Mechanical properties of carbon nanotube–PMMA based hybrid coatings: the importance of surface chemistry
Fayna
Mammeri
*a,
Joan
Teyssandier
a,
Carole
Connan
a,
Eric
Le Bourhis
b and
Mohamed M.
Chehimi
a
aUniv Paris Diderot, Sorbonne Paris Cité, ITODYS, UMR 7086, Bâtiment Lavoisier, F-75205, Paris Cedex 13, France. E-mail: fayna.mammeri@univ-paris-diderot.fr.
bInstitut P', CNRS-Université de Poitiers-ENSMA, UPR 3346, Bd Pierre et Marie Curie, SP2MI, Téléport 2, BP, 30179, 86962, Futuroscope Chasseneuil Cedex, France
First published on 2nd February 2012
Abstract
The poor miscibility of carbon nanotubes (CNTs) in common organic solvents and organic monomers requires their modification by suitable functional (reactive or not) groups prior to their incorporation in thermoplastic polymers. We studied the influence of the functionalization of the surface of CNTs on their miscibility in poly(methyl methacrylate), following two main strategies: (i) the covalent grafting of hydrolyzable Si(OEt)3 groups on oxidized CNTs and (ii) the non-covalent adsorption of a polycation on pristine CNTs, allowing for further reactions by the sol–gel process. The mechanical properties of CNT/polymer-based nanocomposite thin films were studied using the nanoindentation technique. The hardness and the elastic indentation modulus were found to improve using modified CNTs, with more sensitivity observed on the elastic response.
1. Introduction
Carbon nanotubes 1 (CNTs) exhibit interesting mechanical2 and electrical3 properties and are consequently extremely attractive for applications in materials science e.g. for designing new advanced materials such as nanocomposites with improved performance4 or new properties.5 Due to their exceptionally high strength and axial elastic modulus, multiwalled carbon nanotubes (MWCNTs) have been commonly considered as interesting nanofillers for reinforcing various organic polymers.6–18 A key issue for designing nanocomposites based on a thermoplastic matrix and carbon nanotubes is the dispersion state since it is known to govern the resulting morphology and the physical properties of the materials. Homogeneously dispersed organic–inorganic (O–I) hybrid nanocomposites can be obtained by increasing the interfacial interactions between both components via the formation of hydrogen or covalent bonds, by mixing various polymers or via the choice of suitable surface chemistry processes.19–23 The mechanical response of such advanced functional materials is crucial when industrial applications are targeted. It is obvious and well-known that appropriate interfacial bonding and interactions between inorganic and organic components are necessary for improving the mechanical properties of the composites. Nevertheless, up to now, elastic modulus, ultimate strengths and general mechanical properties of CNT based composites were reported to increase, but less than expected, considering the exceptional properties of individual CNTs.2The understanding of the mechanical properties of O–I hybrids has largely increased by testing the materials under different conditions (static and dynamic, low and large deformations up to fracture) and using specific techniques.24 The nanoindentation technique is now a very well established probe for the mechanical study of polymers and organic–inorganic hybrid materials even prepared as thin films.25 Nevertheless, as the mechanical properties of O–I hybrid materials are strongly dependent not only on their micro- and nanostructures but also on the extent of the O–I interfaces, predictable mechanical properties for hybrids still represent a major challenge in the field of hybrid materials science.
The graphitic structure of a CNT surface is known to considerably limit its miscibility in any common organic solvents or monomers with a result of great limitations to their use. Thus, two main approaches, commonly envisaged for the modification of the surface of CNTs have been compared in this study: (a) the covalent attachment of chemical groups (on oxidized CNTs or through the use of diazonium salts26,27) and (b) the non-covalent adsorption of various functional molecules28,29 or polymers.30,31 To the best of our knowledge, no systematic study has been done to compare the influence of the chemistry conducted on the surface of CNTs on the properties of resulting polymer based nanocomposite thin films, using always the same polymer. Almost all studies were designed to increase the overall mechanical properties of various thermoplastics.13 Hence, we propose to study the influence of the size of the CNTs (through the use of oxidized or pristine nanotubes) and the influence of the surface of the modified nanotubes on their state of dispersion in a preformed polymer p(MMA-co-MPTMS). First, CNTs were oxidized in a strong acid mixture before being functionalized by a coupling agent, aminopropyltriethoxysilane (APTES), bearing hydrolysable Si(OEt)3 groups able to react by a sol–gel process. Then, pristine CNTs were embedded in an aqueous solution of a polycation, poly(allylamine hydrochloride), before being reacted by hydrolysis–condensation with tetraethoxysilane (TEOS) under acidic conditions to design a thin layer of sol–gel silica, more versatile for further reactions with functional molecules. The modification of CNTs by diazonium salts will be presented in a forthcoming paper. All pristine and modified CNTs were characterized by X-ray photoelectron spectroscopy (XPS) and transmission electron microscopy (TEM) before being introduced in a matrix of p(MMA-co-MPTMS). Hybrid thin films (containing 2 wt% of CNTs) were prepared by spin-coating and their mechanical properties were determined by nanoindentation. The influence of surface modification on the state of dispersion of the CNTs in the polymer is discussed and correlated to the mechanical properties of the O–I hybrid thin films.
2. Experimental part
Multiwalled carbon nanotubes were purchased from Sigma-Aldrich (diameter 1–10 nm, length 0.1–10 μm) and used as received. THF was dried by distillation over Na/benzophenone before use. Poly(allylamine hydrochloride), aminopropyltriethoxysilane and tetraethoxysilane were obtained from Alfa-Aesar, Acros and Fluka respectively and used as received. Methyl methacrylate (Sigma-Aldrich) was purified by distillation over KOH and under reduced pressure; acetonitrile was dried over molecular sieves.2.1. Preparation of Si(OEt)3 functionalized CNTs: CNT–COOH–APTES
Carboxylated carbon nanotubes (CNT–COOH) were produced by strong oxidation in a mixture of HNO3 and H2SO4. 10 mg of CNTs were weighed and placed in a flask; 9.4 mL of HNO3 (69%), 8 mL of H2SO4 (98%) and 0.6 mL of deionized water were added, corresponding to a solution of 8.0 M HNO3 and 8.0 M H2SO4, with a total volume of about 18.0 mL.32 Then, the solution was stirred in an ultrasonic bath for 2 h at 60 °C. Finally, CNTs were isolated by centrifugation, washed several times with deionized water and dried overnight at 60 °C.5 mg of CNT–COOH were dispersed in 20 mL of dimethylformamide (DMF) and sonicated for 1 h before the introduction of 1.1 mL of aminopropyltriethoxysilane (APTES) and 10 mL of 1 M N,N′-dicyclohexylcarbodiimide (DCC) in N-methylpyrrolidone (NMP); the mixture was sonicated for 72 h at 40 °C. Then, the CNTs were precipitated by acetone, isolated by filtration, washed in DMF and ethanol (by several cycles of dispersion–centrifugation) and dried under vacuum.
2.2. Preparation of CNT–PAH–SiO2
30 mg of CNTs were dispersed by sonication for 2 h in 200 mL of 1 wt% aqueous solution of poly(allylamine hydrochloride) (PAH) to give a concentration of 150 mg L−1. Excess PAH was removed by centrifugation. The product CNT–PAH was dried overnight at 60 °C.4.16 g (0.02 mol) of tetraethoxysilane (TEOS) were stirred for 24 h with 1.43 g (0.08 mol) of acidic aqueous solution (pH = 1, HCl). 10 mg of CNT–PAH were introduced in the sol–gel solution and sonicated for 4 h. Then, CNT–PAH–SiO2 were isolated by centrifugation, washed with ethanol and dried overnight at 60 °C.
2.3. Preparation of the PMMA–CNT hybrid coatings
In this work, we implemented sol–gel syntheses involving modified PMMA, containing about 5 mol% units bearing pendant hydrolyzable groups arising from methacryloxypropyltrimethoxysilane (MPTMS) monomer. The functionalized polymer P(MMA-co-MPTMS), quoted as f-PMMA, was first synthesized according to a procedure described by Mammeri et al. by free radical copolymerization of MMA and MPTMS, using a feed molar ratio [MMA]/[MPTMS] = 20 and 1 wt% of AIBN initiator in respect to monomers.25 After 24 h refluxing in acetonitrile at 75 °C, under argon, the polymer f-PMMA was recovered by precipitation in cold methanol. The molecular weight of this polymer was determined by size exclusion chromatography (SEC) in tetrahydrofurane (THF) and a bimodal distribution was observed: Mw = 44![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 eq. PMMA (PI = 1.02) and Mw = 23500 g mol−1 eq. PMMA (PI = 1.09). The glass-transition temperature was determined by differential calorimetric enthalpy (DSC) and was found to be Tg = 108 °C. On the basis of 1H NMR spectroscopy, the ratio of the CH2 protons next the carboxylate groups in MPTMS moieties (5 protons) over the CH3 protons of COOCH3 groups in MMA moieties (10 protons) indicated a MPTMS content in the copolymer close to 7.1 mol%.
000 g mol−1 eq. PMMA (PI = 1.02) and Mw = 23500 g mol−1 eq. PMMA (PI = 1.09). The glass-transition temperature was determined by differential calorimetric enthalpy (DSC) and was found to be Tg = 108 °C. On the basis of 1H NMR spectroscopy, the ratio of the CH2 protons next the carboxylate groups in MPTMS moieties (5 protons) over the CH3 protons of COOCH3 groups in MMA moieties (10 protons) indicated a MPTMS content in the copolymer close to 7.1 mol%.
Hybrid solutions were obtained by adding 2 wt% of modified-CNTs to a solution of f-PMMA in THF (concentration of 0.1 g L−1). The sols were vigorously sonicated at RT for 6 h before deposition by spin-coating on standard soda-lime-silica glass. Before deposition, the substrates were cleaned using a soap solution followed by rinsing in demineralised water to obtain a hydrophilic surface. After deposition, the samples were cured at 50 °C overnight to complete Si–OH condensation.
2.4. Characterization of the nanocomposite layers
All the pristine and modified carbon nanotubes were characterized by XPS. XPS spectra were recorded using a ThermoVG ESCALAB 250 (UK) with the monochromatic Al Kα X-ray source (hν = 1486.6 eV). The pass energy was set at 150 eV and 40 eV for the survey and the narrow regions (C 1s, O 1s, N 1s and Si 2p) respectively. The step sizes were 1 and 0.1 eV for the survey and the narrow regions respectively. The thicknesses of all films were determined on cross-sectional images obtained by scanning electron microscopy (SEM) and the state of dispersion of CNTs in f-PMMA was characterized by optical microscopy (LabRam HR800, Jobin-Yvon). Thermogravimetric analysis (TGA) was performed on hybrid thick films in an air atmosphere; all samples were heated to 800 °C at a heating rate of 10 °C min−1 on a SETARAM T692-12 instrument.The samples were indented by a Berkovich diamond pyramid using a nano hardness tester (NHT) from the Swiss Centre for Electronic and Microtechnology (CSEM, Switzerland). The tests were performed at room temperature (RT) in the force-control mode of the machine. The calibration procedure suggested by Oliver and Pharr33 was used to correct the load frame compliance of the apparatus and the imperfect shape of the indenter tip; the calibration was done on silica, whose elastic modulus and Poisson coefficient are well known (E = 72 GPa and ν = 0.17). We used a load-hold-rapid unloading procedure in order to limit the influence of the viscoelastic behaviour of the films during the rapid unloading. Indeed, viscous flow affects the contact stiffness S (dF/dh) of the sample and has to be considered when the indentation modulus and the hardness of the material are extracted by the Oliver and Pharr method.33 A correction was proposed by Tang and Ngan,34 which combines the penetration rate  at the end of the holding period rate and the unloading rate
at the end of the holding period rate and the unloading rate  . The corrected stiffness, Sc, is thus related to the measured stiffness S by equation:
. The corrected stiffness, Sc, is thus related to the measured stiffness S by equation:
 | (1) |
3. Results and discussion
3.1. Characterization of modified CNTs
The preservation of the electronic properties (by maintaining the sp2 structure) of the carbon nanotubes (CNTs) is the main advantage of non-covalent adsorption of a polyelectrolyte at the surface of the CNTs (in comparison with other modification techniques such as defect-side chemistry or covalent grafting on the side walls). The polyelectrolyte (a polycation in our case) is a solubilizing agent able to wrap around the carbon nanotube. Poly(allylamine hydrochloride) (PAH) is a polycation, soluble in water, non-toxic and already used for biomedical applications. The acidic method of coating individual nanotubes is more efficient with cationic surfactants, such as PAH, because anionic surfactants (e.g. SDS) become unstable in acidic media. Indeed, a high density of primary amines (free or protonated) can be found along the carbon skeleton and can interact with the surface of the CNTs most likely by cation–π interactions. A better dispersion of CNTs was observed in the aqueous solution of PAH than in pure water; this can be explained by long-range repulsive interactions between the polymer chains which prevent the possible aggregation of the CNTs. Then, modified CNTs (CNT–PAH) were found to be compatible and dispersible in a silica gel. Fig. 1 shows photographs of silica gels in which pristine and CNT–PAH CNTs were dispersed; while pristine CNTs led systematically to phase separation, CNT–PAH were well-dispersed in the gels. Therefore, the addition of a silica precursor, tetraethoxysilane (TEOS), in acidic medium, promotes the encapsulation of CNTs in a thin layer of sol–gel silica (CNT–PAH–SiO2). The overall synthesis pathway is presented in Scheme 1. | ||
| Fig. 1 Dispersion of (a) 0.08 wt% of pristine CNTs, (b) 0.8 wt% of CNT–PAH, (c) 0.4 wt% of CNT–PAH, (d) 0.08 wt% of CNT–PAH in (e) a silica gel prepared by hydrolysis–condensation of 4.16 g of TEOS with 1.44 g of water (pH = 1, HCl). | ||
 | ||
| Scheme 1 Two main strategies to modify the surface of CNTs. | ||
The surface chemical composition of CNTs modified by PAH was determined by XPS (Fig. 2). The survey spectrum of pristine carbon nanotubes exhibits a very intense C 1s peak, centred on 285.0 eV, and one of very low intensity peak, centred on 533 eV, revealing the presence of oxygen (O 1s) and suggesting that the pristine CNTs were slightly oxidized. The C 1s peak could be fitted with two components, centred at 284.6 eV and 285.4 eV corresponding to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (graphene) and CO bonds respectively. A broader peak centred on 290.7 eV was also observed and was attributed to π–π* interactions arising from the aromatic nature of CNTs.
C (graphene) and CO bonds respectively. A broader peak centred on 290.7 eV was also observed and was attributed to π–π* interactions arising from the aromatic nature of CNTs.
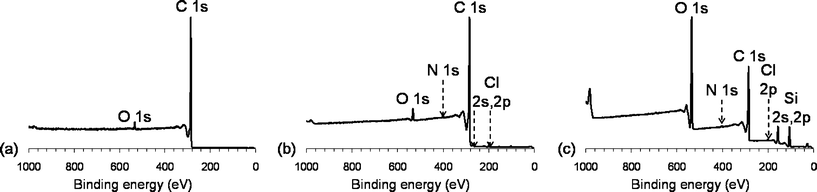 | ||
| Fig. 2 XPS survey spectra for (a) pristine CNTs, (b) CNT–PAH, (c) CNT–PAH–SiO2. | ||
The survey spectrum of CNT–PAH exhibits two new peaks: one centred on 400,6 eV, characteristic of the presence of nitrogen and a second centred at 198.8 eV, characteristic of chlorine (2p); these two peaks confirmed the presence of the protonated amine groups (NH3+) of the polycation and their counter-ions Cl−. The N 1s peak could be fitted with two components: one centred on 402.5 eV, corresponding to protonated amine groups and the second, centred on 400.6 eV, corresponding to free amine groups.
The proportion of protonated amines was estimated to be about 84% from the ratio of the relative intensities of both components of the N 1s peak (Fig. 3). Elemental analyses indicated that the atomic percentage of nitrogen (from NH3+) was about (1.0 ± 0.1) matching the atomic percentage of chlorine (1.2 ± 0.1) eV.
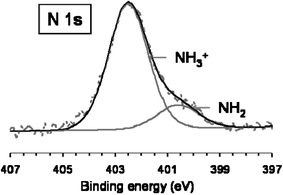 | ||
| Fig. 3 High resolution scan of N 1s region for CNT–PAH. | ||
The survey spectrum of CNT–PAH–SiO2 exhibits two new peaks characteristic of Si 2s and Si 2p (at 154.7 eV and 103.6 eV respectively). One can also notice that the O 1s peak is more intense than for pristine CNT and CNT–PAH. Elemental analyses indicated a silicon atomic percentage of about 15% whereas the oxygen atomic percentage was about 29%. The O/Si ratio was consequently 2, confirming the presence of sol–gel silica SiO2 on the surface of the CNTs. Moreover, the peaks C 1s, N 1s and Cl 2p were still observed, showing that CNT–PAHs were still detected through the silica ad layer. The increase of the intensity of the background led us to conclude that a homogeneous and non-negligible coating was obtained on the carbon nanotubes and formed by PAH and SiO2.
On the other hand, strong oxidation of carbon nanotubes can also be of interest; indeed, carboxylate groups are generated at the ends of the nanotubes and are more reactive towards many functional molecules such as organosilanes. However, oxidation also leads to a shortening of the CNTs and sometimes to some defects at the surface (perforation of the side-walls). Such an alteration (and more specifically the shortening) was observed on TEM micrographs.
Fig. 4 depicts the survey spectra of pristine CNTs, CNT–COOH as prepared and reacted with APTES. After oxidation, the peak centred on 533 eV and corresponding to O 1s became more intense. Furthermore, the peak C 1s could be fitted with two components, one corresponding to sp2carbon (285.0 eV) and the other corresponding to O–CO groups (289.0 eV). The appearance of the latter peak results from the oxidation of pristine CNTs, generating COOH functional groups. The peak corresponding to π–π* interactions, centred on 290.7 eV and resulting from the aromatic character of the CNTs was still observed although slightly attenuated.
 | ||
| Fig. 4 XPS survey spectra for (a) pristine CNTs, (b) CNT–COOH, (c) CNT–COOH–APTES. | ||
The survey spectrum of CNT–COOH-APTES presents three new peaks, attributed to nitrogen (N 1s, 400.5 eV) and silicon (Si 2s and Si 2p, 153.7 and 103.7 eV respectively) while the C 1s peak has become more intense. However, the high atomic percentages of silicon and oxygen suggested that the CNTs were coated by a thick amino-functionalized sol–gel silica layer, responsible for the attenuation of the signal relative to pristine CNTs.
3.2. Characterization of the PMMA–CNT hybrid layers
All the modified nanotubes could be dispersed in THF before being introduced in the solution of f-PMMA. Then, the hybrid layers were deposited by spin-coating on hydrophilic glass substrates. The resulting layers looked very different in terms of the state-of-dispersion of the various CNTs prepared in this study. Pristine CNTs were dispersed poorly in the polymer solution and led to a significant phase separation during the coating process. Similarly, the adsorption of PAH was not sufficient to improve the dispersion and obtain a homogeneous suspension of CNT in the solution of f-PMMA; some aggregates were formed and characterized by Raman spectroscopy. Finally, the introduction of a sol–gel silica layer on the surface of CNTs led to a fairly homogeneous distribution of CNT–PAH–SiO2 in the polymer f-PMMA.Similarly, CNT–COOH were poorly dispersed in the solution of f-PMMA whereas CNT–COOH–APTES led to the most homogeneous hybrid films (Fig. 5). Thus, we could conclude the success of a suitable surface chemistry to disperse efficiently the carbon nanotubes in a thermoplastic polymer such as PMMA or in an oxide such as silica (SiO2).
 | ||
| Fig. 5 Optical micrographs of hybrid films made from (a) f-PMMA, (b) f-PMMA and CNT–COOH–APTES and (c) f-PMMA and CNT–PAH–SiO2. | ||
Thermogravimetric analysis (TGA) performed on thick hybrid films revealed that the organic/inorganic ratios were in good agreement with the solution compositions.
3.3. Mechanical properties of the hybrid layers
The mechanical properties, namely hardness and elastic modulus, of f-PMMA and the hybrid layers made from CNT–COOH–APTES and CNT–PAH–SiO2 were determined by nanoindentation. The largest indentations into the hybrid coatings were observed by optical microscopy. Neither cracking nor delamination was observed at higher loads (150 or 300 mN). Fig. 6 presents loading–unloading curves of the three samples obtained under a load of 1 mN. These contrast dramatically with the one obtained on the glass substrate. The elastic and plastic resistances of the specimens were extracted from the unloading curves.33 | ||
| Fig. 6 Load–displacement curves (after applying 1 mN) for glass substrate and (a) CNT–COOH–APTES based hybrid, (b) CNT–PAH–SiO2 based hybrid and (c) neat f-PMMA. | ||
Fig. 7 presents the evolution of the indentation reduced modulus and the hardness as a function of the contact indentation depth hc (scaled with the coating thickness) for the studied PMMA–CNT hybrid materials. The increase in the mechanical properties as the indentation depth increases results from the strong influence of the substrate. This means that the mechanical responses are dominated by thin films properties at low indentation depths whereas those determined at larger indentation depths are substrate dependent. Various models were suggested to describe the combined influence of both the substrate and the coating allowing the calculation of the sole mechanical response of the thin film. Concerning the determination of the elastic indentation modulus, the most suitable was found to be the reciprocal exponential function, following Doerner and Nix’s suggestion to consider the film and substrate as two springs in series;35 it is given by:
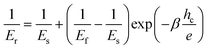 | (2) |
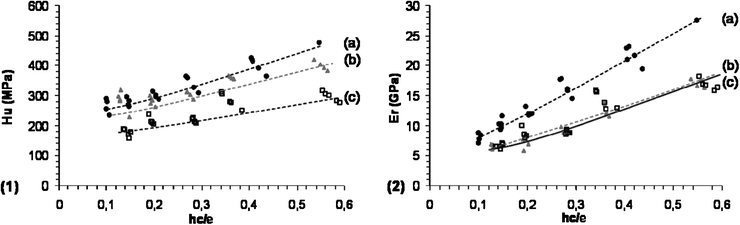 | ||
| Fig. 7 (1) Hardness (Hu) and (2) indentation modulus (Er) as a function of the indentation depth, scaled with the coating thickness (hc/e) for samples: (a) CNT–COOH–APTES based hybrid (○), (b) CNT–PAH–SiO2 based hybrid (△) and (c) neat f-PMMA (□). | ||
A similar procedure was applied to determine the hardness, Hf, of the films. However, at equal indentation depth, the influence of the substrate on the measured hardness was lower than on indentation elastic modulus (Fig. 7); indeed, the plastic strain field is much less extended spatially than the elastic strain field. A previous work on SiO2–PMMA hybrid films showed that the best model is the exponential law proposed by Bhattacharya and Nix:36
 | (3) |
It is to be noted that these equations could be applied to the studied films because CNT–PMMA materials did not present any fracture. Thicknesses, calculated values of indentation modulus and hardness for the different samples are gathered in Table 1.
These results show an improvement in the mechanical behaviour of the hybrid materials (as compared to neat f-PMMA). It is clearly apparent that the elastic modulus is more sensitive than the hardness to the surface modification of the CNTs. It emphasizes that the influence of the incorporation of CNTs is to be considered with care with the respective deformation modes (elastic or plastic). Indeed, while similar hardness values are recorded by incorporating 2 wt% of CNT–PAH–SiO2 and CNT–COOH–APTES, a higher elastic modulus was determined when using CNT–COOH–APTES. The latter are smaller in size, since they were produced by oxidation of pristine nanotubes, and bear functional Si–OH groups only at the ends of the nanotube whereas the former kept their initial length and are theoretically functionalized by Si–OH along the whole of the side walls. Therefore, it appears that the smallest nanofillers are more efficient at enhancing the mechanical properties of the poly(methylmethacrylate) matrix.
Mechanical properties of a composite material (here organic–inorganic hybrids) can generally be modelled using the volume fraction and the mechanical properties of each component. In the present case, we show that it is difficult to predict the mechanical properties of the hybrid materials from each component since the properties are strongly dependent on the nature of the hybrid interface.
4. Conclusions
The control of the nanotube morphology coupled to the versatility of silica chemistry through the sol–gel process leads to interesting structures which can be used as new reactive platforms to design polymer–CNT based hybrid materials. This work demonstrates that both commonly considered pathways (oxidation or non-covalent adsorption of a polycation) led to a better miscibility of CNTs in common organic solvents and in polar polymers like PMMA. Nanoindentation data collected on thin films prepared from p(MMA-stat-MPTMS) and modified CNTs demonstrated unambiguously that CNTs reinforce PMMA and how a suitable surface chemistry, conducted on the surface of the CNTs can improve the mechanical properties of the resulting nanocomposites.Acknowledgements
FM thanks Dr Frédéric Herbst and David Montero for SEM and TEM analysis, Dr Johan Grand for optical microscopy and Raman measurements and Mohamed El Ghazzal for TGA experiments.References
- S. Ijima, Nature, 1991, 354, 56 CrossRef.
- M. M. J. Treacy, T. W. Ebbesen and J. M. Gibson, Nature, 1996, 381, 678 CrossRef CAS.
- T. W. Odom, J. L. Huang, P. Kim and C. M. Lieber, Nature, 1998, 391, 62 CrossRef CAS.
- J. Kim, S. M. Hong, S. Kwak and Y. Seo, Phys. Chem. Chem. Phys., 2009, 11, 10851 RSC.
- S. Piovesan, P. A. Cox, J. R. Smith, D. G. Fatouros and M. Roldo, Phys. Chem. Chem. Phys., 2010, 12, 15636 RSC.
- K. E. Prasad, B. Das, U. Maitra and C. N. R. Rao, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 13186 CrossRef CAS.
- M. Olek, K. Kempa, S. Jurga and M. Giersig, Langmuir, 2005, 21, 3146 CrossRef CAS.
- J. N. Coleman, U. Khan and Y. K. Gun'ko, Adv. Mater., 2006, 18, 689 CrossRef CAS.
- M. Cadek, J. N. Coleman, K. P. Ryan, V. Nicolosi, G. Bister, A. Fonseca, J. B. Nagy, K. Szostak, F. Béguin and W. J. Blau, Nano Lett., 2004, 4, 353 CrossRef CAS.
- R. Andrews and M. C. Weisenberg, Curr. Opin. Solid State Mater. Sci., 2004, 8, 31 CrossRef CAS.
- J. N. Coleman, U. Khan, W. J. Blau and Y. Gun'ko, Carbon, 2006, 44, 1624 CrossRef CAS.
- L. D. Perez, M. A. Zuluaga, T. Kyu, J. E. Mark and B. L. Lopez, Polym. Eng. Sci., 2009, 49, 866 CAS.
- M. T. Byrne and Y. K. Gun'ko, Adv. Mater., 2010, 22, 1672 CrossRef CAS.
- P. C. Ma, N. A. Siddiqui, G. Marom and J. K. Kim, Composites, Part A, 2010, 41, 1345 CrossRef.
- Z. Spitalsky, D. Tasis, K. Papagelis and C. Galiatis, Prog. Polym. Sci., 2010, 35, 357 CrossRef CAS.
- S. Tawfick, X. P. Deng, A. J. Hart and J. Lahann, Phys. Chem. Chem. Phys., 2010, 12, 4446 RSC.
- Y. Lin, M. J. Meziani and Y. P. Sun, J. Mater. Chem., 2007, 17, 1143 RSC.
- H. Qian, E. S. Greenhalgh, M. S. P. Shaffer and A. Bismark, J. Mater. Chem., 2010, 20, 4751 RSC.
- T. Matrab, J. Chancolon, M. Mayne l'Hermite, J. N. Rouzaud, J. P. Boudou, M. M. Chehimi and M. Delamar, Colloids Surf., A, 2006, 287, 217 CrossRef CAS.
- J. B. Kim, T. Premkumar, K. Lee and K. E. Geckeler, Macromol. Rapid Commun., 2007, 28, 276 CrossRef CAS.
- M. Grzelczak, M. A. Correa-Duarte and L. M. Liz-Marzàn, Small, 2006, 2, 1174 CrossRef CAS.
- M. Bottini, L. Tautz, H. Huynh, E. Monosov, N. Bottini, M. I. Dawson, S. Bellucci and T. Mustelin, Chem. Commun., 2005, 758 RSC.
- J. T. Sun, C. Y. Hong and C. Y. Pan, Polym. Chem., 2011, 2, 998 RSC.
- F. Mammeri, E. Le Bourhis, L. Rozes and C. Sanchez, J. Mater. Chem., 2005, 15, 3787 RSC.
- F. Mammeri, L. Rozes, E. Le Bourhis and C. Sanchez, J. Sol-Gel Sci. Technol., 2003, 26, 413 CrossRef CAS.
- P. R. Marcoux, P. Hapiot, P. Batail and J. Pinson, New J. Chem., 2005, 28, 302 RSC.
- S. Mahouche-Chergui, A. Ledebt, F. Mammeri, F. Herbst, B. Carbonnier, H. Ben Romdane, M. Delamar and M. M. Chehimi, Langmuir, 2010, 26, 16115 CrossRef CAS.
- D. Tasis, N. Tagmatarchis, A. Bianco and M. Prato, Chem. Rev., 2006, 106, 1105 CrossRef CAS.
- G. Van Lier, G. Van Assche, H. E. Miltner, N. Grossiord, C. E. Koning, P. Geerlings and B. Van Mele, Phys. Chem. Chem. Phys., 2009, 11, 11121 RSC.
- A. Satti, A. Perret, J. E. Mc Carthy and Y. K. Gun'ko, J. Mater. Chem., 2010, 20, 7941 RSC.
- D. Tuncel, Nanoscale, 2011, 3, 3545 RSC.
- X. Y. Xing, J. Phys. Chem. B, 2004, 108, 19255 CrossRef.
- W. C. Oliver and G. M. Pharr, J. Mater. Res., 1992, 7, 1564 CrossRef CAS.
- B. Tang and A. H. W. Ngan, J. Mater. Res., 2003, 18, 1141 CrossRef CAS.
- M. F. Doerner and W. D. Nix, J. Mater. Res., 1986, 1, 601 CrossRef.
- A. K. Bhattacharya and W. Nix, Int. J. Solids Struct., 1988, 24, 1287 CrossRef.
| This journal is © The Royal Society of Chemistry 2012 |