Ethanol organosolv lignin-based rigid polyurethane foam reinforced with cellulose nanowhiskers
Yang
Li
and
Arthur J.
Ragauskas
*
Institute of Paper Science and Technology, School of Chemistry and Biochemistry, Georgia Institute of Technology, Atlanta, GA 30332, USA. E-mail: arthur.ragauskas@ipst.gatech.edu; Fax: (+1)404-894-4778
First published on 24th February 2012
Abstract
An ethanol organosolv lignin polyol, prepared by reacting lignin with propylene oxide catalyzed by potassium hydroxide, was used to synthesize rigid polyurethane foam which was further reinforced by cellulose nanowhiskers (CNWs) up to 5 wt%. The resulting nanocomposites have shown significantly improved mechanical and thermal properties primarily attributed to the phenolic structure and high functionality of lignin as well as the rigidity of CNWs and crosslinking introduced by CNWs.
With an ever-increasing societal focus on environmental and economical sustainability, biorenewable energy and materials from non-food bioresources, especially wood, are drawing increasing attention from consumers, governments, industries, and research institutes. Cellulose and lignin existing in wood cell walls are abundant biopolymers in nature, corresponding to an annual industrial production of 1.5 × 1012 and 5 × 107 tons, respectively.1,2 Due to their renewable and biodegradable nature, extremely wide availability, non-agricultural based economy, and reactivity attributed to various functional groups, cellulose and lignin have been exploited in many applications.3–15
Cellulose nanowhiskers (CNWs) are a group of needle-like nano particles hydrolyzed from cellulose fibers. It has a wide size range and is normally 100 to 300 nm in length and 3–15 nm in width for wood or cotton-based CNWs.16 Its tensile strength and modulus are approximately 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 MPa and 150 GPa, respectively.17,18 There has been a growing interest in CNWs reinforced nanocomposites in the last decade, including polyurethane (PU). Up to 5 wt%, 30 wt%, and 1 wt% of CNWs reinforced thermoplastic PU films were studied by Marcovich et al.,19 Cao et al.,20,21 and Auad et al.22,23 SEM images of the resulting nanocomposites indicated well-dispersed CNWs in the polymer, and the FT-IR spectra revealed strong hydrogen bonding and chemical reactions between CNWs and isocyanates. Tensile property of the nanocomposites were shown to be significantly improved with increasing CNWs content, primarily attributed to the rigidity of the additives and the higher crosslinking density compared to the neat film. It was also found that CNWs addition favored a phase separation of soft and hard domains which led to an upward shift in melting temperature of crystalline phase, an increase in Young's modulus, and a decrease in deformation at break.22,23
000 MPa and 150 GPa, respectively.17,18 There has been a growing interest in CNWs reinforced nanocomposites in the last decade, including polyurethane (PU). Up to 5 wt%, 30 wt%, and 1 wt% of CNWs reinforced thermoplastic PU films were studied by Marcovich et al.,19 Cao et al.,20,21 and Auad et al.22,23 SEM images of the resulting nanocomposites indicated well-dispersed CNWs in the polymer, and the FT-IR spectra revealed strong hydrogen bonding and chemical reactions between CNWs and isocyanates. Tensile property of the nanocomposites were shown to be significantly improved with increasing CNWs content, primarily attributed to the rigidity of the additives and the higher crosslinking density compared to the neat film. It was also found that CNWs addition favored a phase separation of soft and hard domains which led to an upward shift in melting temperature of crystalline phase, an increase in Young's modulus, and a decrease in deformation at break.22,23
With the growing application of organosolv pretreatment as an environmentally benign biomass pretreatment technology for biofuels, large amounts of ethanol organosolv lignin (EOL) is anticipated to be produced by treatment of biomass with ethanol. EOL is usually high-purity, low molecular weight and sulfur-free lignin which has less condensed products and a narrower molecular weight distribution than lignin obtained from other methods.24–26 The large amounts of hydroxyl groups in lignin structure, as well as the potential benefits it can provide to PU namely antioxidative and fire retardant properties, make it a promising candidate for PU synthesis. This research was initially focused on direct incorporation of lignin.27 However, further development of a lignin polyol could not only improve the lignin content in PU but also result in easier processing and better performance of PU. Oxypropylation of lignin, first studied by Glasser and co-workers28 and further developed by other researchers,29,30 has been recognized as the most promising method to derive solid lignin into a liquid polyol.
A formulation optimization study of Kraft lignin-based rigid PU foam showed that the optimal mechanical property was obtained by using only Kraft lignin polyol without the assistance of any other polyols.31 Based on the above result, herein, EOL-based rigid PU foams were synthesized by directly reacting EOL polyol with polymeric methylene diphenyl diisocyanate (MDI) which was catalyzed by dimethylcyclohexaneamine (DMCHA) and a Mannich base catalyst. Silicone surfactant was added to reduce the surface tension of closed cells. Pentane was used as a physical blowing agent. Foams reinforced with 0, 1 and 5 wt% of CNWs were prepared by a one-shot method.32 A water suspension of CNWs was directly mixed with EOL polyol followed by the removal of water under high vacuum. Compared to its dimethylformamide (DMF) suspension,33 this method avoided freeze drying of CNWs which would cause the agglomeration of CNWs and a difficulty in re-dispersion as well as the large use of DMF.
EOL was completely rendered into a liquid polyol without any insoluble residues. Pure oxypropylated EOL was separated from poly(propylene oxide) (PPO) by extracting the mixture three times with hot hexane under reflux. It accounted for ∼55 wt% of the product. Oxypropylated EOL has an increased molecular weight (Mw = 3.7 × 103 g mol−1) due to the graft of PPO and a narrower polydispersity (Mw/Mn = 2.2) compared to EOL (Mw = 2.6 × 103 g mol−1, Mw/Mn = 2.5) as revealed by GPC results. FT-IR spectra of EOL before and after oxypropylation were normalized to the intensity of the lignin aromatic ring vibrations at 1600 cm−1. Stronger bond intensities at ∼2970 cm−1 and 1000–1100 cm−1 were observed for oxypropylated EOL (Fig. 1b) than the original EOL (Fig. 1a). Those changes are attributed to CH3, CH2, CH and CO stretching vibrations of PPO grafts. The 31P NMR spectrum (Fig. 2a) of phosphitylated EOL shows phosphorus signals corresponding to aliphatic (144.8–150.8 ppm), condensed phenolic (141.6–144.5 ppm), guaiacyl (136.4–141.6 ppm) and a small amount of carboxylic (134.1–136.2 ppm) hydroxyl groups. In contrast, the phosphitylated EOL polyol (Fig. 2b) exhibited only aliphatic (143.0–148.8 ppm) and carboxylic (133.6–136.9 ppm) hydroxyl signals. All results suggest a successful chain extension reaction of EOL. The hydroxyl index of EOL polyol was calculated to be 380 mg KOH per g based on the integrated analysis of 31P NMR spectrum data, which is a suitable value between 300 and 800 for rigid PU foam preparation.30
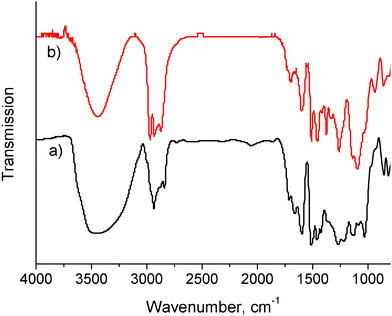 | ||
| Fig. 1 FT-IR spectra of (a) EOL and (b) oxypropylated EOL. | ||
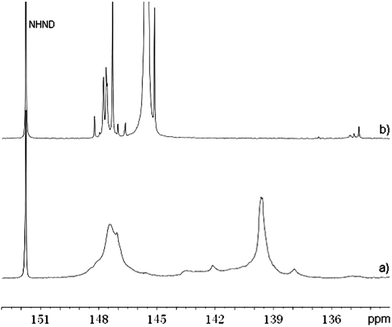 | ||
| Fig. 2 31P NMR spectra of (a) EOL and (b) oxypropylated EOL. | ||
The FT-IR spectra of prepared rigid PU foams are relatively the same where typical linkages of PU can be readily seen in Fig. 3, e.g., N–H stretching and bending vibrations (∼3310 cm−1 and ∼1513 cm−1), aliphatic C–H (∼2932 cm−1), OC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (∼1729 cm−1), CO–NH (∼1612 cm−1), O–CO (∼1227 cm−1) and C–O (∼1079 cm−1) stretching vibrations. PU foams showed reduced average cell diameters from 320 ± 20, 269 ± 28, to 191 ± 16 μm with increasing CNWs contents from 0, 1, to 5 wt% as indicated by SEM images (Fig. 4), the same observation as in our previous study.33 It is presumably because CNWs served as nucleation sites to facilitate the cell nucleation process, and the increased number of nucleation sites led to a finer cell structure.34,35 The density was calculated as an average value of mass divided by volume of three samples from each foam (Table 1). The density of rigid PU foam used for thermal insulation in buildings normally ranges between 30 kg m−3 and 45 kg m−3. However, it can reach 100 kg m−3 for some applications.36 In the present study, the density of the prepared foams increased due to the smaller cell size at higher CNWs content but still within this range.
O (∼1729 cm−1), CO–NH (∼1612 cm−1), O–CO (∼1227 cm−1) and C–O (∼1079 cm−1) stretching vibrations. PU foams showed reduced average cell diameters from 320 ± 20, 269 ± 28, to 191 ± 16 μm with increasing CNWs contents from 0, 1, to 5 wt% as indicated by SEM images (Fig. 4), the same observation as in our previous study.33 It is presumably because CNWs served as nucleation sites to facilitate the cell nucleation process, and the increased number of nucleation sites led to a finer cell structure.34,35 The density was calculated as an average value of mass divided by volume of three samples from each foam (Table 1). The density of rigid PU foam used for thermal insulation in buildings normally ranges between 30 kg m−3 and 45 kg m−3. However, it can reach 100 kg m−3 for some applications.36 In the present study, the density of the prepared foams increased due to the smaller cell size at higher CNWs content but still within this range.
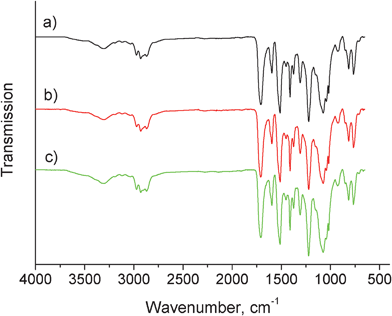 | ||
| Fig. 3 FT-IR spectra of rigid PU foams reinforced with (a) 0 wt%, (b) 1 wt%, and (c) 5 wt% of CNWs. | ||
 | ||
| Fig. 4 SEM images of rigid PU foams reinforced with (a) 0 wt%, (b) 1 wt%, and (c) 5 wt% of CNWs. Scale bar: 500 μm. | ||
| Polyol(s) | Sucrose and glycerol polyol | Sucrose and glycerol polyol | EOL polyol | EOL polyol | EOL polyol |
|---|---|---|---|---|---|
| CNW (wt%) | 033 | 031 | 0 | 1 | 5 |
| Density (kg m−3) | 53.8 ± 0.5 | 29.3 ± 0.4 | 33.6 ± 0.6 | 37.1 ± 0.6 | 62.3 ± 0.5 |
| Strength (MPa) | 0.15 ± 0.05 | 0.10 ± 0.01 | 0.20 ± 0.01 | 0.26 ± 0.03 | 0.52 ± 0.01 |
| Modulus (MPa) | 3.3 ± 0.9 | 1.5 ± 0.1 | 4.1 ± 0.2 | 5.4 ± 0.2 | 12.8 ± 0.1 |
| T g (°C) | — | — | 89 ± 3 | 104 ± 3 | 104 ± 2 |
| T d (°C) | — | — | 247 ± 2 | 263 ± 3 | 296 ± 1 |
Mechanical properties of the resulting rigid PU foams were investigated by compression testing generating a series of stress–strain curves shown in Fig. 5. The compressive strength of all foams is higher than 100 kPa which is a sufficient value for many rigid PU foam applications (Table 1).36 The control foam (0 wt% CNWs) showed enhanced mechanical properties compared to those synthesized from commercial sucrose and glycerol polyols.31,33 Additionally, the yield strength and compressive modulus were slightly increased for the 1 wt% of CNWs reinforced nanocomposite and a significant improvement in the nanocomposite prepared with addition of 5 wt% of CNWs was observed. It can be primarily attributed to the high mechanical properties of CNWs, the rigidity of the lignin phenolic structure, and the higher density due to the decrease of cell size. Moreover, CNWs have been proved to be covalently bonded to polyurethane molecular chains through the reaction of hydroxyl groups of CNWs with isocyanate groups of MDI.37 It could possibly result in a higher crosslinking density which is a potential reason for the improvement in the compressive properties.
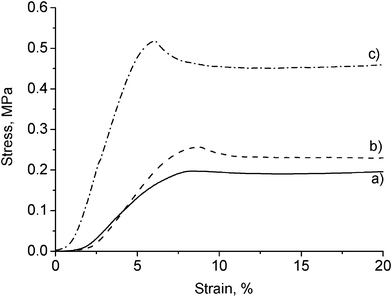 | ||
| Fig. 5 Compressive stress–strain curves of rigid PU foams reinforced with (a) 0 wt%, (b) 1 wt%, and (c) 5 wt% of CNWs. | ||
Thermal properties of the prepared rigid PU foams were investigated by DSC and TGA (Fig. 6) in terms of glass transition temperature (Tg) and decomposition temperature (Td). The DSC and TGA results clearly indicated enhanced thermal stability (Table 1). As mentioned above, a reaction took place between CNWs and MDI which reduced the mobility of the polyurethane chains and caused the increment in Tg and Td. The same observation and explanation can be found elsewhere regarding cellulose micro/nanocrystals reinforced polyurethane.19
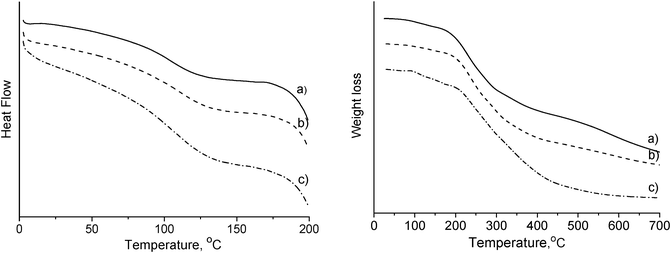 | ||
| Fig. 6 DSC (left) and TGA (right) curves of rigid PU foams reinforced with (a) 0 wt%, (b) 1 wt%, and (c) 5 wt% of CNWs. Both of the DSC and TGA curves were shifted showing a relative value of heat flow or weight loss for easy illustration. | ||
In summary, EOL polyol has been proved as a viable and promising polyol to prepared rigid PU foams. The incorporation of CNWs significantly improves both the mechanical and thermal properties of rigid PU nanocomposite foams at a low content of 5 wt%. The combined application of lignin and cellulose in PU synthesis explores a new and economical way to utilize the most abundant natural biopolymers, and at the same time, results in a greener and more competitive biobased rigid PU foam.
Experimental
EOL preparation
Pine wood chips (160 g) were first grounded into sawdust and extracted with toluene and ethanol (2:1, v/v, 2 L) for 48 h. Pretreatment was carried out in a Parr reactor filled with extracted sawdust, ethanol (65%, 7:1, v/w), and concentrated sulfuric acid (1.1 wt%) at 170 °C for 60 min.38 The pretreated sawdust was washed with distilled water and separated from the liquid portion by filtration. Water was then added in the liquid to precipitate EOL which was then filtered, washed, and oven dried for future use.
Oxypropylation of EOL
Oxypropylation of EOL was performed according to a published method.30 EOL (20.00 mg) was mixed with PO (80.00 mL) and KOH (1.00 mg) in a 250 mL Parr reactor which was then closed and heated under stirring till 160 °C. Temperature and pressure started to increase progressively from this point. After reaching the maximum value, pressure decreased rapidly due to the propylene oxide consumption. The return of the relative pressure to zero indicated the end of the reaction. The reactor was cooled and the ensuing polyol was recovered.Rigid PU foam preparation
EOL polyol (40.00 g) was premixed with DMCHA (1.80 g), Mannich base catalyst (0.90 g), and surfactant (1.50 g) for 1 min, pentane (10.00 g) was then added together with polymeric MDI (41.59 g) under strong stirring for 25 s. Foams were cured at RT for at least 48 h.Characterization
EOL/oxypropylated EOL (∼20 mg) was acetylated by stirring in a mixture of acetic anhydride and pyridine (1:1, v/v, 2 mL) at RT for 72 h. The solvents were then removed under reduced pressure at 50 °C. The acetylated product was dissolved in chloroform (50 mL) and washed with water (20 mL). The chloroform phase was dried over anhydrous MgSO4 and concentrated under reduced pressure. GPC was accomplished using an Agilent Technologies 1200 series analysis system consisting of an autosampler, a UV detector, and four columns of Styragel HR 0.5, HR 2, HR4, and HR 6 linked in series using THF as the eluent. The acetylated sample was dissolved in THF (1 mg mL−1), filtered through a 0.45 μm filter, injected (20 μL) into the GPC system, and detected by a UV detector at 280 nm. Standard polystyrene samples were used to construct a calibration curve.
FT-IR signals were recorded between 4000 and 600 cm−1 with a resolution of 4.0 cm−1 and 64 scans on a Magna 550 FT-IR Spectrometer. KBr powder was used to obtain pellets with a thickness of 1 mm.
2-Chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane (TMDP) was used as a phosphitylation reagent for the 31P NMR analysis. Briefly, a relaxation reagent solution (chromium(III) acetylacetonate, 3.6 mg mL−1) and an internal standard solution (N-hydroxy-5-norborene-1,3-dicarboximide (NHND), 4 mg mL−1) were prepared in a mixed solvent of pyridine and CDCl3 (1.6:1, v/v). 20–25 mg of EOL/EOL polyol was dissolved in 0.5 mL solvent in a vial, followed by TMDP (0.1 mL) addition. Spectra were recorded with a 400 MHz Bruker system at 25 °C with a 25 s pulse delay and typically ∼100 acquisition transients.
SEM images were taken by a Hitachi S-800 FE-SEM under an accelerating voltage of 10 kV and magnification of 40. Samples were coated with gold to prevent charge build up during imaging. Five images from different parts of each foam were taken for an average cell size analysis.
Mechanical testing
Compression testing was carried out on an MTS Insight 2 universal test machine with a crosshead speed of 4 mm min−1 and a specimen size of 25 mm × 25 mm × 15 mm according to ASTM C365/C365M-05. The values of yield strength and compressive modulus were determined as an average by testing 5 specimens from each foam.Thermal analysis
DSC analysis was performed on a Q200 TA instrument. Approximately 10 mg sample was heated from RT to 200 °C under a nitrogen gas atmosphere at a heating rate of 10 °C min−1. TGA was performed on a Q5000 TA instrument with samples weighing 7–10 mg from RT to 700 °C at a heating rate of 10 °C min−1 under nitrogen gas flux of 25 mL min−1. Both the Tg and Td values were determined as the average value of three performances for each foam sample.Acknowledgements
The authors would like to acknowledge the financial support of Paper Science and Engineering fellowship program at Institute of Paper Science and Technology at Georgia Institute of Technology.References
- D. Klemm, H.-P. Schmauder and T. Heinze, in Biopolymers, ed. E. Vandamme, S. De Beats and A. Steinbuchel, Wiley-VCH, Weinheim, 2002, pp. 290–292 Search PubMed.
- D. Klemm, B. Heublein, H.-P. Fink and A. Bohn, Angew. Chem., Int. Ed., 2005, 44, 3358 CrossRef CAS.
- B. L. Holt, S. D. Stoyanov, E. Pelan and V. N. Paunov, J. Mater. Chem., 2010, 20, 10058 RSC.
- H. Sehaqui, M. Allais, Q. Zhou and L. A. Berglund, Compos. Sci. Technol., 2011, 71, 382 CrossRef CAS.
- I. Siro and D. Plackett, Cellulose, 2010, 17, 459 CrossRef CAS.
- H. Liu, D. Liu, F. Yao and Q. Wu, Bioresour. Technol., 2010, 101, 5685 CrossRef CAS.
- J. Tan, H. Kang, R. Liu, D. Wang, X. Jin, Q. Li and Y. Huang, Polym. Chem., 2011, 2, 672 RSC.
- J. Gajdziok, B. Martina, C. Zuzana and R. Miloslava, Drug Dev. Ind. Pharm., 2010, 39, 1115 CrossRef.
- Y. Ikeuchi, F. Z. Khan, N. Onishi, M. Shiotsuki, T. Masuda, Y. Nishio and F. Sanda, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 3986 CrossRef CAS.
- S. Vlad, D. Filip, D. Macocinschi, I. Spiridon, A. Nistor, L. M. Gradinaru and V. E. Musteata, Optoelectron. Adv. Mater., 2010, 4, 407 CAS.
- J. M. Lavoie, W. Bare and M. Bilodeau, Bioresour. Technol., 2011, 102, 4917 CrossRef CAS.
- Q. Shen, T. Zhang, W. Zhang, S. Chen and M. Mezgebe, J. Appl. Polym. Sci., 2011, 121, 989 CrossRef CAS.
- E. M. Nour-Eddine, Q. Yuan and F. Huang, in Research Progress in Paper Industry and Biorefinery (4th ISETPP), ed. R. Sun and S. Fu, South China University of China, Guangzhou, 2010, pp. 53–56 Search PubMed.
- S. Liu, X. Cheng, F. Jin, Q. Zhou, B. Wu and L. Xie, Proc. Int. Symp. Aqua Sci. Water Resour. Low Carbon Energ., 2010, 1251, 344 CAS.
- G. Sivasankarapillai and A. G. McDonald, Biomass Bioenergy, 2011, 35, 919 CrossRef CAS.
- Y. Habibi, L. A. Lucia and O. J. Rojas, Chem. Rev., 2010, 110, 3479 CrossRef CAS.
- S. Kamel, Express Polym. Lett., 2007, 1, 546 CrossRef CAS.
- A. Sturcova, G. R. Davies and S. J. Eichhorn, Biomacromolecules, 2005, 6, 1055 CrossRef CAS.
- N. E. Marcovich, M. L. Auad, N. E. Bellesi, S. R. Nutt and M. I. Aranguren, J. Mater. Res., 2006, 21(4), 870 CrossRef CAS.
- X. Cao, D. Hua and C. M. Li, Biomacromolecules, 2007, 8, 899 CrossRef CAS.
- X. Cao, Y. Habibi and L. A. Lucia, J. Mater. Chem., 2009, 19, 7137 RSC.
- M. L. Auad, V. S. Contos, S. Nutt, M. I. Aranguren and N. E. Marcovich, Polym. Int., 2008, 57, 651 CrossRef CAS.
- M. L. Auad, M. A. Mosiewicki, T. Richardson and M. I. Aranguren, J. Appl. Polym. Sci., 2010, 115, 1215 CrossRef CAS.
- J. Zakzeski, P. C. A. Brujinincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552 CrossRef CAS.
- R. E. Hage, N. Brosse, L. Chrusciel, C. Sanchez, P. Sannigrahi and A. Ragauskas, Polym. Degrad. Stab., 2009, 94, 1632 CrossRef.
- P. Sannigrahi, A. J. Ragauskas and S. J. Miller, Energy Fuels, 2010, 24, 683 CrossRef CAS.
- H. Yoshida, R. Morck, K. P. Kringstad and H. Hatakeyama, J. Appl. Polym. Sci., 1987, 34, 1187 CrossRef CAS.
- L. C.-F. Wu and W. G. Glasser, J. Appl. Polym. Sci., 1984, 29, 1111 CrossRef CAS.
- W. G. Glasser, J. F. Selin, P. L. Hall and S. W. Drew, Proc. Int. Symp. Wood Pulping Chem., 1981, 4, 39 CAS.
- C. A. Cateto, M. F. Barreiro, A. E. Rodrigues and M. N. Belgace, Ind. Eng. Chem. Res., 2009, 48, 2583 CrossRef CAS.
- Y. Li and A. J. Ragauskas, J. Wood Chem. Technol. DOI:10.1080/02773813.2011.652795.
- M. Thirumal, D. Khastgir, N. K. Singha, B. S. Manjunath and Y. P. Naik, J. Appl. Polym. Sci., 2008, 108, 1810 CrossRef CAS.
- Y. Li and A. J. Ragauskas, J. Nanosci. Nanotechnol., 2011, 11, 6904 CrossRef CAS.
- M. Alexandre and P. Dubois, Mater. Sci. Eng., R, 2000, 28(1), 1 CrossRef.
- L. J. Lee, C. Zeng, X. Cao, X. Han, J. Shen and G. Xu, Compos. Sci. Technol., 2005, 65(15–16), 2344 CrossRef CAS.
- PU Europe Technical Report on Thermal insulation materials made of rigid polyurethane foam (PUR/PIR), PU Europe report No 1, Brussels, Belgium, 2006 Search PubMed.
- A. Pei, J.-M. Malho, J. Ruokolainen, Q. Zhou and L. A. Berglund, Macromolecules, 2011, 44, 4422 CrossRef CAS.
- X. Pan, D. Xie, R. W. Yu, D. Lam and J. N. Saddler, Ind. Eng. Chem. Res., 2007, 46, 2609 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |