Additional bioactive guanidine alkaloids from the Mediterranean sponge Crambe crambe†
Stéphanie
Bondu
a,
Grégory
Genta-Jouve
a,
Marta
Leirόs
b,
Carmen
Vale
b,
Jean-Marie
Guigonis
c,
Luis M.
Botana
b and
Olivier P.
Thomas
*a
aNice Institute of Chemistry UMR 7272 CNRS, University of Nice Sophia-Antipolis, Faculté des Sciences, Parc Valrose 06108 Nice, France. E-mail: olivier.thomas@unice.fr; Fax: +33 49207 6151; Tel: +33 49207 6134
bDepartment of Pharmacology, Facultad Veterinaria, Universidad de Santiago de Compostela, 27002, Lugo, Spain. E-mail: Luis.Botana@usc.es; Fax: +34 982822233
cPlate forme Bernard ROSSI Protéome et Métabolome, University of Nice Sophia-Antipolis, Faculté de Médecine, 06107, Nice, France. E-mail: jean-marie.guigonis@unice.fr; Tel: +33 493377708
First published on 9th January 2012
Abstract
The full chemical reinvestigation of the Mediterranean marine sponge Crambe crambe led to the isolation and structural characterization of 11 crambescin derivatives, including 8 new compounds, together with the known crambescidin 816. HRMS/MS studies allowed the complete assignment of the alkyl chain lengths of these guanidine alkaloids while the absolute configurations of all compounds were inferred from the comparison between experimental and theoretical circular dichroism spectra. Crambescidin 816 was proven to be more cytotoxic against neuronal cell lines than crambescin C1.
Introduction
Marine sponges of the order Poecilosclerida are known to produce a large array of structurally diverse bioactive polycyclic guanidine alkaloids.1 This family of sponge marine natural products is today considered as a chemotaxonomic marker of the Crambeidae family.2Crambe crambe (Schmidt, 1862) is a red encrusting marine sponge, widely distributed in the Western Mediterranean Sea but also in the Macaronesian archipelagos. The first chemical studies on this sponge date back to the early 90s where crambines A, B, C1 and C2 were first isolated by the group of Braekman.3 At the same time, the polycyclic crambescidins 800, 816, 830, 844, isocrambescidin 800 and crambidine, which are oxidized analogues of the previously described ptilomycalin A,4 were reported by the groups of Braekman and Rinehart in the same sponge.5 We decided to reinvestigate the entire secondary metabolome of this sponge because several structural revisions have been added later for the crambines, mainly on the basis of chemical synthetic studies.6 Our interest in these compounds also arose from the vast array of biological activities mainly associated with the polycyclic crambescidin derivatives.7As proposed earlier by the group of Rinehart,6b we will change the name crambines to crambescins because crambin has been previously ascribed to a peptide.8 While crambescidins were patented due to their highly interesting cytotoxic and antiviral activities,9 there are only limited data on the pharmaceutical potential of crambescins.6b We describe herein the isolation and structural elucidation of 11 crambescin derivatives (1–11) together with crambescidin 816 (12) from the sponge C. crambe, including for the first time the determination of the absolute configurations deduced from comparisons between experimental and theoretical electronic circular dichroism (ECD) spectra. Four of these compounds have been reported earlier but usually in a mixture.3a A further biological evaluation of these alkaloids was performed against cortical neurons.
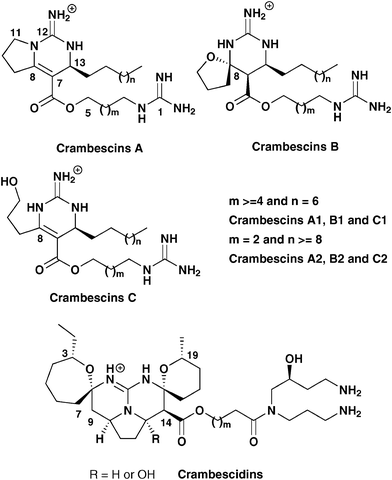
According to the proposition of the group of Rinehart, we will classify the crambescins into three groups according to the structural features of the left propyl side chain at C-8 of the guanidinium core.6b The structure of all crambescins A includes a pyrrolidine ring, while crambescins B are characterized by a spiroaminal, and crambescins C by a linear 3-hydroxypropyl side-chain at C-8 (Fig. 1). Controversies on the structure of crambescins came from the number of methylenes present in the upper alkyl side-chain (n+2) and in the lower guanidinoalkyl side-chain (m+2) but also from the relative configurations of the substituents around the cyclic core. Using chemical synthesis the group of Snider inverted the relative configuration of crambescin B at C-8 initially proposed by the group of Braekman.3b,6c While crambescin B (first named crambine B) was first described with n = 8 and m = 3, the groups of Rinehart and Snider then demonstrated independently using mass spectrometry and chemical synthesis respectively that the chain lengths should be revised as n = 6 and m = 5.6a,6b In the same manner, the structure of crambescin C1 (previously named crambine C1) was revised and the initial values of n = 8 and m = 3 were here also replaced by n = 6 and m = 5.6a,6b
 | ||
| Fig. 1 Crambescins and crambescidin skeletons. | ||
The crambescins C2 (previously named crambine C2) will correspond to a shorter guanidino alkyl side chain with m = 2. In the same manner, we propose to use crambescin A2 and crambescin B2 for the analogues with m = 2 because their structures strongly differ from the usual crambescins with m higher than four. Thus, most batzelladines, a large family of crambescin ‘dimers’, all incorporate a crambescin A2 unit.10 The biological evaluations and full structural assignments (including the absolute configurations of these compounds) were hampered by the difficulties associated with the purification processes, as crambescins were usually isolated as a complex mixture of homologues with added methylene units. We then decided to purify all compounds to give additional data on the pure crambescins.
Results and discussion
After an organic extraction of the lyophilized material with CH2Cl2/MeOH (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), the extract was first submitted to fractionation by reversed phase Vacuum Liquid Chromatography. The methanolic fraction was further purified by two successive HPLC on semi-preparative C18 and analytical C3Phenyl reversed phase to yield 11 pure crambescin derivatives and crambescidin 816. The 1H NMR and HRMS spectra were used to confirm the high purity of each isolated compound (>95%).
:1), the extract was first submitted to fractionation by reversed phase Vacuum Liquid Chromatography. The methanolic fraction was further purified by two successive HPLC on semi-preparative C18 and analytical C3Phenyl reversed phase to yield 11 pure crambescin derivatives and crambescidin 816. The 1H NMR and HRMS spectra were used to confirm the high purity of each isolated compound (>95%).
Each compound was then assigned to a crambescin family on the basis of characteristic signals on their 1H NMR spectra. The non-equivalent methylenes resonating at δH 2.99 and 3.33 (H-9), 2.11 and 2.23 (H-10) and 3.67 and 3.82 (H-11) ppm were reminiscent of a crambescin A skeleton for compounds 1–4.
Compounds 6–8 were members of the crambescin B group due to the absence of the signal at 4.40 (dd, H-13) ppm and the presence of a characteristic signal at 2.99 (d, H-7) ppm. Compounds 9–11 were part of the crambescin C family because of the characteristic signals of the hydroxypropyl side chain methylenes at 2.80 and 2.83 (H2-9), 1.81 (H2-10) and 3.61 (H2-11) ppm (Table 1). Compound 5 exhibited unusual signals in its 1H NMR spectrum which required deeper NMR analyses.
| Position | Crambescin | ||||
|---|---|---|---|---|---|
| A2 2 | A1 4 | 5 | B1 7 | C1 10 | |
| 2 | 3.23 | 3.17 | 3.16 | 3.16 | 3.17 |
| 3 | 1.68 | 1.60 | 1.61 | 1.59 | 1.60 |
| 3A | 1.42 | 1.41 | 1.38 | 1.38 | |
| 3B | 1.42 | 1.41 | 1.38 | 1.41 | |
| 3C | 1.42 | 1.41 | 1.38 | 1.43 | |
| 4 | 1.75 | 1.69 | 1.80 | 1.67 | 1.71 |
| 5 | 4.25 | 4.21 | 4.36 | 4.17 | 4.20 |
| 4.22 | 4.17 | 4.13 | 4.17 | ||
| 7 | 2.99 | ||||
| 9 | 3.33 | 3.31 | 3.57 | 2.12 | 2.83 |
| 2.99 | 2.95 | 2.07 | 2.80 | ||
| 10 | 2.23 | 2.23 | 2.39 | 2.10 | 1.81 |
| 2.11 | 2.10 | 2.05 | |||
| 11 | 3.82 | 2.82 | 4.26 | 4.02 | 3.61 |
| 3.67 | 3.66 | 3.93 | |||
| 13 | 4.40 | 4.39 | 3.86 | 4.42 | |
| 14 | 1.56 | 1.60 | 3.07 | 1.59 | 1.60 |
| 15 | 1.41 | 1.41 | 1.72 | 1.47 | 1.41 |
| 16 | 1.30 | 1.30 | 1.41 | 1.30 | 1.30 |
| 17-(15+n) | 1.25–1.35 | 1.25–1.35 | 1.25–1.35 | 1.25–1.35 | 1.25–1.35 |
| 16+n | 0.90 | 0.90 | 0.90 | 0.90 | 0.90 |
Comparison of the 1H NMR spectra for compounds 1–4 revealed clear differences in the intensity of the broad signals around δH 1.30 and 1.40 ppm, assigned to the aliphatic side chains protons of both the alkyl side chain at C-13 (n+2) and the guanidinoalkyl chain at C-6 (m+2). These data were inferred to differences in the lengths of both alkyl side chains. This explanation may also account for the differences in the chemical shifts at δH 4.25 and 4.22 (H2-5) ppm for 2, while these value were δH 4.21 and 4.17 ppm for compound 4.
However, it was not possible to give clear data on the chain lengths by NMR spectroscopy, due to signal overlapping and imprecision in the signal integrations. As previously reported by the group of Rinehart, mass spectrometry proved to be the method of choice to assess the side-chain lengths.6b To be more confident with our results, we performed (+)-HRESIMS/MS analyses of each pure compound. All four crambescidin A analogues differ by one or two methylene units (differences of 14 or 28 Da) (Table 2). Fragmentation analyses on compound 1 at m/z 449.36017 [M+H]+ yielded two major fragments at m/z 336.26456 and 241.16585 corresponding to the molecular formulae C19H34N3O2 and C11H21N4O2, respectively and were consistent with the fragmentation scheme shown in Fig. 2.
 | ||
| Fig. 2 Fragmentation pattern of crambescins. | ||
| Compound | Crambescin | m/z [M+H]+ | m | n |
|---|---|---|---|---|
| 1 | A2 | 449.36017 | 2 | 8 |
| 2 | A2 | 463.37552 | 2 | 9 |
| 3 | A2 | 477.39197 | 2 | 10 |
| 4 | A1 | 463.37531 | 5 | 6 |
| 5 | (Didehydro) A1 | 461.35976 | 5 | 6 |
| 6 | B1 | 467.37051 | 4 | 6 |
| 7 | B1 | 481.38629 | 5 | 6 |
| 8 | B1 | 495.40207 | 6 | 6 |
| 9 | C1 | 467.37027 | 4 | 6 |
| 10 | C1 | 481.38635 | 5 | 6 |
| 11 | C1 | 495.40182 | 6 | 6 |
The fragment peak at m/z 336.26456 [M–C5H11N3]+ indicated the loss of a guanidine alkyl chain with m = 2 (Fig. 2). The fragment at m/z 241.16285 [M–C13H24N2]+ was consistent with an upper alkyl chain length of n = 8. The occurrence of the analogous fragments at m/z 350.28018 and m/z 364.39654 in the (+)-HRESIMS/MS spectra of compounds 2 and 3, respectively, implied that both compounds had the same guanidinoalkyl chain length (m = 2). The additional methylene units were then located on the upper alkyl side chain which was confirmed by the presence of the same fragments for all three compounds at m/z 241. Consequently, compounds 1–3 are all members of the crambescin A2 family. The (+)-HRESIMS/MS mass fragmentation pattern of the molecular ion corresponding to compound 4 at m/z 463.37531 was completely different from those observed for compounds 1–3. The presence of an intense fragment ion at m/z 283.21280 was only consistent with a longer guanidinoalkyl chain (m = 5). The length of the upper alkyl chain was then deduced to be n = 6. According to this study, compound 4 is the first isolated member of the crambescin A1 family and was proven to be an isomer of compound 2, a member of the crambescin A2 family, which underlined the importance of the HRMS/MS study. In the original paper of the group of Braekman only crambescins A2 (crambine A) were described in a mixture.3b
The molecular formula of compound 5 was determined to be C25H44N6O2 by HRESIMS (Δ −0.2 ppm by internal calibration), indicating seven degrees of unsaturation, one more than compounds 2 and 4. The characteristic signals of crambescin A were absent in the 1H NMR spectrum of 5 and the signals of the methylenes at C-5 (δH, 4.36, t), C-9 (δH, 3.57, t) and C-11 (δH, 4.26, t) became equivalent and deshielded comparing to the data obtained for 2 and 4. Furthermore, the upper alkyl chain was connected to a quaternary carbon at C-13 (δC 180 ppm) due to the H-14/C-13 HMBC. In consequence the additional unsaturation was located at C-13 which induced an aromatized planar pyrimidine cycle. The length of the upper alkyl chain was estimated as being n = 6 from the integration of signals at δH 1.31 ppm (10H) assigned to H2-17 to H2-21 (Table 1). The length of the guanidinoalkyl chain was assessed by the integration of the signal at 1.41 ppm (8H) which corresponds to H2-3A, H2-3B and H2-3C and H2-16, thus suggesting m = 5. The lengths of the alkyl chains at C-13 and C-6 were further confirmed by (+)-HRESIMS/MS fragmentation pattern of the molecular peak at m/z 231.18352 [M+2H]2+ which gave two majors fragments at m/z 306.21762 and m/z 156.14943, in accordance with the molecular formulae C17H26N2O3 and C8H18N3, respectively.
The fragment peak at m/z 306.21762 [M–C8H17N3]+ corresponds to the loss of a molecular ion of 155 Da, a 7–guanidino-1-heptene. This result is consistent with m = 5 and n = 6 for both alkyl chains and we could conclude that 5 is an oxidized form of compound 4, belonging to the crambescin A1 family. 5 is then a didehydrocrambescin A1 derivative. A dehydrocrambine A has already been reported from an unknown sponge of the genus Monanchora in Palau.11 The described dehydrocrambine A is an oxidized equivalent of crambescin A2 (named crambine A) they also isolated from that sponge. Aromatization into a pyrimidine is not rare in this family of compounds.12 Just like for the crambescin A2 derivative found in Monanchora sp., compound 4 was also isolated together with its oxidized form 5 which reinforced the idea that an oxidation could partially take place during the purification process, especially in the acidic conditions required for the HPLC purification.
The 1H NMR spectra of compounds 6–8 first indicated that they belong to the crambescin B family, especially for the occurrence of the small doublet at 2.99 (J = 4 Hz, H-7) ppm. The chain lengths were assigned on the basis of HRMS/MS data. The fragmentation pattern differed from crambescins A and both guanidines remain intact during the fragmentation whereas the presence of the pyrrolidine in crambescins A induced a fragmentation of the cyclic guanidine (Fig. 2).
Fragments were then observed at m/z 270.18124, 284.19693 and 298.21896 for compounds 6, 7 and 8 respectively, which indicated a change in the chain lengths for the guanidino alkyl chain with m = 4, 5 and 6 respectively and a common upper alkyl chain with n = 6 for all crambescin B1 derivatives. The relative configurations for compounds 6–8 were assigned by interpretation of the coupling constant between H-7 and H-13 which was reported as 4–4.2 Hz in the cis isomers and 11.5–12 Hz in the trans isomer.6a All isolated crambescins B1 had JH7–H13 close to 4.2 Hz revealing a cis configuration between H-7 and H-13. Comparison of the chemical shifts for the proton of the spiro cycle with the previously described crambescins B1 (crambine B) led us to propose the same relative configurations at C-8 than those proposed by Snider.6a
The 1H NMR spectra of compounds 9–11 indicated that they belong to the crambescin C family, especially for the occurrence of the triplet at 3.61 (H2-11) ppm. The same fragmentation patterns as those obtained for crambescins B were observed which allowed us to assign the chain lengths of all three crambescin C derivatives. Here also, differences between the three compounds 9–11 were identified in the length of the lower guanidine alkyl chain with m values of 4, 5 and 6 while the upper alkyl chain at C-13 remain unchanged with the same n = 6. In consequence these compounds are all crambescin C1 analogues. At this point, it is interesting to note that most of the crambescin A derivatives are from the crambescin A2 family while crambescins B1 and C1 are only found in the sponge C. crambe. In contrast to the second study of the group of Braekman,3a we did not manage to identify a sufficient quantity of crambescin C2. The presence of short guanidino alkyl chains with m = 2 in crambescin A2 is remarkable and much more if we observe that all batzelladine derivatives incorporate only this monomer.12
This observation raises the question of the biosynthetic pathways leading to these compounds, as studied first by the group of Snider,6a and then by the group of Quinn for the mirabilins, which are tricyclic monomers, analogues of the mono or bicyclic crambescins.13 The addition of a guanidine to a unique polyketide chain is questionable as no enzyme has been described to perform such transformation. We rather propose the involvement of an arginine or a homologue for the guanidines. Indeed, we previously shown that the 2-aminoimidazole side chain of oroidin originates from homoarginine,14 and, recently, the group of Molinski proposed the same origin for a guanidinated derivative of the polyacetylene family.15 We will address this issue for crambescins by in vivo biosynthetic experiments.
With all pure crambescins in hand, we were able to assign the absolute configurations of the three families A, B and C using ECD and comparison with theoretical values as already reported by us for other families of marine natural products.16 We anticipated strong Cotton effects due to the presence of chromophoric groups (n→π* transition of the enone) placed in the vicinity of asymmetric centers as for the asymmetric center at C-13 always substituted by the guanidine and an enone in the cases of crambescins A and C. Experimental ECD spectra were then compared to the calculated ECD spectra of both enantiomers. As illustrated on Fig. 3, there is a good overlapping between both experimental and theoretical ECD spectra for one enantiomer of the three different kinds of crambescins. All our results converge to suggest an absolute S configuration at C-13 which is in accordance with the only result obtained on the biosynthetic congener crambidine using asymmetric chemical synthesis.17 The only discrepancy we observed for these 11 compounds was for the ECD spectrum of one crambescin C1 (9) where the signs of the Cotton effects opposite to the one measured for 10 and 11. Nevertheless in this case the intensities of the bands were very low and the other results obtained for 10 and 11 seemed more reliable.
 | ||
| Fig. 3 Experimental and calculated CD spectra for crambescins A2 (1), B1 (7) and C1 (11). | ||
While crambescidins were largely evaluated for their biological properties, crambescins were much less studied because of the difficulties to obtain pure compounds. For the first time, we decided to test the activity of one representative of each family isolated in sufficient amount on cortical neurons. In the MTT assay, we observed that 24 h of exposure of the cortical neurons to different concentrations (0.001 to 1 μM) of crambescidin 816 (12) caused a dose-dependent increase on the cytotoxic effect and the almost complete cell death at 1 μM (86.3 ± 6.8%), whereas crambescin C1 (10) only lowered cellular viability by 22.6 ± 4.1% at 1 μM (Fig. 4). The effects after long term treatment were similar, with an IC50 value of 432 nM for 12, and a decrease in cell viability of 40.8 ± 12.6% for 10 at 1 μM. Crambescin C1 (10) and crambescidin 816 (12), tested at different concentrations (0.001 to 1 μM) caused reductions in the number of cortical living cell at 1 μM compared to the cells control. However no significant differences were observed for the several concentrations of 10. There is not much literature about crambescidin 816 (12) cytotoxicity. In a previous publication in IC2 murine mast cell lines the crambescidin acid presents an IC50 value of 25.0 μg ml−1,18 whereas our value in cortical neurons is 0.354 μg ml−1. A full cytotoxic evaluation of crambescidin analogues was also reported but crambescidin 816 (12) was not evaluated.19 This observation suggests that cell type is a crucial determinant for the potency of the compound. Therefore, the toxic or therapeutic effect of 12 must the carefully studied in each system, given the extremely high potency of the compound on the nervous model.
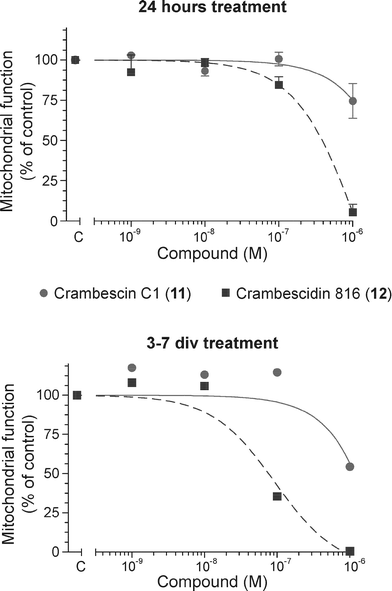 | ||
| Fig. 4 Effects of crambescin C1 (10) and crambescidin 816 (12) on cell viability in primary cultures of cortical neurons. | ||
Experimental
General experimental procedures
HPLC purifications were carried out on a Waters 600 system equipped with a Waters 717 plus autosampler, a Waters 996 photodiode array detector, and a Sedex 55 evaporative light-scattering detector (SEDERE, France). Detection wavelengths were set at 214, 254 and 280 nm. 1H and 13C NMR spectra were recorded on a 500 MHz Bruker Avance NMR spectrometer. Chemical shifts (δ) are recorded in ppm with CD3OD (δH 3.31 ppm and δC 49.00 ppm) as internal reference with multiplicity (s singlet, d doublet, t triplet, m multiplet, br broad). LCESIMS analyses were carried out on a Waters 2695 system equipped with a Waters 2487 dual λ absorbance detector and a Bruker Esquire 3000 Plus spectrometer (Ion Trap). High-resolution mass spectra ESIMS (HRESIMS/MS) were conducted on a LTQ Orbitrap mass spectrometer (Thermo Fisher Scientific). The MSI system of the LTQ-Orbitrap hybrid mass spectrometer was operated in the positive mode at a voltage of 4.5 kV, with no sheath or auxiliary gas and in maintaining the ion transfer tube at 275 °C. The compounds were then infused at a flow rate of 0.5 μL min−1. The Orbitrap mass analyzer was calibrated according to the manufacturer's directions using a mixture of caffeine, MRFA peptide, and Ultramark for positive ionization mode. Selected ion monitoring profile MS data (10 Da window and 40 scans) were acquired with resolving power setting of 100 K. The injection time was from 50 to 350 ms with a target of 100000 ions adjusted by automatic gain control. To achieve the highest possible mass accuracy, the lock mass function was enabled with the pollutant ion at an m/z of 391.28429 in the positive ionization mode used for real-time internal recalibration. MS data acquisition and processing were performed using Xcalibur software (version 2.0.7; Thermo Fisher Scientific). Spectral accuracy was calculated by MassWorks using sClips software (version 2.0; Cerno Bioscience). The mass tolerance for the sClips searches was 2 ppm. Optical rotations were measured on a Jasco P-1020 polarimeter. UV and CD spectra were measured using a JASCO J-810 spectropolarimeter.
Collection
The sponge specimens of Crambe crambe were collected in March 2011 by hand using SCUBA diving at Villefranche-Sur-Mer (France), at depths ranging from 10 to 20 m, and kept frozen until used.Bioassays
Extraction and isolation
The samples of Crambe crambe were freeze dried and then ground to obtain a dry powder (15 g) which was exhaustively extracted with a mixture of MeOH/CH2Cl2 (1:1, v/v) to yield 2.4 g of crude extract after concentration under reduced pressure. The crude extract was fractionated by Reverse Phase Vacuum Liquid Chromatography over RP-18 silica gel (200 g) with a step gradient of H2O (F1 fraction, 581 mg), MeOH/H2O 2/1 v/v (F2 fraction 286 mg), MeOH/H2O 3/1 v/v (F3 fraction, 284 mg), MeOH (F4 fraction, 767 mg) and DCM (F5 fraction, 450 mg) (500 mL each). The F2 (140 mg, 100 mg ml−1) and F3 (160 mg, 100 mg ml−1) fractions were then subjected to semi-preparative HPLC purification (Phenomenex Luna C6-Phenyl, 250 × 10 mm, 5 μm) eluted with a gradient of H2O/ACN/TFA : isocratic step from 0 min to 5 min (90:10:0.1) and then gradient steps from 5 min to 6 min (from 90:10:0.1 to 70:30:0.1) and from 6 to 36 min (from 70:30:0.1 to 55:45:0.1) (flow rate: 3.0 mL min−1, injection volume: 100 μL) to give 9 and 13 fractions respectively, named from F2P1 to F2P9 and F3P1 to F3P13. The fractions F2P2 (tr 21.5 min), F2P3 (tr 23.0 min), F2P4 (tr 26.0 min), F2P5 (tr 28.0 min) and F2P9 (tr 36.0 min) were further subjected to analytical HPLC purification (XSelect C6-Phenyl, 250 × 4.6 mm, 5 μm). Fractions were eluted with a gradient of H2O/ACN/TFA: isocratic step from 0 min to 5 min (90:10:0.1) and then gradient steps from 5 min to 6 min (from 90:10:0.1 to 68:32:0.1) and isocratic step from 6 to 30 min (68:32:0.1) (flow rate: 0.80 mL min−1, injection volume: 30 μL). This step yielded the major components of the F2P2, F2P3, F2P4 and F2P9 fractions and so to yield compound 9 (1.2 mg), 10 (11.1 mg), 11 (2.4 mg), 12 (10.2 mg), respectively, and, in the case of F2P5 fraction, to afford 5 (tr 24.5 min, 0.8 mg), 6 (tr 25.2 min, 0.9 mg), 4 (tr 26.5 min, 0.9 mg) and 7 (tr 28.2 min, 1.1 mg). Similar analytical HPLC analyses were performed on fractions F3PP7 (tr 29.0 min), F3P8 (tr 30.5 min), F3P9 (tr 32.0 min) and F3P12 (tr 37.0 min) and let to afford 1 (2.0 mg), 8 (1.1 mg), 2 (2.0 mg), 3 (1.3 mg), respectively. At the total, twelve guanidine alkaloids were purified. All of them were identified by a combination of spectroscopic methods (1D and 2D NMR, HRESIMS).
Norcrambescin A2 (1)
[α]20D +7.1 (c 0.2, MeOH); UV (MeOH) λmax (log ε) 288 nm (2.7); CD (MeOH, c 4.46 × 10−4 M) Δε (λmax nm) +0.66 (209) –2.22 (248) –0.99 (291); ESIMS m/z 449.3 [M+H]+; HRESIMS m/z 449.36017 [M+H]+ (Calc for C24H45O2N6, 449.35985, Δ −0.70 ppm). 1H NMR (500 MHz) and 13C NMR (125 MHz, CD3OD) (lH, J, 13C) C-1 (158.6), H2C-2 (3.23 t, 6.8; 42.1), H2C-3 (1.68 m; 26.6), H2C-4 (1.75 m; 27.1), H2C-5 (4.22 m, and 4.23 m; 65.1), C-6 (166.1), C-7 (103.2), C-8 (152.7), H2C-9 (2.99 ddd, 9.5/ 9.8/18.3 and 3.30; 31.9), H2C-10 (2.11 m and 2.23 m; 22.9), H2C-11 (3.67 ddd, 7.2/9.7/9.93 and 3.82 ddd, 2.8/ 9.1/9.4; 49.2), C-12 (153.0), HC-13 (4.40 dd, 7.1; 51.3), H2C-14 (1.56 m; 37.5), H2C-15 (1.41 m; 25.2), H2C-16 to H2C-21 (1.25–1.35 br s; 30.0–31.0), H2C-22 (1.25–1.35 br s; 33.1), H2C-23 (1.29 br s; 23.8), H3C-24 (0.90 t, 6.8; 14.3).Crambescin A2 (2)
[α]20D +12.1 (c 0.18, MeOH); UV (MeOH) λmax 287 nm (log ε 2.7); CD (MeOH, c 4.33 × 10−4 M) Δε (λmax nm) –1.86 (249) –0.81 (288); ESIMS m/z 463.3 [M+H]+; HRESIMS m/z 463.37552 [M+H]+ (Calc for C24H45O2N6, 463.37550, Δ −0.04 ppm).Homocrambescin A2 (3)
(3) [α]20D +35.9 (c 0.1, MeOH); UV (MeOH) λmax 288 nm (log ε 2.3); CD (MeOH, c 4.20 × 10−4 M) Δε (λmax nm) +0.58 (207) –1.93 (249) –0.85 (288); ESIMS m/z 477.0 [M+H]+; HRESIMS m/z 477.39197 [M+H]+ (Calc for C26H49O2N6, 477.39115, Δ –1.71 ppm).Crambescin A1 (4)
[α]20D +8.9 (c 0.06, MeOH); UV (MeOH) λmax (log ε) 289 nm (1.5); CD (MeOH, c 4.84 × 10−4 M) Δε (λmax nm) −0.10 (218) −0.06 (288) +0.08 (330); ESIMS m/z 463.2 [M+H]+; HRESIMS m/z 463.37531 [M+H]+ (Calc for C25H47O2N6, 463.37550, Δ –0.42 ppm). 1H NMR (500 MHz) and 13C NMR (125 MHz, CD3OD) (lH, J, 13C) C-1 (158.6), H2C-2 (3.17 t, 6.9; 42.5), H2C-3 (1.60 m; 29.8), H2C-3A (1.42 br s; 27.6), H2C-3B (1.42 br s; 29.9), H2C-3C (1.42 br s; 27.1), H2C-4 (1.69 m; 29.8), H2C-5 (4.17 m, 17.4 and 4.21 m, 17.4; 65.6), C-6 (166.9), C-7 (100.4), C-8 (155.1), H2C-9 (2.95 ddd, 9.1/ 9.5/18.9 and 3.31; 31.9), H2C-10 (2.10 m and 2.23 m; 22.9), H2C-11 (3.66 ddd, 7.5/9.5/10.3 and 3.82 ddd, 2.8/9.5/10.3; 49.5), C-12 (154.4), HC-13 (4.39 m; 51.4), H2C-14 (1.60 m; 37.6), H2C-15 (1.30 br m; 25.2), H2C-16 to H2C-19 (1.30 br m; 30.35, 30.44, 30.60, 30.64), H2C-20 (1.30 br m; 33.1), H2C-21 (1.30 br m; 23.8), H3C-22 (0.90 t, 7.4; 14.5).Didehydrocrambescin A1 (5)
ESIMS m/z 461.3 [M+H]+; HRESIMS m/z 461.35976 [M+H]+ (Calcd for C25H45N6O2 461.35985 found 461.35976, Δ –0.20 ppm). 1H NMR (500 MHz) and 13C NMR (125 MHz, CD3OD) (lH, J, 13C) C-1 (158.5), H2C-2 (3.16 t, 6.6; 42.5), H2C-3 (1.61 m; 30.1), H2C-3A (1.41 br s; 28.9), H2C-3B (1.41 br s; 29.9), H2C-3C (1.41 br s; 27.3), H2C-4 (1.80 m; 29.9), H2C-5 (4.36 t, 7.6; 67.0), C-6 (165.9), C-7 (111.7), C-8 (170.6), H2C-9 (2.57 t, 8.6 and 3.30; 35.6), H2C-10 (2.05 m and 2.39 q; 20.8), H2C-11 (4.26 t, 9.1; 53.1), C-13 (180.2), H2C-14 (3.07 t, 9.5; 38.2), H2C-15 (1.72 m; 30.0), H2C-16 (1.41 br m; 27.3), H2C-17 to H2C-19 (1.31 br m; 30.0-31.0), H2C-20 (1.31 br m; 32.8), H2C-21 (1.31 br m; 23.6), H3C-22 (0.90 t, 7.5; 14.5).Norcrambescin B1 (6)
[α]20D −114.0 (c 0.05, MeOH); UV (MeOH) λmax (log ε) 285 nm (1.8), 326 nm (1.9); CD (MeOH, c 4.03 × 10−4M) Δε (λmax nm) −0.53 (211) +0.75 (247) +0.55 (326); ESIMS m/z 467.3 [M+H]+; HRESIMS m/z 467.37051 [M+H]+ (Calc for C24H47O3N6, 467.37042, Δ +0.21 ppm).Crambescin B1 (7)
[α]20D −116.3 (c 0.04, MeOH); UV (MeOH) λmax (log ε) 286 nm (1.9), 325 nm (1.9); CD (MeOH, c 3.58 × 10−4 M) Δε (λmax nm) –0.51 (211) +0.74 (247) +0.53 (328); ESIMS m/z 481.2 [M+H]+; HRESIMS m/z 481.38629 [M+H]+ (Calc for C25H49O3N6, 481.38607, Δ +0.47 ppm). 1H NMR (500 MHz) and 13C NMR (125 MHz, CD3OD) (lH, J, 13C) C-1 (158.6), H2C-2 (3.16 t, 7.3; 42.6), H2C-3 (1.59 m; 29.7), H2C-3A (1.38 br s; 27.8), H2C-3B (1.38 br s; 30.3), H2C-3C (1.38 br s; 27.1), H2C-4 (1.67 m; 29.9), H2C-5 (4.13 m and 4.17 m; 66.3), C-6 (169.9), HC-7 (2.99 d, 4.3; 50.2), C-8 (89.9), H2C-9 (2.07 m and 2.12, m; 36.1), H2C-10 (2.10 m; 25.7), H2C-11 (3.93 m and 4.02 m; 68.9), C-12 (155.2), HC-13 (3.86 ddd, 4.3/6.7/7.3; 49.7), H2C-14 (1.59 m; 33.1), H2C-15 (1.47 m; 26.5), H2C-16 to H2C-19 (1.31 br m; 30.0–31.0), H2C-20 (1.31 br m; 32.8), H2C-21 (1.31 br m; 23.7), H3C-22 (0.90 t, 7.1; 14.4).Homocrambescin B1 (8)
[α]20D −145.5 (c 0.07, MeOH); UV (MeOH) λmax (log ε) 284 nm (1.8), 326 nm (1.8); CD (MeOH, c 1.34 × 10−4 M) Δε (λmax nm) –0.90 (211) +1.42 (247) +0.90 (326); ESIMS m/z 495.3 [M+H]+; HRESIMS m/z 495.40207 [M+H]+ (Calc for C26H51O3N6, 495.40172, Δ −0.71 ppm).Norcrambescin C1 (9)
[α]20D +76.3 (c 0.08, MeOH); UV (MeOH) λmax (log ε) 279 nm (2.3); CD (MeOH, c 5.15 × 10−4 M) Δε (λmax nm) +0.08 (207) –0.20 (247); ESIMS m/z 467.5 [M+H]+; HRESIMS m/z 467.37027 [M+H]+ (Calc for C24H47O3N6, 467.37042, Δ −0.31 ppm).Crambescin C1 (10)
[α]20D +33.1 (c 0.39, MeOH); UV (MeOH) λmax (log ε) 278 nm (2.8); CD (MeOH, c 1.39 × 10−4 M) Δε (λmax nm) −0.48 (213) +2.24 (247); ESIMS m/z 481.5 [M+H]+; HRESIMS m/z 481.38635 [M+H]+ (Calc for C25H49N6O3, 481.38607, Δ 0.59551). 1H NMR (500 MHz) and 13C NMR (125 MHz, CD3OD) (lH, J, 13C) C-1 (159.1), H2C-2 (3.17 t, 7.0; 42.8), H2C-3 (1.60 m; 30.9), H2C-3A (1.41 br s; 27.9), H2C-3B (1.41 br s; 30.4), H2C-3C (1.41 br s; 27.4), H2C-4 (1.71 m; 30.2), H2C-5 (4.17 m and 4.20 m; 66.3), C-6 (166.5), C-7 (106.4), C-8 (149.4), H2C-9 (2.80 m and 2.83, m; 29.1), H2C-10 (1.81q; 32.5), H2C-11 (3.61 t, 6.5; 62.5), C-12 (153.9), HC-13 (4.42 dd, 7.5/5.0; 51.5), H2C-14 (1.60 m; 37.4), H2C-15 (1.41 m; 25.4), H2C-16 to H2C-19 (1.30 br m; 30.0-32.0), H2C-20 (1.30 br m; 33.4), H2C-21 (1.30 br m; 24.7), H3C-22 (0.90 t, 6.5; 15.1).Homocrambescin C1 (11)
[α]20D +62.7 (c 0.13, MeOH); UV (MeOH) λmax (log ε) 279 nm (2.9); CD (MeOH, c 0.68 × 10−4 M) Δε (λmax nm) –0.58 (214) +3.03 (249); ESIMS m/z 495.0 [M+H]+; HRESIMS m/z 495.40182 [M+H]+ (Calc for C26H51O3N6, 495.40172, Δ +0.22 ppm).Crambescidin 816 (12)
As reported in ref. 8.Theoretical calculations of the electronic dichroism spectra
Quantum chemical calculations have been performed for all examined compounds 1–11. Conformational analysis was performed using the conformer research algorithm implemented in the Conflex-Barista software.23 Given the high degree of conformational freedom of the side chains, the conformational analysis led to a large number of structures (n > 500). The Gaussian03W package24 has been used for the electronic circular dichroism calculations on the most stable conformer of each compound. Density functional theory (DFT) with B3LYP functional25 and Pople's 6.31++G(d,p)26 basis set was used on the lowest energy conformers. TDDFT was employed to calculate excitation energy (in eV) and rotatory strength R in dipole velocity (Rvel) and dipole length (Rlen) forms. The calculated rotatory strengths were simulated in ECD curve by using a Gaussian function: | (1) |
where Δ is half the width of the band at 1/e peak height expressed in energy units. The parameters ΔE0a and R0a are the excitation energies and the rotatory strengths for transition from the ground state 0 to an excited state a, respectively, Δ = 0.1 eV and Rvel were used.
Conclusions
The full chemical study of the Mediterranean sponge C. crambe led to the isolation and structural characterization of 11 crambescins A1, A2, B1 and C1 in their pure forms. Eight of the 12 isolated compounds are described for the first time and the lengths of the alkyl chains were deduced from the careful analyses of HRMS/MS data. The presence of the new didehydrocrambescin A1 (5) derivative supports the assumption that aromatization could occur in this family during the acidic purification process. Our studies allowed the assignment of the absolute configurations of all compounds and revealed a high cytotoxic activity of crambescidin 816 (12) against cortical neurons.Acknowledgements
This work was supported by the European Coordinative Project FP7-KBBE-2010-4-BAMMBO (265896) and the French ECIMAR project (ANR-06-BDIV-01). We are grateful to Thierry Perez (Centre d'Océanologie d'Endoume) for taxonomic identification and to Marc Gaysinski (PFTC Nice) for recording the NMR spectra. Assistance for submarine collection was kindly given by David Luquet (Observatoire Océanologique de Villefranche sur Mer).References
- (a) R. G. S. Berlinck, A. C. B. Burtoloso, A. E. Trindade-Silva, S. Romminger, R. P. Morais, K. Bandeira and C. M. Mizuno, Nat. Prod. Rep., 2010, 27, 1871–1907 RSC; (b) R. G. S. Berlinck and M. H. Kossuga, in Modern Alkaloids, ed. E. Fattorusso and O. Taglialatela-Scafati, Wiley-VCH Verlag GmbH & Co, KGaA, Weinheim, Germany, 2008, pp 305–337 Search PubMed.
- D. Erpenbeck and R. W. M. van Soest, Mar. Biotechnol., 2007, 9, 2–19 CrossRef CAS.
- (a) R. G. S. Berlinck, J. C. Braekman, D. Daloze, I. Bruno, R. Riccio, D. Rogeau and P. Amade, J. Nat. Prod., 1992, 55, 528–532 CrossRef CAS; (b) R. G. S. Berlinck, J. C. Braekman, D. Daloze, K. Hallenga, R. Ottinger, I. Bruno and R. Riccio, Tetrahedron Lett., 1990, 31, 6531–6534 CrossRef CAS.
- Y. Kashman, S. Hirsh, O. J. McConnell, I. Ohtani, T. Kusumi and H. Kakisawa, J. Am. Chem. Soc., 1989, 111, 8925–8926 CrossRef CAS.
- (a) R. G. Berlinck, J. C. Braekman, D. Daloze, I. Bruno, R. Riccio, S. Ferri, S. Spampinato and E. Speroni, J. Nat. Prod., 1993, 56, 1007–1015 CrossRef CAS; (b) E. A. Jares-Erijman, R. Sakai and K. L. Rinehart, J. Org. Chem., 1991, 56, 5712–5715 CrossRef CAS.
- (a) B. B. Snider and Z. Shi, J. Org. Chem., 1993, 58, 3828–3839 CrossRef CAS; (b) E. A. Jares-Erijman, A. A. Ingrum, F. Sun and K. L. Rinehart, J. Nat. Prod., 1993, 56, 2186–2188 CrossRef CAS; (c) B. B. Snider and Z. Shi, J. Org. Chem., 1992, 57, 2526–2528 CrossRef CAS.
- D. S. Dalisay, J. P. Saludes and T. F. Molinski, Bioorg. Med. Chem., 2011, 19, 6654–6657 CrossRef CAS.
- C. H. Van Etten, H. C. Nielsen and J. E. Peters, Phytochemistry, 1965, 4, 467–473 CrossRef CAS.
- (a) A. Olszewski, K. Sato, Z. D. Aron, F. Cohen, A. Harris, B. R. McDougall, W. E. Robinson Jr., L. E. Overman and G. A. Weiss, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 14079–14084 CrossRef CAS; (b) K. L. Rinehart and E. A. Jares-Erijman, US Pat. 5756734A, 1998 Search PubMed.
- A. D. Patil, N. V. Kumar, W. C. Kokke, M. F. Bean, A. J. Freyer, C. de Brosse, S. Mai, A. Truneh, B. Carte, A. L. Breen, R. P. Hertzberg, R. K. Johnson, J. W. Westley and B. C. M. Potts, J. Org. Chem., 1995, 60, 1182–1188 CrossRef CAS.
- L. Chang, N. F. Whittaker and C. A. Bewley, J. Nat. Prod., 2003, 66, 1490–1494 CrossRef CAS.
- R. Laville, O. P. Thomas, F. Berrué, D. Marquez, J. Vacelet and P. Amade, J. Nat. Prod., 2009, 72, 1589–1594 CrossRef CAS.
- M. El-Naggar, M. Conte and R. J. Capon, Org. Biomol. Chem., 2010, 8, 407–412 CAS.
- G. Genta-Jouve, N. Cachet, S. Holderith, F. Oberhänsli, J.-L. Teyssié, R. Jeffree, A. Al Mourabit and O. P. Thomas, ChemBioChem, 2011, 12, 2298–2301 CrossRef CAS.
- B. I. Morinaka and T. F. Molinski, Org. Lett., 2011, 13, 6338–6341 CrossRef CAS.
- (a) E. L. Regalado, C. Jimenez-Romero, G. Genta-Jouve, D. Tasdemir, P. Amade, C. Nogueiras and O. P. Thomas, Tetrahedron, 2011, 67, 1011–1018 CrossRef CAS; (b) N. Cachet, G. Genta-Jouve, E. L. Regalado, R. Mokrini, P. Amade, G. Culioli and O. P. Thomas, J. Nat. Prod., 2009, 72, 1612–1615 CrossRef CAS.
- L. E. Overman and Y. H. Rhee, J. Am. Chem. Soc., 2005, 127, 15652–15658 CrossRef CAS.
- K. M. Meragelman, T. C. McKee and J. B. McMahon, J. Nat. Prod., 2004, 67, 1165–1167 CrossRef CAS.
- Z. D. Aron, H. Pietraszkiewicz, L. E. Overman, F. Valeriote and C. Cuevas, Bioorg. Med. Chem. Lett., 2004, 14, 3445–3449 CrossRef CAS.
- C. Vale, E. Alonso, J. A. Rubiolo, M. R. Vieytes, F. M. La Ferla, L. Gimenez-Llort and L. M. Botana, Cell. Mol. Neurobiol., 2010, 30, 577–590 CrossRef CAS.
- C. Vale, K. C. Nicolaou, M. O. Frederick, B. Gomez-Limia, A. Alfonso, M. R. Vieytes and L. M. Botana, J. Med. Chem., 2007, 50, 356–363 CrossRef CAS.
- T. Varming, J. Drejer, A. Frandsen and A. Schousboe, J. Neurosci. Res., 1996, 44, 40–46 CrossRef CAS.
- (a) H. Goto, K. Ohta, T. Kamakura, S. Obata, N. Nakayama, T. Matsumoto and E. Osawa, Conflex Corp: Tokyo-Yokohama, 2004 Search PubMed; (b) H. Goto and E. Osawa, J. Chem. Soc., Perkin Trans. 2, 1993, 187–198 RSC; (c) H. Goto and E. Osawa, Tetrahedron Lett., 1992, 33, 1343–1346 CrossRef CAS; (d) H. Goto and E. Osawa, J. Am. Chem. Soc., 1989, 111, 8950–8951 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, A. Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03, 2003 Search PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR and HRMS spectra are given for compounds 1–11. See DOI: 10.1039/c2ra00045h |
| This journal is © The Royal Society of Chemistry 2012 |