Mesostructure-controlled synthesis of chiral norbornane-bridged periodic mesoporous organosilicas†
Ting Ting
Hao
a,
Jiao Yi
Shi
a,
Ting Yan
Zhuang
b,
Wei David
Wang
a,
Fu Chong
Li
a and
Wei
Wang
*a
aState Key Laboratory of Applied Organic Chemistry, College of Chemistry and Chemical Engineering, Lanzhou University, Lanzhou, Gansu 730000, P. R. China. E-mail: wang_wei@lzu.edu.cn; Fax: +86 931 8915557; Tel: +86 931 8912282
bCenter Testing International (Shenzhen) Corporation, Building C, Hongwei Industrial Park, Bao'an 70 District, Shenzhen, Guangdong 518101, P. R. China
First published on 11th January 2012
Abstract
In this contribution, chiral periodic mesoporous organosilica (PMO) materials with three-dimensional body-centered (Im3m) and face-centered (Fm3m) cubic mesostructures were synthesized for the first time. Under acidic conditions and with triblock copolymer F127 as the template, an Im3m-type chiral PMO was obtained with spherical morphology from the self-condensation of chiral norbornane-bridged precursor 1. In contrast, using triblock copolymer P123 as the template and with the aid of a co-surfactant, Gemini surfactant C14-6-14, an Fm3m-type chiral PMO was obtained with an unprecedented pumpkin-like morphology from the self-condensation of chiral precursor 1. The synthetic conditions such as the stirring, standing, and aging time are crucial for the mesostructure and morphology of the synthesized chiral PMO materials.
Introduction
Embedding the bridging organic group R within the framework efficiently and uniformly, periodic mesoporous organosilica (PMO) materials1 are elaborately constructed by the self-assembly of bis-alkoxysilane (R′O)3Si–R–Si(OR′)3 precursors in the presence of certain structural templates. The unique pore-wall functionality together with the ordered mesoporous structure and uniform morphology have rendered this new type of organic–inorganic hybrid mesoporous materials with high potential in various applications.2–4 To meet the demand for practical use, new efforts have been made recently towards the mesostructure-controlled synthesis of PMOs.5 However, the PMOs synthesized so far possess almost exclusively the two-dimensional (2D) channel-like hexagonal (p6mm) mesostructure. Three-dimensional (3D) cubic PMO materials with cage-like pores (Pm3n,6Im3m,7Fm3m,8 or Ia3d5b), although having advantages for accommodation and transportation of various guest molecules, could rarely be obtained and, are limited to small and achiral bridging groups such as ethane,5a benzene,6c,9 and thiophene.7b Moreover, no report is available so far on the synthesis of 3D cubic chiral PMO materials.In addition to the issues of mesostructure control, it is also highly desirable to synthesize the PMO materials with well-defined morphology to satisfy the needs for their applications as supports, catalysts, adsorbents and templates. For example, their potential application as stationary phases in chromatography (such as HPLC) requires the synthesized PMOs as spherical particles with an average size of ca. 10 μm.10 Accordingly, various attempts have been made to manipulate the PMO materials with controlled morphologies, such as of sphere,11 rod,12 film,13 and mesoporous single crystals,6b,6d,7c which could explore further possibilities for various applications of these PMOs.
On the other hand, increasing attention has been paid to chiral PMO materials,14 which are potentially applicable in heterogeneous asymmetric catalysis and chiral separation. However, multi-step and laborious work is often involved in the synthesis of chiral bis-alkoxysilane precursors. Moreover, only a few chiral precursors are rigid enough to construct the PMO mesostructure alone without the co-condensation with tetraethyl orthosilicate (TEOS) or other rigid precursors. As a result, the chiral PMO materials, if synthesized, often lack long-range meso-order, and suffer from partial cleavage of the Si–C bonds. Recently, we reported the facile synthesis of a novel chiral norbornane-bridged precursor with 91% yield and 99% ee via a two-step chemical transformation from norbornadiene.15 Accordingly, the chiral norbornane-bridged PMO material was successfully synthesized with a highly-ordered 2D channel-like hexagonal (p6mm) mesostructure via the self-condensation of the chiral precursor. To generate practical applications, especially for size- and shape-selective separation, heterogeneous catalysis, and optoelectronic devices, the mesophase structure and macroscopic morphology of chiral PMO materials should be further controlled, which still remains a challenging task.
Based on our previous work,15 we report herein for the first time the mesostructure-controlled synthesis of chiral PMO materials from the single source of chiral precursor 1 (Scheme 1). Under acidic conditions, the chiral PMOs with 3D cubic body-centered (Im3m) and face-centered (Fm3m) mesostructures were successfully obtained by using nonionic surfactant (triblock copolymer F127) and co-structure directing agents (triblock copolymer P123 and Gemini surfactant C14-6-14), respectively. In addition to the control of mesostructure, the morphology of the chiral PMOs was also adjusted by varying the stirring, standing, and aging time. The chiral PMO material with cubic Im3m-type mesostructure exhibits well-defined spherical morphology; the PMO particles are monodispersed with most sizes around 10 μm. Moreover, the chiral PMO material with cubic Fm3m-type mesostructure possesses an unprecedented pumpkin-like morphology.
 | ||
| Scheme 1 Schematic description for the mesostructure-controlled synthesis of chiral PMO materials from the single source of chiral precursor 1. | ||
Experimental
Chemical reagents
The chiral precursor (1R,2S,4R,5S)-exo,exo-2,5-bis(trimethoxysilyl)bicycle-[2,2,1]heptane 1 was synthesized according to the literature.15,16 Pluronic P123 (EO20PO70EO20) and Pluronic F127 (EO106PO70EO106) were purchased from Aldrich. Gemini surfactant [C14H29N+(CH3)2(CH2)6N+(CH3)2C14H29] 2Br− (C14-6-14) was synthesized from the reaction of 1-bromotetradecane (Alfa Aeser) with N,N,N′,N′-tetramethyl-1,6-diaminohexane (Alfa Aeser). All chemical reagents were used as received.Synthesis
The chiral PMO material with cubic Im3m symmetry was synthesized from the self-condensation of chiral norbornane-bridged precursor 1 in the presence of triblock copolymer F127 as the template under acidic conditions. In a typical preparation, 0.25 g of F127 was dissolved in 11 mL of deionised water and 0.3 mL of 2.0 M HCl. Afterwards, 0.59 g of the chiral precursor was added to the solution and stirred at 40 °C for 2.5 h, then kept for another 24 h under static conditions. Finally the suspension was aged at 100 °C for 24 h in a closed PTFE autoclave. After filtration and washing thoroughly with water, a white powder was obtained.The chiral PMO material with cubic Fm3m symmetry was synthesized from the self-condensation of 1 in the presence of triblock copolymer P123 and Gemini surfactant C14-6-14 as a co-structure directing agent under acidic conditions. The typical procedure is described as follows: 0.30 g of P123 and 0.04 g of C14-6-14 was dissolved in 10 mL of deionised water and 0.3 mL of concentrated HCl (36 wt.%). Afterwards, 0.50 g of the chiral precursor 1 was added to the solution and stirred at 40 °C for 2.5 h, then kept for another 24 h under static conditions. Finally the suspension was aged at 100 °C for 24 h in a closed PTFE autoclave. After filtration and washing thoroughly with water, a white powder was obtained.
The surfactant was removed twice using a 100 mL mixture of ethanol/HCl (conc.) (100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5, v/v) per 1 g of sample or using 100 mL acetone per 1 g of sample. Changing the stirring, standing, or aging time results in the formation of solids with low-ordered or amorphous structure. The detailed results are described in the ESI.†
:5, v/v) per 1 g of sample or using 100 mL acetone per 1 g of sample. Changing the stirring, standing, or aging time results in the formation of solids with low-ordered or amorphous structure. The detailed results are described in the ESI.†
Characterisation
Powder X-ray diffraction (PXRD) measurements were taken with a PANalytical X′Pert Pro using Cu-Kα (λ = 1.5406) radiation (40 kV, 40 mA), with a step size of 0.008°/2θ and scan step time of 38.32 s. Transmission electron microscopy (TEM) experiments were performed on a JSM-6701F electron microscope (JEOL, Japan) with an acceleration voltage of 200 kV. For TEM analysis, the samples were dispersed in ethanol, and then dipped and dried on a micro-grid. Scanning electron microscope (SEM) images were obtained with a Hitachi S-4800 instrument operating at 2 or 5 kV. The samples were deposited on a sample holder and sputtered with gold prior to imaging. The nitrogen adsorption and desorption isotherms were obtained on a Micromeritics ASAP 2020M analyser at 77 K. Before the measurements, all samples were outgassed at 120 °C for 8 h. Surface areas were analysed by using a Brunauer–Emmett–Teller (BET) method. The pore size distributions were calculated from the absorption data using the Barrett–Joyner–Halenda (BJH) method. The solid-state 13C cross-polarisation magic-angle spinning (CP/MAS) and 29Si high-power proton-decoupling (HPDEC) MAS NMR spectra were performed on a Bruker WB Avance II 400 MHz spectrometer using a 4-mm double-resonance MAS probe with a sample spinning rate of 10 KHz. The 13C CP/MAS NMR spectra were measured with a contact time of 2 ms (ramp 100) and pulse delay of 3 s. The 29Si MAS NMR spectra were measured with π/4 single-pulse excitation of 2.0 μs and pulse delay of 30 s.Results and discussion
3D cubic Im3m-type chiral PMO material
In Fig. 1 is shown the powder XRD pattern of the chiral PMO material prepared with F127 as the template (Scheme 1). The obtained sample shows three well-resolved diffraction peaks in the region of 2θ = 0.6–1.8°. These peaks with lattice spacings of d = 108, 77, and 63 Å can be indexed as [110], [200], and [211] facets associated with a body-centered cubic (Im3m) symmetry. The successful synthesis of 3D cubic Im3m-type PMO material was further confirmed by the TEM analysis. From the TEM images of the synthesized PMO material (Fig. 2), the regular arrangement of bright spots recorded along the [100] and [111] directions reveal that the material has 3D cage-type mesostructure with high long-range order. As shown in Fig. 3, the obtained chiral PMO material has spherical morphology; most of the PMO particles have diameters in the range of 5–12 μm. | ||
| Fig. 1 Powder XRD pattern of the 3D cubic Im3m-type chiral PMO material synthesized with F127 as the template (Scheme 1). | ||
![TEM images of the 3D cubic Im3m-type chiral PMO material recorded along the [100] (a) and [111] (b) directions.](/image/article/2012/RA/c2ra00805j/c2ra00805j-f2.gif) | ||
| Fig. 2 TEM images of the 3D cubic Im3m-type chiral PMO material recorded along the [100] (a) and [111] (b) directions. | ||
 | ||
| Fig. 3 SEM images of the as-synthesized 3D cubic Im3m-type chiral PMO material. | ||
The nitrogen adsorption–desorption isotherms of the extracted chiral PMO material (Fig. 4) exhibit type-IV isotherm characteristics with a type-H2 hysteresis loop. A well-defined step in the adsorption branch at a relative pressure (P/P0) of 0.4 is typical for uniform mesoporous solids, and a clear H2 hysteresis loop indicates that the material has a cage-like pore-structure. The BET surface area, pore volume, and BJH pore diameter were determined as 1080 m2 g−1, 0.69 cm3 g−1, and 5.6 nm, respectively.
 | ||
| Fig. 4 N2 adsorption–desorption isotherms and BJH pore size distribution (inset) for the 3D cubic Im3m-type chiral PMO material. | ||
Solid-state 13C CP/MAS and 29Si MAS NMR spectra recorded for the cubic Im3m-type chiral PMO material are shown in Fig. 5, offering information on the basic structural units at the molecular level. The 13C CP/MAS NMR signals at 26, 36, and 38 ppm (Fig. 5a) are characteristic for the norbornane-bridged groups.15 The very weak signals at 16 and 70 ppm are attributed to the residual template molecules inside the extracted PMO sample. The additional small peak at 58 ppm corresponds to the carbon atom of surface ethoxy groups which were formed in the process of solvent extraction using ethanol. The three NMR peaks at −51, −59, and −67 ppm in the 29Si MAS NMR spectrum (Fig. 5b), can be assigned to T-type organosilica species of T1 [RSi(OSi)(OH)2], T2 [RSi(OSi)2(OH)], and T3 [RSi(OSi)3], respectively. In comparison with the case of 2D p6mm-type chiral material,15 the ratio of signal intensities of T1 and T3 species varies significantly. Moreover, 29Si MAS NMR signals for Q species are totally absent in Fig. 5b, indicating that no cleavage of Si–C bonds occurs during our synthetic procedure under acidic conditions. As confirmed by these solid-state NMR measurements, therefore, all organic moieties keep intact in the framework of the synthesized chiral PMO material.
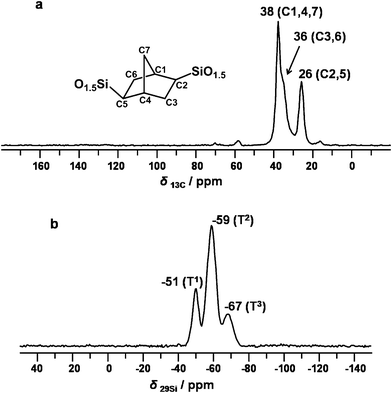 | ||
| Fig. 5 13C CP/MAS (a) and 29Si MAS (b) solid-state NMR spectra of the 3D cubic Im3m-type chiral PMO material after the extraction of surfactant. | ||
As is known, the formation of PMO mesostructures can be influenced by various parameters of the synthetic conditions, such as the type and concentration of the surfactant, the presence of co-surfactant, pH value, the concentration and temperature of the reaction solution, and so on. We also examined the influence of reaction conditions on the cubic Im3m-type mesostructure of chiral PMO material. With the same reactant components, but changing the stirring time or without keeping the suspension under static conditions, amorphous gels or lower mesostructure-ordered materials were obtained. The investigation details can be found in Fig. S1, ESI.†
3D cubic Fm3m-type chiral PMO material
Fig. 6 shows the powder XRD patterns of the chiral PMO material prepared (Scheme 1) using triblock copolymer P123 as a soft template (Fig. 6a) or using P123 and Gemini surfactant C14-6-14 as a co-structure directing agent (Fig. 6b). As shown in Fig. 6a, the synthesized PMO material has three peaks with d-spacings of 108, 54, and 41 Å, respectively, which can be indexed as [100], [200], and [210] facets contributed to a p6mm type mesostructure. Remarkably, with the same reactant components and under the same reaction conditions, when 0.04 g of Gemini surfactant was additionally added, the mesophase was changed from a p6mm to Fm3m-type structure. Fig. 6b shows the powder XRD pattern of the sample prepared with Gemini surfactant C14-6-14 as a co-structure directing agent. Different from those in Fig. 6a, the three peaks in the low-angle region have the lattice spacings of 96, 59 and 51 Å, respectively. These three peaks can be indexed as [111], [220], and [311] reflections of an Fm3m-type structure, indicating that the chiral PMO material possesses a 3D cubic symmetry. | ||
| Fig. 6 Powder XRD patterns of the chiral PMO materials prepared with P123 (a) and with the co-template of P123 and C14-6-14 (b). | ||
The formation of a highly-ordered 3D cubic Fm3m-type mesostructure was also confirmed by the TEM analysis. Large highly-ordered areas with confined pore arrangement can be clearly observed from different incident directions as shown in Fig. 7 ([100] in Fig. 7a, and [110] in Fig. 7b), indicating a perfect crystallisation of the PMO sample. The hexagonal close-packed (hcp) mesostructure may co-exist with the cubic close-packed phase,8c but in our case, no evidence for the intergrowth phase could be found. Therefore, the chiral PMO prepared with P123 and C14-6-14 as a co-structure directing agent has a pure face-centered cubic (Fm3m-type) mesostructure.
![TEM images of the 3D cubic Fm3m-type chiral PMO recorded along the [100] (a) and [110] (b) directions.](/image/article/2012/RA/c2ra00805j/c2ra00805j-f7.gif) | ||
| Fig. 7 TEM images of the 3D cubic Fm3m-type chiral PMO recorded along the [100] (a) and [110] (b) directions. | ||
The SEM images (Fig. 8) showed that the morphology of the cubic Fm3m-type chiral PMO material exhibits pumpkin-like spheres with an average particle size of ca. 10 μm. The pumpkin-like spheres have 12 arris on their surface. To the best of our knowledge, this is the first example of high-quality 3D cubic mesoporous organosilicas with the special pumpkin-like morphology.
 | ||
| Fig. 8 SEM images of the 3D cubic Fm3m-type chiral PMO material with pumpkin-like morphology synthesized with the co-structure directing agent of P123 and C14-6-14. | ||
The N2 adsorption–desorption isotherms (Fig. 9) of the cubic Fm3m-type chiral PMO material show a type-IV isotherm, typical for mesoporous materials, while the clear type-H2 hysteresis loop is characteristic for materials with cage-like pore-structures. The BET surface area, pore volume, and BJH pore diameter were determined as 756 m2 g−1, 0.68 cm3 g−1, and 5.3 nm respectively. Solid-state 13C CP/MAS and 29Si MAS NMR measurements for the cubic Fm3m-type chiral PMO are shown in Fig. S4, ESI,† which reveal that all organic moieties remain intact in the PMO framework and no cleavage of Si–C bonds occurs during the synthetic procedure under acidic conditions.
 | ||
| Fig. 9 N2 adsorption–desorption isotherms and BJH pore size distribution (inset) for the synthesized chiral PMO material. | ||
The synthetic conditions, such as the stirring, standing, and aging time also play important roles in our synthetic processes for the 3D cubic Fm3m-type chiral PMO material. The investigation details can be found in Fig. S2 and S3, ESI.†
Conclusions
In summary, chiral PMO materials with 3D body-centered (Im3m) and face-centered (Fm3m) cubic mesostructures were synthesized for the first time. Meanwhile, with the control of synthetic parameters, the chiral PMO materials with well-defined morphologies could be obtained. Under acidic conditions, the chiral PMO with 3D cubic Im3m symmetry was prepared by using Pluronic F127 triblock copolymer as the template. A spherical morphology was obtained and most of the monodispersed PMO particles have sizes in the range of 5–12 μm. Using triblock copolymer P123 and Gemini surfactant C14-6-14 as co-structure directing agents, an Fm3m-type chiral PMO with special pumpkin-like morphology was obtained. The pumpkin-like PMO particles with 12 arris have uniform sizes of ca. 10 μm. This is the first example of synthesizing chiral PMO materials with mesostructure-control, which may provide new possibilities for the practical applications of chiral PMO materials, such as for selective adsorption and separation, heterogeneous catalysis, and chiral supports. Research towards these applications is currently underway in this laboratory.Acknowledgements
The National Natural Science Foundation of China (No. 20972064) and the Fundamental Research Funds for the Central Universities are gratefully acknowledged for financial support.References
- (a) T. Asefa, M. J. MacLachlan, N. Coombs and G. A. Ozin, Nature, 1999, 402, 867 CAS; (b) B. J. Melde, B. T. Holland, C. F. Blanford and A. Stein, Chem. Mater., 1999, 11, 3302 CrossRef CAS; (c) S. Inagaki, S. Guan, Y. Fukushima, T. Ohsuna and O. Terasaki, J. Am. Chem. Soc., 1999, 121, 9611 CrossRef CAS.
- (a) F. Hoffmann, M. Cornelius, J. Morell and M. Fröba, J. Nanosci. Nanotechnol., 2006, 6, 265 CAS; (b) F. Hoffmann, M. Cornelius, J. Morell and M. Fröba, Angew. Chem., Int. Ed., 2006, 45, 3216 CrossRef CAS; (c) F. Hoffmann and M. Fröba, Chem. Soc. Rev., 2011, 40, 608 RSC.
- (a) B. Hatton, K. Landskron, W. Whitnall, D. Perovic and G. A. Ozin, Acc. Chem. Res., 2005, 38, 305 CrossRef CAS; (b) W. J. Hunks and G. A. Ozin, J. Mater. Chem., 2005, 15, 3716 RSC; (c) W. D. Wang, J. E. Lofgreen and G. A. Ozin, Small, 2010, 6, 2634 CrossRef CAS.
- (a) S. Fujita and S. Inagaki, Chem. Mater., 2008, 20, 891 CrossRef CAS; (b) Q. Yang, J. Liu, L. Zhang and C. Li, J. Mater. Chem., 2009, 19, 1945 RSC.
- (a) Y. Liang, E. S. Erichsen, M. Hanzlik and R. Anwander, Chem. Mater., 2008, 20, 1451 CrossRef CAS; (b) H. I. Lee, C. Pak, S. H. Yi, J. K. Shon, S. S. Kim, B. G. So, H. Chang, J. E. Yie, Y. U. Kwon and J. M. Kim, J. Mater. Chem., 2005, 15, 4711 RSC; (c) R. Atluri, Y. Sakamoto and A. E. Garcia-Bennett, Langmuir, 2009, 25, 3189 CrossRef CAS.
- (a) A. Sayari, S. Hamoudi, Y. Yang, I. L. Moudrakovski and J. R. Ripmeester, Chem. Mater., 2000, 12, 3857 CrossRef CAS; (b) S. Guan, S. Inagaki, T. Ohsuna and O. Terasaki, J. Am. Chem. Soc., 2000, 122, 5660 CrossRef CAS; (c) M. P. Kapoor, M. Yanagi, Y. Kasama, T. Yokoyama, S. Inagaki, T. Shimada, H. Nanbu and L. R. Juneja, J. Mater. Chem., 2006, 16, 3305 RSC; (d) M. P. Kapoor and S. Inagaki, Chem. Mater., 2002, 14, 3509 CrossRef CAS.
- (a) W. Guo, I. Kim and C. S. Ha, Chem. Commun., 2003, 2692 RSC; (b) E. B. Cho, D. Kim, J. Górka and M. Jaroniec, J. Mater. Chem., 2009, 19, 2076 RSC; (c) J. Li, Y. Wei, Y. H. Deng, D. Gu, X. D. Yang, L. J. Zhang, B. Tu and D. Y. Zhao, J. Mater. Chem., 2010, 20, 6460 RSC; (d) R. M. Grudzien, S. Pikus and M. Jaroniec, J. Phys. Chem. B, 2006, 110, 2972 CrossRef CAS.
- (a) Y. Liang, M. Hanzlik and R. Anwander, Chem. Commun., 2005, 525 RSC; (b) Y. Liang, M. Hanzlik and R. Anwander, J. Mater. Chem., 2005, 15, 3919 RSC; (c) X. Zhou, S. Qiao, N. Hao, X. Wang, C. Yu, L. Wang, D. Y. Zhao and G. Q. Lu, Chem. Mater., 2007, 19, 1870 CrossRef CAS; (d) L. Zhao, G. Zhu, D. Zhang, Y. Di, Y. Chen, O. Terasaki and S. Qiu, J. Phys. Chem. B, 2005, 109, 764 CrossRef CAS.
- (a) H. Y. Wu, C. T. Chen, I. M. Hung, C. H. Liao, S. Vetrivel and H. M. Kao, J. Phys. Chem. C, 2010, 114, 7021 CrossRef CAS; (b) Y. Goto and S. Inagaki, Microporous Mesoporous Mater., 2006, 89, 103 CrossRef CAS.
- V. Rebbin, R. Schmidt and M. Fröba, Angew. Chem., Int. Ed., 2006, 45, 5210 CrossRef CAS.
- (a) V. Rebbin, M. Jakubowski, S. Potz and M. Fröba, Microporous Mesoporous Mater., 2004, 72, 99 CrossRef CAS; (b) Y. D. Xia, Z. X. Yang and R. Mokaya, Chem. Mater., 2006, 18, 1141 CrossRef CAS; (c) M. P. Kapoor and S. Inagaki, Chem. Lett., 2004, 33, 88 CrossRef CAS.
- S. Z. Qiao, C. Z. Yu, W. Xing, Q. H. Hu, H. Djojoputro and G. Q. Lu, Chem. Mater., 2005, 17, 6172 CrossRef CAS.
- (a) S. S. Park and C. S. Ha, Chem. Mater., 2005, 17, 3519 CrossRef CAS; (b) S. S. Park and C. S. Ha, Chem. Commun., 2004, 1986 RSC.
- (a) A. Thomas, Angew. Chem., Int. Ed., 2010, 49, 8328 CrossRef CAS; (b) M. Beretta, J. Morell, P. Sozzani and M. Fröba, Chem. Commun., 2010, 46, 2495 RSC; (c) A. Ide, R. Voss, G. Scholz, G. A. Ozin, M. Antonietti and A. Thomas, Chem. Mater., 2007, 19, 2649 CrossRef CAS; (d) S. Inagaki, S. Guan, Q. Yang, M. P. Kapoor and T. Shimada, Chem. Commun., 2008, 202 RSC; (e) A. Kuschel and S. Polarz, J. Am. Chem. Soc., 2010, 132, 6558 CrossRef CAS; (f) A. Kuschel, H. Sievers and S. Polarz, Angew. Chem., Int. Ed., 2008, 47, 9513 CrossRef CAS; (g) S. MacQuarrie, M. P. Thompson, A. Blanc, N. J. Mosey, R. P. Lemieux and C. M. Crudden, J. Am. Chem. Soc., 2008, 130, 14099 CrossRef CAS; (h) J. Morell, S. Chatterjee, P. J. Klar, D. Mauder, I. Shenderovich, F. Hoffmann and M. Fröba, Chem.–Eur. J., 2008, 14, 5935 CrossRef CAS; (i) S. Polarz and A. Kuschel, Adv. Mater., 2006, 18, 1206 CrossRef CAS; (j) X. Wu, T. Blackburn, J. D. Webb, A. E. Garcia-Bennett and C. M. Crudden, Angew. Chem., Int. Ed., 2011, 50, 8095 CrossRef CAS.
- T. Y. Zhuang, J. Y. Shi, B. C. Ma and W. Wang, J. Mater. Chem., 2010, 20, 6026 RSC.
- C. Cai and C. G. Xia, Synthesis, 2006, 14, 2297 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and characterisation details. See DOI: 10.1039/c2ra00805j |
| This journal is © The Royal Society of Chemistry 2012 |