New chiral coordination polymers constructed from well elaborated achiral and chiral ligands†‡
Yun-Wu
Li
,
Ying
Tao
,
Li-Fu
Wang
,
Tong-Liang
Hu
and
Xian-He
Bu
*
Department of Chemistry, and TKL of Metal- and Molecule-Based Material Chemistry, Nankai University, Tianjin, 300071, P. R. China. E-mail: buxh@nankai.edu.cn; Fax: +86-22-23502458
First published on 5th March 2012
Abstract
The elaboration of structurally related tetrazolate-based ligands from achiral to chiral led to the formation of two new chiral coordination polymers (CCPs). Compound 1 contains an infinite 1D zigzag Hg chain and a 1D helical water chain, its chirality is induced by the symmetry breaking of an achiral ligand while compound 2 is a 3D framework with a new 3-connected structural topology constructed from a homochiral ligand. The chiral natures of the two CCPs were both confirmed by circular dichroism (CD) spectra. The luminescent properties of compounds 1 and 2 were also investigated. Their strong luminescence suggest that both compounds may be excellent candidates as potential photoactive materials.
Introduction
Chirality plays important roles in chemical and biological processes. It is one of the most compelling challenges to design and synthesize crystalline materials possessing chiral features and properties, as systematically discussed in several comprehensive reviews.1 Chiral coordination polymers (hereafter abbreviated as CCPs) with accessible chiral functionalities, have accordingly arrived at the forefront of scientific research and pharmaceutical industries.2 Up to now, current research has focused on two effective strategies to construct CCPs. One is directly introducing chiral sources including chiral organic ligands,3 metal-organic moieties,4 or even cluster-based building blocks to the skeletons of CCPs via chemical modification,5 or chiral organic and metal-organic templates into their porous networks as structure-directing reagents,6 in order to facilitate chirality transfer to the whole framework. The other strategy is to build CCPs from achiral precursors through spontaneous resolution without any chiral auxiliaries.7–10 The second process often leads to bond length alteration or structure distortion, so-called symmetry breaking.9 Due to the achiral nature of the precursors, the target bulk sample tends to be a conglomerate (an equal racemic mixture of chiral crystals), although the single crystals are chiral. As reported in previous literature, various long chain-like N- or O-donor achiral ligands with diverse flexibility are good candidates for the assembly of CCPs, mainly because of their propensity to form versatile conformation and coordination modes to provide more opportunities for the formation of chiral frameworks.10 In comparison, using achiral rigid ligands to construct CCPs is still a challenging project due to their fixed and monotonic conformation.11 Aiming at further exploring the possible ways of synthesizing CCPs, our group have devoted much effort to the second approach using achiral rigid ligands and have obtained several chiral complexes.8,12b Typical of such complexes is a robust three-dimensional zeolite-like chiral metal-organic framework (MOF) constructed from ZnII and an achiral ditopic tetrazolate-based ligand, 2,3-di-1H-tetrazol-5-ylpyrazine (H2dtp). The chirality of the complex is due to the screw coordination arrangement of the achiral H2dtp ligands around the metal ZnII centers,8b which is similar to 1,1′-binaphthyl derivative complexes.1b,1c,3a,16 This heuristic work and others prompted us to further explore H2dtp-based ligands, and elaborate a family of derivatives from achiral to chiral, attempting to obtain more significant results. We thus deliberately designed four achiral ligands and one chiral ligand, namely, H2dtp, H2edtp, H2bdtp, H2pdtp (2,3-bis(pyridine-2-yl)-5,6-di-1H-tetrazol-5-ylpyrazine) and H2cdtp (2,3-camphoryl-5,6-di-1H-tetrazol-5-ylpyrazine) (Scheme S1, ESI†), and subsequently obtained nine achiral coordination polymers (ACPs) and five chiral coordination polymers (CCPs).8b,12 In our previous research, we have reported three CCPs built from achiral ligands H2dtp and H2pdtp using the second strategy via symmetry breaking.8b,12b In this work, we also obtained a new CCP {[Hg(pdtp)Cl2](H2O)2}n (1) by this approach. Based on these successful examples, we recently attempted to design a chiral H2dtp-based ligand (H2cdtp) for fabrication of the desired CCPs in a controllable way. Following this strategy, a targeted CCP {[Zn3(cdtp)3(H2O)](H2O)}n (2) was isolated. Upon deliberate ligand elaboration from achiral to chiral we have successfully constructed a number of CCPs. Such an approach leads to a greater degree of efficient and targetable synthetic routes to devise and construct CCPs in a designed manner, as demonstrated here in the construction of two new CCPs 1 and 2. Herein the synthesis, crystal growth and structural characterization of these two CCPs is presented and discussed. Furthermore, the solid-state circular dichroic (CD) spectra and luminescent properties of the two compounds have also been investigated.Experimental section
Materials and methods
All chemicals were commercially purchased and used as received. The ligands H2pdtp and H2cdtp were synthesized by a modified literature method.12,13Elemental analyses (C, H, and N) were performed on a Perkin-Elmer 240C analyzer (Perkin-Elmer, USA). X-Ray powder diffraction (XRPD) patterns were recorded on a Rigaku D/Max-2500 diffractometer at 40 kV, 100 mA for a Cu-target tube and a graphite monochromator. Simulation of the XRPD spectra were carried out by single-crystal data and diffraction-crystal module of the Mercury (Hg) program available free of charge via the Internet at http://www.iucr.org. IR spectra were measured in the range of 400–4000 cm−1 on a Tensor 27 OPUS FT-IR spectrometer using KBr pellets (Bruker, German). Fluorescence spectra were recorded at room temperature on a Varian Cary Eclipse fluorescence spectrometer (Varian, USA). The solid-state circular dichroism (CD) spectra were recorded on a JASCO J-715 spectropolarimeter with KCl pellets.
Synthesis of complexes
X-Ray crystallography
Crystallographic data for 1 and 2 were collected on a Rigaku SCX-mini diffractometer at 293(2) K with Mo-Kα radiation (λ = 0.71073 Å). The program SAINT14 was used for integration of the diffraction profiles. The crystal data for 1 and 2 were solved by direct methods and refined by full-matrix least-square method on F2 using the SHELXL-97 crystallographic software package.15 Metal atoms in the complexes were located from the E-maps, and other non-H atoms were located in successive difference Fourier syntheses and refined with anisotropic thermal parameters on F2. The H atoms of the ligands were generated theoretically on the specific atoms and refined isotropically with fixed thermal factors. H-atoms of water molecules in 1 were added according to the difference Fourier maps. H-atoms of water molecules in 2 could not be modeled properly so they were directly included in the final molecular formula. For 2, the thermal restraint command ‘isor’ was used to restrain five non-H atoms with ADP problems (C42, N17, N18, O1 and O2) to make the displacement parameters more reasonable. This command leads to a relatively high restraint value (30). Further details of crystal data and structure refinement for 1 and 2 are summarized below. Selected bond lengths and angles of 1 and 2 are given in Table S1 (ESI†).Results and discussion
Description of the structure
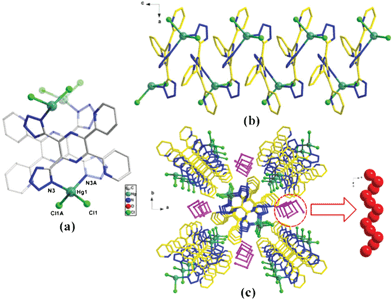 | ||
| Fig. 1 Ball-and-stick view of (a) the coordination environment of Hg2+ ions in 1; (b) the infinite 1D zigzag Hg chain; (c) the 1D helical water chain arranged in a interlaced fashion in parallel with the Hg chain. | ||
There is only one bidentate coordinated environment of H2pdtp ligands, which is a bridging mode with two N atoms from two tetrazole ring groups connecting two different Hg2+ ions. Based on the above coordination modes, all the Hg2+ centers are bridged by tetrazolate groups of H2pdtp ligands into a 1D zigzag Hg chain running along the c-direction (Fig. 1b). In such a structure, the intra-chain Hg⋯Hg distance is 8.721 Å. Furthermore, the pyridine rings and the pyrazine rings of the H2pdtp ligands only act as non-coordinating groups decorating both sides of a 1D zigzag Hg chain. More interestingly, the uncoordinated H2O molecules formed another 1D helical water chain by H⋯O–H hydrogen-bonding interaction, which is arranged in a interlaced fashion in parallel with the Hg chain along the c-direction (Fig. 1c). The chirality of the molecular units is induced by the coordination of the H2pdtp ligands in a twisted chiral conformation. As shown in Fig. S3 (ESI†), due to the asymmetric rotation about the C–C single bond, the two tetrazole rings are in fact staggered with respect to each other along the plane direction of the pyrazine ring. Consequently, this produces a huge torsion dihedral angle of ca. 66.265(11)° (Fig. S3a, ESI†).16 At the same time, the two pyridine rings also produce asymmetric rotation about the C–C single bond along the plane direction of the pyrazine ring and form a torsion dihedral angle of ca. 63.973(13)° (Fig. S3b, ESI†). When the two tetrazoles ligate to two metal ion of Hg2+, this locks the ligand in a twisted chiral conformation and produces atropoisomeric chirality. Obviously, the symmetry breaking of the ligands plays a crucial role in the initial generation of molecular chirality.
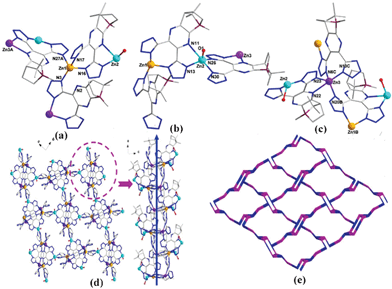 | ||
| Fig. 2 Ball-and-stick view of (a)–(c) the coordination environment of three independent Zn2+ centers; (d) the 3D homochiral framework; (e) the 3-connected topological network of CCP 2. | ||
Furthermore, there are two different coordination environments for H2cdtp ligands. One type of H2cdtp ligand (Lcdtp-1) chelates one Zn2+ center using two 4-position N atoms from two tetrazolate ring and bridges another Zn2+ center through one 1-position N atom of one tetrazolate ring, showing a chelating–bridging mode (mode I, Scheme S3, ESI†). The other type of H2cdtp ligand (Lcdtp-2) has a complicated coordination mode, that is, one 1-position N atom from one tetrazolate ring and one N atom from the pyrazine ring chelates one Zn2+ center, while another two 4-position N atoms from two tetrazolate rings chelate the second Zn2+ center, and at the same time, the 5-position N atom from one tetrazolate ring bridges the third Zn2+ center, generating a pentadentate chelating–chelating–bridging mode (mode II, Scheme S3, ESI†). As a result, these two kinds of Lcdtp ligands connect with different metal ions of Zn1 and Zn3 along the a-axis to afford an infinite double “S” type 1D chain (Fig. 1d). Based on this 1D chain as a 21 screw axis, all Zn3 ions run along this axis at the two side of it, resulting in a 3D homochiral framework (see Fig. 1d).
From the topological point of view, all the Zn2+ centers and Lcdtp ligands can be simplified as 3-connected nodes. Based on this simplification, the overall 3D networks of 2 can be rationalized as a new structural topology with Schläfli symbol {7·102}{72·10}.17,18
Solid-state circular dichroism (CD) spectrum
In order to confirm the chiral nature of CCPs 1 and 2, solid-state circular dichroism (CD) spectrum measurements using KCl plates were performed at room temperature.19 The CD spectrum for 1 shows that the bulk sample exhibits negative Cotton effects at λ = 211, 267, 350 nm, and positive Cotton effects at λ = 250, 285 nm, respectively, revealing its homochiral nature (Fig. 3a) while the CD spectrum for the bulk sample 2 shows negative Cotton effects at λ = 222, 264, 300 nm, and positive Cotton effects at λ = 242, 278, 346 nm, respectively, also indicating its homochiral nature (Fig. 3b). The observations are consistent with the structures obtained by single-crystal X-ray diffraction.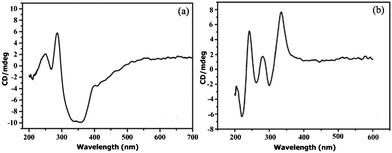 | ||
| Fig. 3 Solid CD spectra of CCPs (a) 1 and (b) 2. | ||
Luminescence properties
The luminescent properties of the two free ligands and CCPs were carried out in the solid state at room temperature. As shown in Fig. 4a and b, the free ligands H2pdtp and H2cdtp both exhibit intense emission bands at 490 nm (λex = 360 nm) and 426 nm (λex = 375 nm), respectively, which could be attributed to intraligand charge transfer (n–π* or π–π*).12,20,21 CCPs 1 and 2 exhibit strong fluorescent emission with maxima at 476 and 406 nm upon excitation at 389 and 342 nm, respectively. Compared with emissions of the two free ligands, CCPs 1 and 2 display similar slight blue shifts of the emission peaks and increased/reduced luminescent densities. From a theoretical viewpoint, the slight blue shifts of 1 and 2 can be attributed to the intraligand transitions (n–π* or π–π*) of the metal-modified ligands, since similar emissions can be observed for the two ligands. As reported previously, since the heteroatoms of the heterocyclic aromatic ligands can decrease their π and π* orbital energies, and the HOMO and LUMO orbitals as well as orbitals with similar energies in the compounds may thus reduce the contributions of the d10 metal atoms, while at the same time, the Hg(II) and Zn(II) ions are difficult to oxidize or to reduce due to their d10 configuration, so that ligand–metal charge-transfer transitions (LMCT) or metal–ligand charge transfer transitions (MLCT) can be excluded from consideration.20,21 The enhancement of luminescence may be attributed to ligand ligation to the electronically inactive metal centers, which effectively increases the rigidity and asymmetry of the ligands and reduces the loss of energy by radiationless decay.20 This observation suggests that 1 and 2 may be excellent candidates for potential photoactive materials.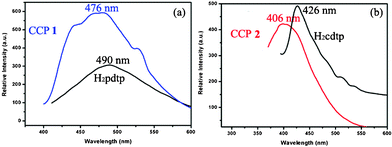 | ||
| Fig. 4 Solid-state photoluminescence spectra of (a) ligand H2pdtp and CCP 1; and (b) ligand H2cdtp and CCP 2 at room temperature. | ||
Conclusions
In conclusion, by ligands elaboration from achiral to chiral, we have successfully realized/achieved the design and isolation of CCPs from two different chiral construction strategies. The achiral and chiral ligands are both structurally related bis(tetrazolate) derivatives bearing different substituting groups. The chirality of compound 1 is induced by the symmetry breaking of an achiral ligand locked in a chiral conformation upon coordination, while 2 is constructed from a homochiral ligand. The present work provides a new paradigm for further design and synthesis of new CCPs by carefully elaborating ligands in an anticipated manner. Further investigations are underway, including the study of their other properties and an extension of the strategy into other systems.Acknowledgements
This work was financially supported by the 973 Program of China (2012CB821700), the NNSF of China (21031002), and the Natural Science Fund of Tianjin, China (10JCZDJC22100).References
- (a) M. E. Davis, Nature, 2002, 417, 813 CrossRef CAS; (b) B. Kesanli and W. B. Lin, Coord. Chem. Rev., 2003, 246, 305 CrossRef CAS; (c) Y. Liu, W. M. Xuan and Y. Cui, Adv. Mater., 2010, 22, 4112 CrossRef CAS.
- (a) M. Yoon, R. Srirambalaji and K. Kim, Chem. Rev., 2012, 112, 1196 CrossRef CAS; (b) K. Biradha, C. Seward and M. J. Zaworotko, Angew. Chem., Int. Ed., 1999, 38, 492 CrossRef CAS.
- (a) Y. Cui, S. J. Lee and W. B. Lin, J. Am. Chem. Soc., 2003, 125, 6014 CrossRef CAS; (b) R. G. Xiong, X. Z. You, B. F. Abrahams, Z. L. Xue and C. M. Che, Angew. Chem., Int. Ed., 2001, 40, 4422 CrossRef CAS.
- (a) D. B. Dang, P. Y. Wu, C. He, Z. Xie and C. Y. Duan, J. Am. Chem. Soc., 2010, 132, 14321 CrossRef CAS; (b) H. Y. An, E. B. Wang, D. R. Xiao, Y. G. Li, Z. M. Su and L. Xu, Angew. Chem., Int. Ed., 2006, 45, 904 CrossRef CAS.
- J. S. Seo, D. Whang, H. Lee, S. I. Jun, J. Oh, Y. J. Jeon and K. Kim, Nature, 2000, 404, 982 CrossRef CAS.
- (a) J. Zhang, R. Liu, P. Y. Feng and X. H. Bu, Angew. Chem., 2007, 119, 8540 CrossRef; (b) Z. Lin, A. M. Z. Slawin and R. E. Morris, J. Am. Chem. Soc., 2007, 129, 4880 CrossRef CAS; (c) Y. D. Han, Y. Li, J. H. Yu and R. R. Xu, Angew. Chem., Int. Ed., 2011, 50, 3003 CrossRef CAS.
- (a) E. Q. Gao, Y. F. Yue, S. Q. Bai, Z. He and C. H. Yan, J. Am. Chem. Soc., 2004, 126, 1419 CrossRef CAS; (b) G. Tian, G. S. Zhu, X. Y. Yang, Q. R. Fang, M. Xue, J. Y. Sun, Y. Wei and S. L. Qiu, Chem. Commun., 2005, 1396 RSC.
- (a) J. R. Li, Q. Yu, Y. Tao, X. H. Bu, J. Ribas and S. R. Batten, Chem. Commun., 2007, 2290 RSC; (b) J. R. Li, Y. Tao, Q. Yu, X. H. Bu, H. Sakamoto and S. Kitagawa, Chem.–Eur. J., 2008, 14, 2771 CrossRef CAS; (c) X. L. Tong, T. L. Hu, J. P. Zhao, Y. K. Wang, H. Zhang and X. H. Bu, Chem. Commun., 2010, 46, 8543 RSC.
- S. T. Wu, Y. R. Wu, Q. Q. Kang, H. Zhang, L. S. Long, Z. P. Zheng, R. B. Huang and L. S. Zheng, Angew. Chem., Int. Ed., 2007, 46, 8475 CrossRef CAS.
- (a) Y. Q. Lan, L. S. Li, X. L. Wang, K. Z. Shao, D. Y. Du, Z. M. Su and E. B. Wang, Chem.–Eur. J., 2008, 14, 9999 CrossRef CAS; (b) D. F. Sun, D. J. Collins, Y. X. Ke, J. L. Zuo and H. C. Zhou, Chem.–Eur. J., 2006, 12, 3768 CrossRef CAS.
- (a) Z. Su, M. S. Chen, J. Fan, M. Chen, S. S. Chen, L. Luo and W. Y. Sun, CrystEngComm, 2010, 12, 2040 RSC; (b) L. Hou, J. P. Zhang, X. M. Chen and S. W. Ng, Chem. Commun., 2008, 4019 RSC.
- (a) Y. Tao, J. R. Li, Z. Chang and X. H. Bu, Cryst. Growth Des., 2010, 10, 564 CrossRef CAS; (b) T. L. Hu, Y. Tao, Z. Chang and X. H. Bu, Inorg. Chem., 2011, 50, 10994 CrossRef CAS.
- (a) F. D. Popp, J. Heterocycl. Chem., 1974, 11, 79 CrossRef CAS; (b) W. Ried and S. Aboul-Fetouh, Tetrahedron, 1988, 44, 3399 CrossRef CAS; (c) Z. P. Demko and K. B. Sharpless, Org. Lett., 2001, 3, 4091 CrossRef CAS; (d) Z. P. Demko and K. B. Sharpless, J. Org. Chem., 2001, 66, 7945 CrossRef CAS.
- Bruker AXS, SAINT Software Reference Manual, Madison, WI, 1998 Search PubMed.
- (a) G. M. Sheldrick, SHELXL97, Program for Crystal Structure Refinement, University of Göttingen, Göttingen, Germany, 1997 Search PubMed; (b) G. M. Sheldrick, SHELXS97, Program for Crystal Structure Solution, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
- L. Pu, Chem. Rev., 1998, 98, 2405 CrossRef CAS.
- S. R. Batten and K. S. Murray, Coord. Chem. Rev., 2003, 246, 103 CrossRef CAS.
- V. A. Blatov, TOPOS, A multipurpose Crystallochemical Analysis with the Program Package, Samara State University, Russia, 2004 Search PubMed.
- R. Feng, F. L. Jiang, L. Chen, C. F. Yan, M. Y. Wu and M. C. Hong, Chem. Commun., 2009, 5296 RSC.
- (a) K. Balasubramanian, Relativistic Effects in Chemistry. Part A: Theory and Techniques; Part B: Applications, Wiley, New York, 1997 Search PubMed; (b) B. Valeur, Molecular Fluorescence: Principles and Applications, Wiley-VCH, Weinheim, 2002 Search PubMed; (c) S. L. Zheng, J. P. Zhang, X. M. Chen, Z. L. Huang, Z. Y. Lin and W. T. Wong, Chem.–Eur. J., 2003, 9, 3888 CrossRef CAS; (d) S. L. Zheng, J. H. Yang, X. L. Yu, X. M. Chen and W.-T. Wong, Inorg. Chem., 2004, 43, 830 CrossRef CAS.
- V. W. W. Yam and K. K. W. Lo, Chem. Soc. Rev., 1999, 28, 323 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Scheme S1: Structures of H2pdtp and H2cdtp. Scheme S2: The route for ligand synthesis. Table S1: Selected bond lengths (Å) and angles (º) of CCPs 1 and 2. Fig. S1 and S2: XRD of CCPs 1 and 2. Fig. S3: Twisted conformations of H2pdtp ligands and their torsion dihedral angles. Scheme S3: Two coordination modes of H2cdtp ligands. CCDC reference numbers 857789 and 857790. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2ra00046f |
| ‡ . |
| This journal is © The Royal Society of Chemistry 2012 |