Fabrication of magnetic drug-loaded polymeric composite nanofibres and their drug release characteristics†
Linge
Wang
*a,
Meng
Wang
b,
Paul D.
Topham
c and
Yong
Huang
*de
aDepartment of Biomedical Science, University of Sheffield, Firth Court, Western Bank, Sheffield, S10 2TN, UK. E-mail: linge.wang@sheffield.ac.uk; Tel: +44 (0)114 222 4662; Fax: +44 (0)114 222 2787
bShenzhen Zhijun Pharmaceutical Co. Ltd, Shenzhen, 518110, China. E-mail: wangmeng@gic.ac.cn
cChemical Engineering and Applied Chemistry, Aston University, Birmingham, B4 7ET, UK. E-mail: p.d.topham@aston.ac.uk; Tel: +44 (0)121 204 3413
dNatural Research Center for Engineering Plastics, Technical Institute of Physics & Chemistry, Chinese Academy of Sciences, Beijing, 100190, China, . E-mail: yhunag@mail.ipc.ac.cn; Tel: +86 (0)10 82543478; Fax: +86 (0)10 62554670
eState Key Laboratory of Polymer Physics and Chemistry, Institute of Chemistry, Chinese Academy of Sciences, Beijing, 100080, China
First published on 2nd February 2012
Abstract
Magnetic polymer nanofibres intended for drug delivery have been designed and fabricated by electrospinning. Magnetite (Fe3O4) nanoparticles were successfully incorporated into electrospun nanofibre composites of two cellulose derivatives, dehydroxypropyl methyl cellulose phthalate (HPMCP) and cellulose acetate (CA), while indomethacin (IDN) and aspirin have been used as model drugs. The morphology of the neat and magnetic drug-loaded electrospun fibres and the release characteristics of the drugs in artificial intestinal juice were investigated. It was found that both types of electrospun composite nanofibres containing magnetite nanoparticles showed superparamagnetism at room temperature, and their saturation magnetisation and morphology depend on the Fe3O4 nanoparticle content. Furthermore, the presence of the magnetite nanoparticles did not affect the drug release profiles of the nanofibrous devices. The feasibility of controlled drug release to a target area of treatment under the guidance of an external magnetic field has also been demonstrated, showing the viability of the concept of magnetic drug-loaded polymeric composite nanofibres for magneto-chemotherapy.
1. Introduction
Electrospinning 1 is a highly versatile technique for generating ultrafine (sub-micron) fibres from a wide variety of polymers, polymer blends and nanoparticle- or drug-impregnated polymers.2–5 Technically, almost any soluble polymer with sufficiently high molecular weight, including highly branched copolymers,6 can be electrospun. With their ultrafine size and huge surface-to-volume ratio, electrospun fibres can be applied in numerous areas, most notably perhaps in biomedical, environmental engineering and energy storage applications.In the biomedical field, drug delivery is one of the most important strands of electrospinning research. The drug-loaded electrospun polymer fibre mats provide the advantage of increased drug release as compared to cast-films due to the increased surface area.7,8 Generally, biopolymers [such as poly(caprolactone) (PCL),9,10 poly(D, L-lactic acid) (PLA),7,8,11–13 poly(vinyl alcohol) (PVA),14–17 cellulose derivates18–20 and selected copolymers21–25] are more popular than synthetic polymers for drug delivery usage because of their biodegradable (or biocompatible) properties and previous FDA approval.
Recently, with the development of materials and nanotechnology, polymeric nanocomposite drug delivery systems (or targeted nanocarriers) have gained significant attention due to their unique advantages in tumour diagnostics and therapeutics. In particular, magnetic micro- and nanoparticles are currently recognized as one of the most promising modalities of such carriers.26–28 This approach (similar to ‘magnetic targeting’ which was proposed some 30 years ago29,30) consists of applying an external magnetic field to trap drug-loaded carriers within a targeted site.31 Such magnetic micro- or nano-drug carriers, are conventionally synthesised by coating magnetic nanoparticles with functionalised ligands,27 or by mixing magnetic nanoparticles with drugs to produce composites.28,32 To date, there are more than 200 papers describing the magnetic properties or electrical conductivity of magnetic nanoparticle/polymer composite nanofibres, and the number of reports on drug delivery from nanofibres is above 400. However, there is only one report focusing on a combination of the two approaches; introducing magnetic particles into electrospun polymeric fibres for the purpose of targeted and controlled drug release. Tan et al.33 used a fluorescent enzyme (albumin with dog-fluorescein isothiocyanate) as the model drug and monitored the drug release properties of the composite fibres using UV-Vis and fluorescence emission spectroscopy. In their brief investigation, it was found that the drug was released within 24 h. Although this work was pioneering, the authors did not provide in-depth details about the drug release profile and the corresponding effect of the magnetic nanoparticles. Our research herein concerns the effect of magnetic particle loading on the drug release process for the first time (for two commonly dispensed drugs, ibuprofen and indomethacin).
Cellulose is one of the most common biopolymers on earth. Consequently, cellulose (and its derivatives) have been widely used in various biomedical applications, such as tissue engineering and drug delivery. Hydroxypropyl methylcellulose phthalate (HPMCP), a cellulose derivative, is a pH-sensitive polymer that can be dissolved at pH ≥ 5.5. This is because the phthalyl substituents of the polymer cause a reversible hydration or coacervate formation (hydrophobic aggregation) depending on the pH of the solution.34HPMCP has been reported by many groups, in various forms, as drug carriers (such as micro-capsules,34 tablets35 and enteric drugs36,37). In our previous work,19 ultrafine fibre mats of HPMCP were electrospun for the first time and explored as drug carriers for targeted release in the intestine. It was found that the drug (erythromycin) release from HPMCP fibre mats proceeded by a slow diffusion process in artificial gastric juice, while the release was almost directly proportional to the incubation time in artificial intestinal juice (approximately 2.5 times faster than in artificial gastric juice) because of the dissolution of the HPMCP fibres. Conversely, work on drug-loaded electrospun CA fibre mats showed that the drug release exhibits a slow diffusion profile in artificial intestinal juice due to swelling of the fibres rather than complete dissolution in the buffer media.18,38
In the present contribution, magnetite (Fe3O4) nanoparticles have been incorporated into drug-loaded polymer composite fibre mats to develop a novel magnetic drug carrier. Two types of cellulose derivative, HPMCP and cellulose acetate (CA), were used as the polymer scaffold, respectively, while indomethacin (IDN, a poorly water-soluble non-steroidal anti-inflammatory drug) and aspirin (acetylsalicylic acid, which is slightly soluble in water; ca. 10 mg ml−1 at 37 °C) were selected as the model drugs. The morphology of the neat and magnetic drug-loaded electrospun fibre mats and the release profiles of the drugs were investigated from these two different systems; (i) water-soluble polymer/hydrophobic drug (HPMCP/IDN) composite fibres and (ii) water-insoluble polymer/hydrophilic drug (CA/aspirin) composite fibres, both in artificial intestinal juice.
2. Experimental section
2.1 Materials
HPMCP (Mn = 7.4 × 104 g mol−1) and CA (Mn = 2.9 × 104 g mol−1) were supplied from Taian Ruitai Cellulose Company (China) and Aldrich, respectively. Fe3O4 nanoparticles, phosphate buffered saline (PBS), aspirin and IDN (99.9%) were purchased from Sigma. Trypsin (tissue culture grade) was supplied by AMRESCO without further purification to prepare the buffer solutions. Ethanol, acetone and dimethylacetamide (DMAc) were all of analytical purity and used as received.2.2 Electrospinning magnetic drug-loaded cellulosic fibre mats
A 12 wt% HPMCP solution in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (w/w) acetone/ethanol19 and a 15 wt% CA solution in 2:1 (w/w) acetone/DMAc39,40 were prepared and used as the original solutions for the drug loading. Briefly, the Fe3O4 nanoparticles were used as received, without any modification. Appropriate amounts of Fe3O4 nanoparticles and model drugs, IDN and aspirin were then added to the HPMCP and CA solutions, respectively, at various polymer/drug/Fe3O4 ratios (as shown in Table 1) under constant stirring for 12 h, followed by sonication for 5 min. Electrospinning was performed at 20 °C in air, using home-made apparatus similar to that used in the literature41 and a schematic of the process is shown in Fig. 1. The mixed polymer/drug/Fe3O4 solution was placed in a 10 ml syringe and was dispensed by a syringe pump (Aladdin-220, World Precision Instruments Ltd., USA) at a rate of 5 ml h−1. A high voltage supply (Genvolt-73030, Genvolt High Voltage Industries Ltd., UK) was connected to the metallic needle (0.8 mm diameter) with a fixed voltage of 16 kV, and a sheet of aluminium foil was placed 15 cm below the tip of the needle to collect the fibres. The electrospun fibre mats were dried in a vacuum oven at room temperature for 24 h to remove any residual solvent.
:1 (w/w) acetone/ethanol19 and a 15 wt% CA solution in 2:1 (w/w) acetone/DMAc39,40 were prepared and used as the original solutions for the drug loading. Briefly, the Fe3O4 nanoparticles were used as received, without any modification. Appropriate amounts of Fe3O4 nanoparticles and model drugs, IDN and aspirin were then added to the HPMCP and CA solutions, respectively, at various polymer/drug/Fe3O4 ratios (as shown in Table 1) under constant stirring for 12 h, followed by sonication for 5 min. Electrospinning was performed at 20 °C in air, using home-made apparatus similar to that used in the literature41 and a schematic of the process is shown in Fig. 1. The mixed polymer/drug/Fe3O4 solution was placed in a 10 ml syringe and was dispensed by a syringe pump (Aladdin-220, World Precision Instruments Ltd., USA) at a rate of 5 ml h−1. A high voltage supply (Genvolt-73030, Genvolt High Voltage Industries Ltd., UK) was connected to the metallic needle (0.8 mm diameter) with a fixed voltage of 16 kV, and a sheet of aluminium foil was placed 15 cm below the tip of the needle to collect the fibres. The electrospun fibre mats were dried in a vacuum oven at room temperature for 24 h to remove any residual solvent.
| HPMCP/IDN/Fe3O4 | CA/aspirin/Fe3O4 | |||||
|---|---|---|---|---|---|---|
| Polymer/drug/Fe3O4 ratio in solution | Solution surface tension (mN m−1) | Solution conductivity (μS cm−1) | AFD (μm) | Solution surface tension (mN m−1) | Solution conductivity (μS cm−1) | AFD (μm) |
| 9:0:0 |
37.7 ± 0.5 | 143 ± 2 | 0.72 ± 0.30 | 40.0 ± 0.5 | 8.5 ± 0.2 | 0.78 ± 0.33 |
| 9:1:0 |
36.0 ± 0.5 | 151 ± 2 | 0.64 ± 0.19 | 38.4 ± 0.5 | 9.0 ± 0.2 | 0.75 ± 0.35 |
| 9:1:1 |
35.9 ± 0.5 | 182 ± 2 | 0.65 ± 0.33 | — | — | — |
| 9:1:3 |
35.1 ± 0.5 | 187 ± 2 | 0.66 ± 0.15 | 36.5 ± 0.5 | 37.3 ± 0.2 | 0.73 ± 0.30 |
| 9:1:9 |
35.3 ± 0.5 | 197 ± 2 | 0.61 ± 0.26 | 36.1 ± 0.5 | 41.6 ± 0.2 | 0.74 ± 0.34 |
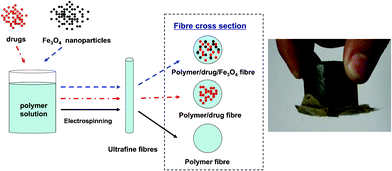 | ||
| Fig. 1 Schematic illustration for the preparation of magnetic drug-loaded polymer nanofibres, and a demonstration of the nanofibres being attracted to a magnet. | ||
2.3 Characterisation of the magnetic drug-loaded cellulosic fibre mats
The fibre mats were gold-coated and the morphology was studied using a scanning electron microscope (SEM, XL-30, Philips, Dutch) operating at 20 kV. A transmission electron microscope (TEM, JEOL-100CX/II, NEC, Japan) was also used to observe the morphology of the fibres, operating at 100 kV. Average fibre diameters (AFD) of the electrospun fibres from each solution were obtained using an UTHSCSA Image Tool Program using at least five SEM images (approximately 40–60 fibres were counted in each image), as shown in Table 1. The magnetic properties of the fibres were measured at room temperature (300 K) using a Magnetic Property Measurement System (MPMS XL-7, QUANTUM DESIGN, USA).2.4 Drug release study
An artificial intestinal juice buffer was made from 0.01 mg ml−1 trypsin solution in PBS buffer and adjusted to pH 6.8 using sodium hydroxide aqueous solution (0.1 M). The Fe3O4 and drug-containing electrospun HPMCP and CA fibre mats were cut into 1 cm diameter round pieces (approximately 100 mg by mass), and each sample was placed in a vial filled with 20 ml of the buffer solution. Subsequently, the vial was incubated at 37 °C in a thermostatic shaker. At appropriate intervals, each mat was transferred to another 20 ml of fresh buffer solution and the released drug (IDN or aspirin) in the original buffer solution was measured by the colorimetric method, as monitored by a UV-Vis spectrophotometer (UV-2550, SHIMADZU, Japan) at the characteristic wavelength of the drug (320 nm for IDN and 296 nm for aspirin). The UV absorbance of the drug was then converted to its concentration according to the predetermined calibration curve of the drug in buffer (see ESI†). Finally, the accumulative weight and relative percentage of the released drug were calculated as a function of incubation time.3. Results and discussion
3.1 Morphology
One of the great advantages of the electrospinning technique is that one can fabricate composite nanofibres of nanoparticle-or drug-impregnated polymer blends in which one or more of the components cannot be processed by electrospinning alone. Novel composite nanofibres can be made by physically mixing the additional component with the polymer solution. The polymer thus acts as a supporting matrix without the necessity of chemical modification of any of the materials. In order to obtain magnetic drug-loaded fibres, Fe3O4 nanoparticles and the drug were dispersed in HPMCP and CA solutions. Fig. 2(a)–(e) show the typical morphologies of the electrospun HPMCP fibres and the magnetic drug-loaded composite fibres at various HPMCP/IDN/Fe3O4 mass ratios (in the as-prepared solution). Typically, the HPMCP fibres and the HPMCP/IDN composite fibres were smooth and tape- or ribbon-like, which indicated that HPMCP and IDN were well incorporated into the composite fibres. The HPMCP/IDN/Fe3O4 composite fibres with low magnetite content also showed a smooth surface and were ribbon in shape. However, when the magnetite content in the HPMCP/IDN/Fe3O4 composite fibres was larger than 9:1:3, the quality of the composite fibres was markedly inferior (compare Fig. 2(d) with (e)), and the fibres became non-uniform along the fibre axis, caused by the aggregation of excessive Fe3O4 nanoparticles in the as-prepared solution and the electrospun fibres (Fig. 2(f)). The maximum loading of Fe3O4 nanoparticles was shown to be the same amount, by mass, as the polymer content of the fibres. Beyond that point, the quality of the composite nanofibres becomes very poor.
 | ||
| Fig. 2
SEM micrographs of HPMCP/IDN/Fe3O4 nanofibres with various respective mass ratios in as-prepared solutions of (a) 9:0:0, (b) 9:1:0, (c) 9:1:1, (d) 9:1:3, and (e) 9:1:9. (f) shows a typical TEM micrograph of HPMCP/IDN/Fe3O4 nanofibres (9:1:9). | ||
Furthermore, the higher the content of Fe3O4 nanoparticles embedded within the composite fibres, the more the Fe3O4 nanoparticles accumulated on the surface of fibres. Interestingly, no significant change was observed in the fibre diameter when altering the content of Fe3O4; the average fibre diameter slightly decreased from 0.72 μm to 0.61 μm after being loaded with IDN and Fe3O4 nanoparticles (as summarised in Table 1). This is attributed to the change in solution properties, such as a decrease in surface tension and an increase in conductivity by the addition of drug, complementing similar results reported in the literature.11,18,19,38
Similarly, electrospun fibres with diameters ranging from 0.78 μm to 0.73 μm were obtained by electrospinning CA/aspirin/Fe3O4 solutions with different drug and magnetite contents. As shown in Fig. 3, the electrospun fibres with a low magnetite content had a smooth surface and were cylindrical in shape, while with high magnetite content the Fe3O4 nanoparticles did not distribute uniformly in the polymeric fibres and the tendency to agglomerate together could not be avoided (see ESI†).
 | ||
| Fig. 3
SEM micrographs of the CA/aspirin/Fe3O4 nanofibres with various respective mass ratios in as-prepared solutions of (a) 9:0:0, (b) 9:1:0, (c) 9:1:3, and (d) 9:1:9. | ||
3.2 Magnetic properties
The HPMCP/IDN/Fe3O4 fibres and the CA/aspirin/Fe3O4 fibres both displayed magnetic properties characteristic of the starting iron oxides. Fig. 4 shows magnetisation curves of the HPMCP/IDN/Fe3O4 composite fibres measured at 300 K. No obvious magnetic hysteresis was observed at this temperature. This is characteristic of superparamagnetic nanoparticles in which thermal fluctuations are sufficient to overcome the anisotropic energy barrier.33,42 The saturation magnetisation of the HPMCP/IDN/Fe3O4 fibres at mass ratios of 9:1:3 and 9:1:9 were 0.28 and 0.52 emu g−1, respectively, clearly dependent on the level of Fe3O4 nanoparticles incorporation in the composite fibres,33 while the saturation magnetisation of CA/aspirin/Fe3O4 fibres at a mass ratio of 9:1:9 was 0.49 emu g−1. For comparison, the saturation magnetisation is 76 emu g−1 for bulk magnetite43 and 1.1 emu g−1 for PLLA/Fe3O4 electrospun nanofibres.33 The difference is attributed to a good distribution of the Fe3O4 nanoparticles.44 Moreover, our magnetic drug-loaded nanofibres could be moved under the guidance of an external magnetic field (see Fig. 1 and ESI†), demonstrating the feasibility of controlled drug release to target specific areas of treatment.
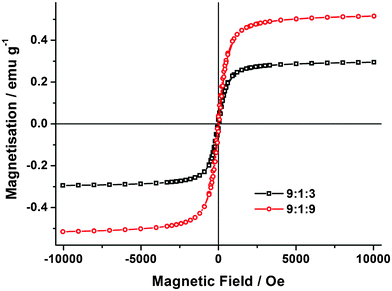 | ||
| Fig. 4 Magnetisation curves of the composite fibres of HPMCP/IDN/Fe3O4 at mass ratios of 9:1:3 and 9:1:9, measured at 300 K. | ||
3.3 Drug release
When IDN-loaded HPMCP fibre mats were placed into the artificial intestinal juice (pH = 6.8, above the critical dissolution pH), the fibres gradually dissolved in the artificial intestinal juice over a long enough period of time, about 11 h (as shown in Fig. 5). Under SEM observation (Fig. 5), the IDN-loaded HPMCP fibres (whether containing magnetite or not) were broken up and cracked after they were placed in the artificial intestinal juice for just a couple of hours (compare with Fig. 2). The cumulative release of the IDN (reported as the percentage of the actual amount of the drug present within the drug-loaded sample) as a function of immersion time from the IDN-loaded fibre mats is shown in Fig. 5. The results show that the release of IDN was almost directly proportional to the incubation time (close to a zero-order kinetic process) from the IDN-containing electrospun HPMCP fibre mats. No burst release was observed. Within 12 h, IDN was completely released from both of the electrospun fibre mat samples studied (HPMCP/IDN/Fe3O4 mass ratio of 9:1:0 and 9:1:9). Clearly, the release rate of IDN and the dissolution rate of the electrospun HPMCP fibres in the artificial intestinal juice occur simultaneously, indicating that the release of IDN in the artificial intestinal juice is dominated by the dissolution of HPMCP. However, no obvious effect of magnetite content on the IDN release profile was observed.
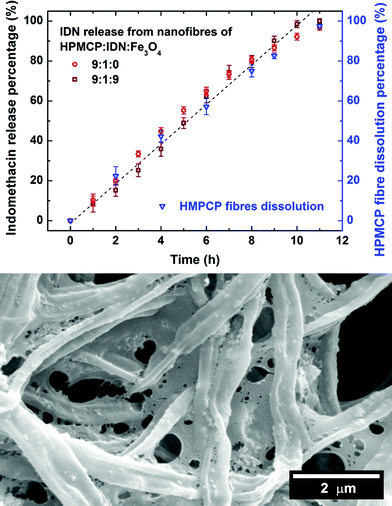 | ||
| Fig. 5 Profiles of IDN released (and HPMCP dissolved) from the HPMCP/IDN/Fe3O4 nanofibres (9:1:9) in artificial intestinal juice buffer, and a corresponding SEM micrograph of the nanofibres after 4 h. | ||
For all of the CA/aspirin/Fe3O4 composite fibres, the release profiles of aspirin were smooth and monotonously increased to reach the maximum value (i.e. ∼95%) after 24 h without an obvious burst release (as shown in Fig. 6). It is important to note that the release mechanism was tested for both first and second order (with respect to drug concentration) with the appropriate kinetic plots (data not shown), but neither model produced a linear progression with time. The SEM image (Fig. 6) shows that the morphology of fibres after 24 h immersion in buffer solution was almost unchanged (compare with Fig. 3(d)). This phenomenon is attributed to the insolubility of CA in aqueous media. Since there was no dissolution of CA, the aspirin was released by diffusion38 from the surface of the electrospun CA/aspirin/Fe3O4 composite fibres. This is a common feature of drug delivery systems of polymer/hydrophobic (or poorly water-soluble) drug composite fibre mats11 and magnetic drug-loaded nanocomposites.45 Also, the cumulative release of aspirin was much faster than that of IDN within 10 h, due to the higher solubility of aspirin in water than that of IDN. Meanwhile, no obvious effect of magnetite content was observed on the aspirin release profile (similar to the HPMCP/IDN/Fe3O4 composite fibre results), indicating that the magnetite nanoparticles did not adversely interfere with the drug release mechanism. The magnetite nanoparticles were thus physically dispersed throughout the composite fibres (see the TEM images in Fig. 2(f) and ESI†). In effect, the fibres operate as a magnetic label for the drugs and readily lead the drug to the area of treatment (e.g. inside the digestive system) under the guidance of an external magnetic field. Therefore, our system allows for a tissue-specific release of drugs that will consequently reduce side effects arising from release of the drug to erroneous areas of the body.
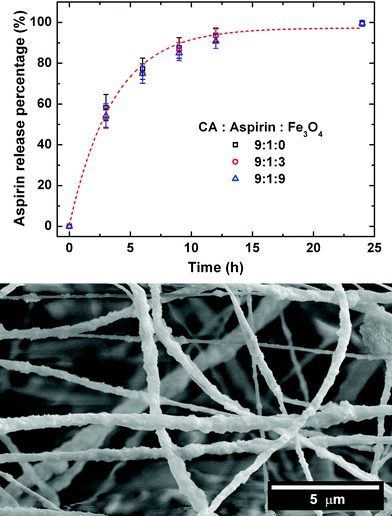 | ||
| Fig. 6 Profiles of aspirin released from the CA/aspirin/Fe3O4 nanofibres in artificial intestinal juice buffer, and a corresponding SEM micrograph of the nanofibres after 24 h. | ||
4. Conclusions
Superparamagnetic biocompatible polymer nanofibres containing magnetite nanoparticles and drugs were successfully prepared by electrospinning. Two series of HPMCP/IDN/Fe3O4 and CA/aspirin/Fe3O4 composite fibres were fabricated, where the morphologies of the nanofibres varied depending on the magnetite nanoparticle content. The neat polymer fibres and the polymer/drug composite fibres had smooth surfaces, as did those containing low magnetite content, indicating that the polymer, drug and the magnetite nanoparticles were well distributed within the composite fibres. High magnetite contents, on the other hand, led to the quality of the composite fibres becoming markedly inferior as the fibres became non-uniform along the fibre axis, caused by the aggregation of the excessive Fe3O4 nanoparticles. However, all composite nanofibres containing magnetite nanoparticles showed superparamagnetism at room temperature, with the saturation magnetisation depending on the amount of Fe3O4 nanoparticles added to the original solution.In both types of magnetic drug-load composite nanofibres, the presence of magnetite nanoparticles did not affect the drug release process, which was found to be primarily dominated by the polymeric carrier matrix properties. Owing to the pH-sensitive properties of HPMCP, IDN was released from the IDN-containing HPMCP fibre mats following close to zero-order kinetics in the artificial intestinal juice (HPMCP dissolves in aqueous media at pH ≥ 5.5). Meanwhile, the release of aspirin from the drug-loaded electrospun composite fibre mats was smooth and monotonously increased to reach a maximum plateau value by a slow diffusion process in the buffer. Based on the results, such magnetic drug-loaded nanofibres are of potential interest for various applications in medicines, especially drug delivery to precise target areas in the digestive system.
Acknowledgements
Financial support by the National Natural Science Foundation of China (No. 50821062) is greatly appreciated. The authors also thank Dr Changzhong Chen for his valuable advice.References
- J. Doshi and D. H. Reneker, J. Electrost., 1995, 35, 151–160 CrossRef CAS.
- S. Agarwal, J. H. Wendorff and A. Greiner, Polymer, 2008, 49, 5603–5621 CrossRef CAS.
- D. Li and Y. N. Xia, Adv. Mater., 2004, 16, 1151–1170 CrossRef CAS.
- Z. M. Huang, Y. Z. Zhang, M. Kotaki and S. Ramakrishna, Compos. Sci. Technol., 2003, 63, 2223–2253 CrossRef CAS.
- S. Ramakrishna, K. Fujihara, W. E. Teo, T. Yong, Z. W. Ma and R. Ramaseshan, Mater. Today, 2006, 9, 40–50 CrossRef CAS.
- L. Wang, C. M. Li, A. J. Ryan and S. P. Armes, Adv. Mater., 2006, 18, 1566–1570 CrossRef CAS.
- E. R. Kenawy, G. L. Bowlin, K. Mansfield, J. Layman, D. G. Simpson, E. H. Sanders and G. E. Wnek, J. Controlled Release, 2002, 81, 57–64 CrossRef CAS.
- X. H. Zong, K. Kim, D. F. Fang, S. F. Ran, B. S. Hsiao and B. Chu, Polymer, 2002, 43, 4403–4412 CrossRef CAS.
- K. Kanawung, K. Panitchanapan, S. O. Puangmalee, W. Utok, N. Kreua-Ongarjnukool, R. Rangkupan, C. Meechaisue and P. Supaphol, Polym. J., 2007, 39, 369–378 CrossRef CAS.
- Z. M. Huang, C. L. He, A. Z. Yang, Y. Z. Zhang, X. J. Hang, J. L. Yin and Q. S. Wu, J. Biomed. Mater. Res., Part A, 2006, 77A, 169–179 CrossRef CAS.
- J. Zeng, L. X. Yang, Q. Z. Liang, X. F. Zhang, H. L. Guan, X. L. Xu, X. S. Chen and X. B. Jing, J. Controlled Release, 2005, 105, 43–51 CrossRef CAS.
- W. G. Cui, X. H. Li, X. L. Zhu, G. Yu, S. B. Zhou and J. Weng, Biomacromolecules, 2006, 7, 1623–1629 CrossRef CAS.
- S. Maretschek, A. Greiner and T. Kissel, J. Controlled Release, 2008, 127, 180–187 CrossRef CAS.
- C. H. Dong, X. Y. Yuan, M. Y. He and K. D. Yao, J. Biomater. Sci., Polym. Ed., 2006, 17, 631–643 CrossRef CAS.
- P. Taepaiboon, U. Rungsardthong and P. Supaphol, Nanotechnology, 2006, 17, 2317–2329 CrossRef CAS.
- D. Z. Yang, Y. N. Li and J. Nie, Carbohydr. Polym., 2007, 69, 538–543 CrossRef CAS.
- J. Zeng, A. Aigner, F. Czubayko, T. Kissel, J. H. Wendorff and A. Greiner, Biomacromolecules, 2005, 6, 1484–1488 CrossRef CAS.
- O. Suwantong, P. Opanasopit, U. Ruktanonchal and P. Supaphol, Polymer, 2007, 48, 7546–7557 CrossRef CAS.
- M. Wang, L. Wang and Y. Huang, J. Appl. Polym. Sci., 2007, 106, 2177–2184 CrossRef CAS.
- O. Suwantong, U. Ruktanonchai and P. Supaphol, Polymer, 2008, 49, 4239–4247 CrossRef CAS.
- Y. K. Luu, K. Kim, B. S. Hsiao, B. Chu and M. Hadjiargyrou, J. Controlled Release, 2003, 89, 341–353 CrossRef CAS.
- K. Kim, Y. K. Luu, C. Chang, D. F. Fang, B. S. Hsiao, B. Chu and M. Hadjiargyrou, J. Controlled Release, 2004, 98, 47–56 CrossRef CAS.
- E. Luong-Van, L. Grondahl, K. N. Chua, K. W. Leong, V. Nurcombe and S. M. Cool, Biomaterials, 2006, 27, 2042–2050 CrossRef CAS.
- J. W. Xie and C. H. Wang, Pharm. Res., 2006, 23, 1817–1826 CrossRef CAS.
- X. R. Li and X. Y. Yuan, Prog. Chem., 2007, 19, 973–981 CAS.
- O. Veiseh, J. W. Gunn and M. Q. Zhang, Adv. Drug Delivery Rev., 2010, 62, 284–304 CrossRef CAS.
- F. H. Chen, L. M. Zhang, Q. T. Chen, Y. Zhang and Z. J. Zhang, Chem. Commun., 2010, 46, 8633–8635 RSC.
- P. Pouponneau, J. C. Leroux, G. Soulez, L. Gaboury and S. Martel, Biomaterials, 2011, 32, 3481–3486 CrossRef CAS.
- K. J. Widder, R. M. Morris, G. Poore, D. P. Howard and A. E. Senyei, Proc. Natl. Acad. Sci. U. S. A., 1981, 78, 579 CrossRef CAS.
- K. J. Widder, R. M. Morris, G. A. Poore, D. P. Howard and A. E. Senyei, Eur. J. Cancer Clin. Oncol., 1983, 19, 135–139 CrossRef CAS.
- R. Fernandez-Pacheco, C. Marquina, J. G. Valdivia, M. Gutierrez, M. S. Romero, R. Cornudella, A. Laborda, A. Viloria, T. Higuera, A. Garcia, J. A. G. de Jalon and M. R. Ibarra, J. Magn. Magn. Mater., 2007, 311, 318–322 CrossRef CAS.
- S. Brule, M. Levy, C. Wilhelm, D. Letourneur, F. Gazeau, C. Menager and C. Le Visage, Adv. Mater., 2011, 23, 787–790 CrossRef CAS.
- S. T. Tan, J. H. Wendorff, C. Pietzonka, Z. H. Jia and G. Q. Wang, ChemPhysChem, 2005, 6, 1461–1465 CrossRef CAS.
- G. Weiss, A. Knoch, A. Laicher, F. Stanislaus and R. Daniels, Int. J. Pharm., 1995, 124, 97–105 CrossRef CAS.
- I. H. Kim, J. H. Park, I. W. Cheong and J. H. Kim, J. Controlled Release, 2003, 89, 225–233 CrossRef CAS.
- Y. I. Jeong, T. Ohno, Z. P. Hu, Y. Yoshikawa, N. Shibata, S. Nagata and K. Takada, J. Controlled Release, 2001, 71, 175–182 CrossRef CAS.
- E. Toorisaka, M. Hashida, N. Kamiya, H. Ono, Y. Kokazu and M. Goto, J. Controlled Release, 2005, 107, 91–96 CrossRef CAS.
- S. Tungprapa, I. Jangchud and P. Supaphol, Polymer, 2007, 48, 5030–5041 CrossRef CAS.
- H. Q. Liu and Y. L. Hsieh, J. Polym. Sci., Part B: Polym. Phys., 2002, 40, 2119–2129 CrossRef CAS.
- C. Z. Chen, L. Wang and Y. Huang, Polymer, 2007, 48, 5202–5207 CrossRef CAS.
- X. H. Wu, L. G. Wang, H. Yu and Y. Huang, J. Appl. Polym. Sci., 2005, 97, 1292–1297 CrossRef CAS.
- R. Mincheva, O. Stoilova, H. Penchev, T. Ruskov, I. Spirov, N. Manolova and I. Rashkov, Eur. Polym. J., 2008, 44, 615–627 CrossRef CAS.
- R. M. Cornell and U. Schwertmann, The Iron Oxides: Structure, Properties, Reactions, Occurrences and Uses, 2nd edn, Wiley-VCH, 2003 Search PubMed.
- Y. Long, Z. Chen, J. L. Duvail, Z. Zhang and M. Wan, Phys. B, 2005, 370, 121–130 CrossRef CAS.
- V. Zavisova, M. Koneracka, O. Strbak, N. Tomasovicova, P. Kopcansky, M. Timko and I. Vavra, J. Magn. Magn. Mater., 2007, 311, 379–382 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2ra00484d |
| This journal is © The Royal Society of Chemistry 2012 |