharge curves of Li4Ti5−xMoxO12 (0 ≤ x ≤ 0.2) at a 0.1 C charge–discharge rate (a) x = 0, (b) x = 0.05, (c) x = 0.1, (d) x = 0.15 and (e) x = 0.2. | |
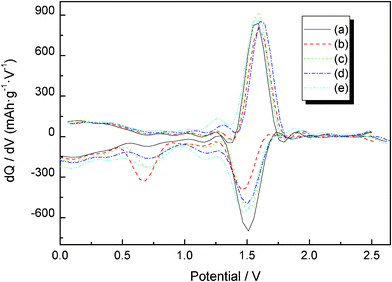 |
| | Fig. 6 Initial charge–discharge characteristics curves and differential capacity vs. voltage plots of Li4Ti5−xMoxO12/Li coin cells carried out galvanostatically between 0 and 2.5 V. | |
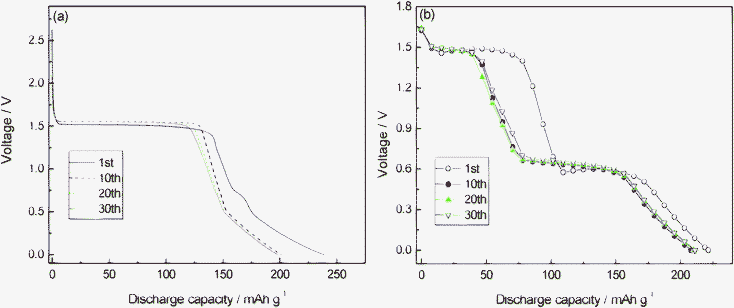 |
| | Fig. 7 Discharge profiles of Li4Ti5O12 at different current densities and cycles (a) 0.1 C; (b) 3.5 C. | |
Fig. 8 shows the reversible capacities of the investigated samples observed in the continuous cycling at rates of 0.1 C, 3.5 C and 5 C at room temperature, respectively. The Figure indicates that the specific capacity and the cycling performance of the Mo-doped Li4Ti5O12 are obviously better than those of the pristine Li4Ti5O12. At low charge–discharge rate (0.1 C), Li4Ti5−xMoxO12 (x = 0.1, 0.15, 0.2) exhibits a high discharge capacity at every cycle as shown in Fig. 8a. The initial specific capacities of Li4Ti5−xMoxO12 (x = 0, 0.05, 0.1, 0.15, 0.2) at 3.5 C rate are 221.5, 222.3, 249.8, 278.3 and 295.7 mAh g−1, respectively. After 100 cycles, their capacities are 210.8, 211.8, 214.3, 220.7 and 202.6 mAh g−1, respectively. This reveals that Mo-doped Li4Ti5O12 (0.05 ≤ x ≤ 0.15) exhibits a higher discharge capacity than that of pure Li4Ti5O12 during cycling. The reason may be as follows: (1) the Mo6+ doping can increase the electro-active Ti3+, and the doped samples allow Li+ ions to be extracted more, and so improve the discharge capacity; (2) the reduction of Mo6+ to Mo4+ can also increase the resulting capacity according to the previous reported work.29 Similar research works, such as Mo6+-doped LiFePO429 and LiNi1/3Co1/3Mn1/3O2,30 have also been proposed by other researchers, but Li4Ti5−xMoxO12 (x = 0.15) shows the highest discharge capacity. Li4Ti5−xMoxO12 (x = 0.1, 0.15) samples maintained considerable capacities until 6 C rates, whereas pristine Li4Ti5O12 shows severe capacity decline at high rates. After 100 cycles, the specific capacities of Li4Ti5O12 and Li4Ti4.9Mo0.1O12 are 195.8 and 210.8 mAh g−1, respectively. As we know, the capacity fading experienced in Li4Ti5O12 electrodes is generally less than that in other electrodes, as Li4Ti5O12 has a unique robust structure and exhibits zero strain properties due to insertion.31 However, the apparently larger capacity fading that occurs at high C-rates is due to the poor electric conductivity of pristine Li4Ti5O12. The capacity fading is relatively lower and can be eventually attributed to the enhancement of electric conductivity due to the Mo doping. Li4Ti5−xMoxO12 (x = 0.2) shows the relatively lower cycling performance, and it may be due to the too much impurity as plotted in Fig. 1. Hence, Mo-doped Li4Ti5O12 could be successfully used as the anode in a hybrid cell suitable for high power applications.
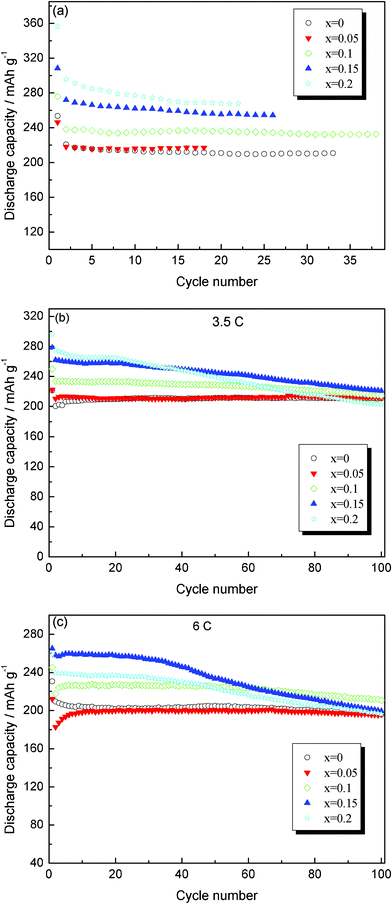 |
| | Fig. 8 Discharge capacity versus cycle number for Li4Ti5−xMoxO12 electrodes at different charge–discharge rates: (a) 0.1 C, (b) 3.5 C and (c) 6 C. | |
Electrochemical impedance spectroscopy (EIS) measurements of Li4Ti5−xMoxO12 (0 ≤ x ≤ 0.2) were carried out in the frequency range from 104 to 0.1 Hz before galvanostatic cycles, and typical Nyquist plots are given in Fig. 9. The equivalent circuit was given in our previous work.20 The inclined lines at low frequency correspond to lithium diffusion within the active material,32 and the capacitive loop is caused by charge transfer resistance (Rct).33 The depressed semicircles at high frequency are reflective of the SEI film.34 Obviously, Li4Ti5O12 has the highest charge transfer resistance, and the charge transfer resistance decreases with increasing Mo doping as shown in Fig. 10. This reveals that the Mo doping improves the electronic conductivity of the electrode in the local environment and significantly boost the electrochemical activity as the polarization of the electrode is greatly reduced, and then results in its relatively higher cycle performance.
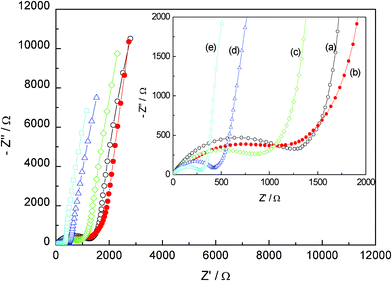 |
| | Fig. 9 EIS curves (Nyquist plots) for Li4Ti5−xMoxO12 electrodes (a) x = 0, (b) x = 0.05, (c) x = 0.1, (d) x = 0.15 and (e) x = 0.2). | |
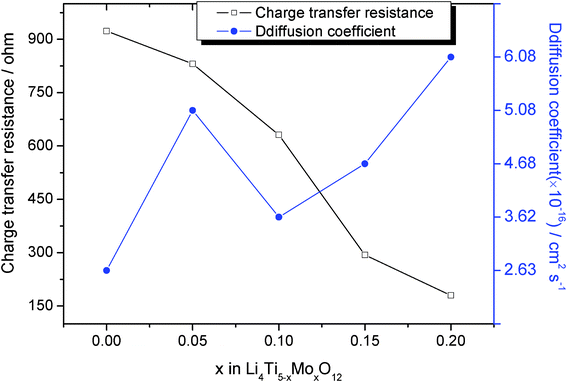 |
| | Fig. 10 Charge transfer resistance and diffusion coefficients of x in Li4Ti5−xMoxO12. | |
The lithium ion diffusion coefficient (DLi) is calculated according to the following equation:35,36
| | 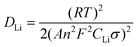 | (1) |
where
n is the number of electrons per molecule during oxidization,
A the surface area of the electrode,
R the gas constant,
T the absolute temperature,
F the Faraday constant,
CLi the concentration of lithium ion, and
σ is the Warburg factor which has relationship with
Zre:
| | 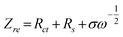 | (2) |
Fig. 11 shows the relationship between Zre and the square root of frequency (ω−1/2) in the low-frequency region. The diffusion coefficient of Li-ions can be calculated based on eqn (1) and (2), and the calculated result is given in Fig. 10. It is obvious that Mo-doped Li4Ti5O12 has a larger diffusion coefficient of lithium ions than that of pristine Li4Ti5O12. This shows that there is an apparent improvement in the kinetics of lithium ions during intercalation and de-intercalation from the doped Mo.
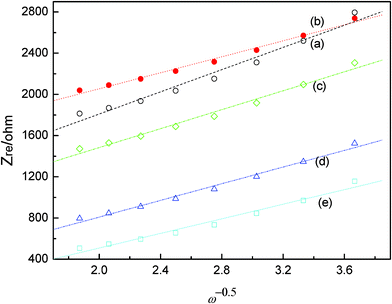 |
| | Fig. 11 Relationship between Zre and ω−1/2 in the low-frequency region of Li4Ti5−xMoxO12 (x = 0, 0.05, 0.1, 0.15, 0.2). | |
4. Conclusions
Li4Ti5−xMoxO12 (x = 0, 0.05, 0.1, 0.15, 0.2) spinel was prepared by a solid-state reaction. Though the particle size is several microns, the Mo-doped Li4Ti5O12 material shows high rate capability and excellent cyclability. A capacity value of 298 mAh g−1 is achieved at the 0.1 C charge-discharge rate, and Mo doping improves the initial discharge capacity. Li4Ti5−xMoxO12 (x = 0.1, 0.15) sample maintained considerable capacities until 6 C rates, whereas pristine Li4Ti5O12 shows a severe capacity decline at high rates. Higher rates affect the moderate decrease in capacity (210.8 mAh g−1) of Li4Ti4.9Mo0.1O12 with more than 70% of the theoretical capacity (298 mAh g−1) retained at 6 C after 100 cycles. This implies to some extent the enhanced electrochemical kinetics of Mo-doped Li4Ti5O12 electrode compared with pure Li4Ti5O12. The main effect of the Mo doping is to enhance the electronic conductivity and the diffusion coefficient of lithium ions. Our obtained results demonstrated that a small amount of Mo substitution in Li4Ti5O12 can improve the performance of this anode material, making it more attractive for future practical application. We have also demonstrated a simple and relatively low cost method for the production of this promising lithium battery anode material.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (no. 50902001), the Key project of Scientific Research Foundation sponsored by Education Department of Anhui Province, China (no. KJ2010A045), and the Foundation for Young Talents in College of Anhui Province, China (no. 2010SQRL033ZD). The work is also supported by the Program for Innovative Research Team in the Anhui University of Technology.
References
- J. Arai, J. Appl. Electrochem., 2002, 32, 1071 CrossRef CAS.
- S. S. Zhang, K. Xu and T. R. Jow, J. Power Sources, 2003, 113, 166 CrossRef CAS.
- S. S. Zhang, J. Power Sources, 2006, 161, 1385–1391 CrossRef CAS.
- T. Ohzuku, A. Ueda and N. Yamamoto, J. Electrochem. Soc., 1995, 142, 1431–1435 CrossRef CAS.
- M. R. Jo, K. M. Nam, Y. Lee, K. Song, J. T. Park and Y.-M. Kang, Chem. Commun., 2011, 47, 11474–11476 RSC.
- G.-N. Zhu, H.-J. Liu, J.-H. Zhuang, C.-X. Wang, Y.-G. Wang and Y.-Y. Xia, Energy Environ. Sci., 2011, 4, 4016–4022 CAS.
- L. Shen, C. Yuan, H. Luo, X. Zhang, K. Xu and Y. Xia, J. Mater. Chem., 2010, 20, 6998–7004 RSC.
- N. Takami, H. Inagaki, T. Kishi, Y. Harada, Y. Fujita and K. Hoshina, J. Electrochem. Soc., 2009, 156, A128–A132 CrossRef CAS.
- B. Scrosati and J. Garche, J. Power Sources, 2010, 195, 2419–2430 CrossRef CAS.
- A. N. Jansen, A. J. Kahaian, K. D. Kepler, P. A. Nelson, K. Amine, D. W. Dees, D. R. Vissers and M. M. Thackeray, J. Power Sources, 1999, 81–82, 902–905 CrossRef CAS.
- P. Reale, A. Fernicola and B. Scrosati, J. Power Sources, 2009, 194, 182–189 CrossRef CAS.
- I. Belharouak, Y.-K. Sun, W. Lu and K. Amine, J. Electrochem. Soc., 2007, 154, A1083–A1087 CrossRef CAS.
- I. Belharouak, G. M. Koenig Jr. and K. Amine, J. Power Sources, 2011, 196, 10344–10350 CrossRef CAS.
- H. F. Xiang, X. Zhang, Q. Y. Jin, C. P. Zhang, C. H. Chen and X. W. Ge, J. Power Sources, 2008, 183, 355–360 CrossRef CAS.
- S. P-Reale, B. Panero and B. Scrosati, J. Electrochem. Soc., 2005, 152, A1949–A1954 CrossRef.
- T.-F. Yi, L.-J. Jiang, J. Shu, C.-B. Yue, R.-S. Zhu and H.-B. Qiao, J. Phys. Chem. Solids, 2010, 71, 1236–1242 CrossRef CAS.
- H. Gao, L. Jiao, W. Peng, G. Liu, J. Yang, Q. Zhao, Z. Qi, Y. Si, Y. Wang and H. Yuan, Electrochim. Acta, 2011, 56, 9961–9967 CrossRef CAS.
- Z. Zhong, Electrochem. Solid-State Lett., 2007, 10, A267–A269 CrossRef CAS.
- J. Wolfenstine and J. L. Allen, J. Power Sources, 2008, 180, 582–585 CrossRef CAS.
- T.-F. Yi, Y. Xie, J. Shu, Z. Wang, C.-B.o Yue, R.-S. Zhu and H.-B. Qiao, J. Electrochem. Soc., 2011, 158, A266–A274 CrossRef CAS.
- Z. Yu, X. Zhang, G. Yang, J. Liu, J. Wang, R. Wang and J. Zhang, Electrochim. Acta, 2011, 56, 8611–8617 CrossRef CAS.
- R. D. Shannon, Acta. Crystallogr., 1976, A32, 751 CrossRef CAS.
- L. Aldon, P. Kubiak, M. Womes, J. C. Jumas, J. Olivier-Fourcade, J. L. Tirado, J. I. Corredor and C. Perez Vicente, Chem. Mater., 2004, 16, 5721 CrossRef CAS.
- R. Baddour-Hadjean and J.-P. Pereira-Ramos, Chem. Rev., 2010, 110, 1278 CrossRef CAS.
- P. Kubiak, A. Garcia, M. Womes, L. Aldon, J. O. Fourcade, P.-E. Lippens and J.-C. Jumas, J. Power Sources, 2003, 119–121, 626 CrossRef CAS.
- W. Lu, I. Belharouak, J. Liu and K. Amine, J. Electrochem. Soc., 2007, 154, A114 CrossRef CAS.
- H. Ge, N. Li, D. Li, C. Dai and D. Wang, Electrochem. Commun., 2008, 10, 719 CrossRef CAS.
- B. Tian, H. Xiang, L. Zhang, Z. Li and H. Wang, Electrochim. Acta, 2010, 55, 5453 CrossRef CAS.
- M. Zhang, L.-F. Jiao, H.-T. Yuan, Y.-M. Wang, J. Guo, M. Zhao, W. Wang and X.-D. Zhou, Solid State Ionics, 2006, 177, 3309–3314 CrossRef CAS.
- S.-H. Park, S. W. Oh and Y.-K. Sun, J. Power Sources, 2005, 146, 622–625 CrossRef CAS.
- T. Ohzuku, A. Ueda, N. Yamamoto and Y. Iwakoshi, J. Power Sources, 1995, 54, 99–101 CrossRef CAS.
- G. T.-K. Fey, C. Z. Lu and T. P. Kumar, J. Power Sources, 2003, 115, 332 CrossRef CAS.
- Y. Wang, H. Liu, K. Wang, H. Eiji, Y. Wang and H. Zhou, J. Mater. Chem., 2009, 19, 6789 RSC.
- L. Yang and L. Gao, J. Alloys Compd., 2009, 485, 93 CrossRef CAS.
- A. Y. Shenouda and H. K. Liu, J. Power Sources, 2008, 185, 1386 CrossRef CAS.
- Q. Cao, H. P. Zhang, G. J. Wang, Q. Xia, Y. P. Wu and H. Q. Wu, Electrochem. Commun., 2007, 9, 1228 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.