A one pot synthesis of [1,3,4]-oxadiazoles mediated by molecular iodine†
Srimanta
Guin
,
Saroj Kumar
Rout
,
Tuhin
Ghosh
,
Nilufa
Khatun
and
Bhisma K.
Patel
*
Department of Chemistry, Indian Institute of Technology, Guwahati, 781 039, Assam, India. E-mail: patel@iitg.ernet.in; Fax: +91-3612690762
First published on 26th January 2012
Abstract
A one-pot synthesis of 3-amino-1,3,4-oxadiazoles has been achieved from the corresponding dithiocarbamate salt, employing the thiophilic property of molecular iodine. The precursor thiosemicarbazides could be derived in situ which underwent an intramolecular cyclodesulfurization in the presence of iodine to afford 3-amino-1,3,4-oxadiazoles exclusively. Apart from being milder and environmentally sustainable, this method involves a simple, reliable approach to give good to excellent yields of the desired products and is compatible with a wide range of functional groups.
Introduction
The N, O, and S heterocycles are of considerable importance due to their prevalence in biologically active compounds.1 Amongst these heterocycles, [1,3,4]-oxadiazole scaffold is established as a member of the privileged structures class particularly from a medicinal point of view. Due to their metabolic profile, improved pharmacokinetics and in vivo performance they are recognized as biologically important pharmacophores.2–5 Their biological activities encompass analgesic, anti-inflammatory, anticonvulsive, diuretic, antineoplastic, antimitotic, antiemetic and muscle relaxant properties.2 Some display insecticidal3a and herbicidal5b properties as well. Besides, 2,5-diaryl-[1,3,4]-oxadiazoles exhibit inhibition toward platelet aggregation,4a while derivatives of 2-hydroxyphenyl-[1,3,4]-oxadiazole possess hypnotic and sedative properties.4b The wide spectrum of biological importance has led to the emergence of a number of marketed medicines bearing a [1,3,4]-oxadiazole core (Fig. 1). Instances of medicinally important compounds containing a [1,3,4]-oxadiazole core are MK-0633 p-toluenesulfonate,5a nesapidil,5b,c ABT-751-oxadiazole,5d and furamizole5e,f (Fig. 1). Apart from their latent potential in medicinal chemistry they also find applications in the field of material science and have received much attention particularly in the field of organic electronics because of their good electron-transporting and hole-blocking abilities.6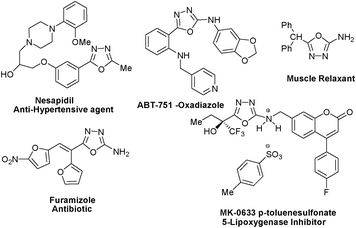 | ||
| Fig. 1 Some pharmaceutical compounds having oxadiazole scaffold. | ||
The widespread applications of 2-amino-[1,3,4]-oxadiazoles have resulted to they being encountered through the development of various protocols. Several methodologies that have been documented in the literature, most commonly employ the cyclodehydration of semicarbazides or the cyclodesulfurization of thiosemicarbazides as two main strategies. The cyclodehydration processes are mediated typically by POCl3,7a–d SOCl2,7e conc. H2SO4,7f–h or tosyl chloride.7i Alternatively, phosphonium salts,8a–d and Burgess-type reagents8e–f are also found to be equally effective in promoting dehydrative cyclization. In the case of thiosemicarbazides as oxadiazole precursors, thiophilic reagents such as salts of mercury9a,b and lead2b have been used extensively to accomplish the desired desulfurative cyclization. Alternatively, activation of sulfur using iodine in the presence of strong base (NaOH)10a–c or tosyl chloride in combination with triethylamine10d or toxic pyridine,2d have been performed under refluxing conditions. Coupling reagents such as EDC11a,b and DCC11c–e as well as alkylating agents such as methyl iodide11f,g and ethyl bromoacetate11h have been exploited to afford various oxadiazoles. The [1,3,4]-oxadiazoles have also been accomplished through a multicomponent reaction involving a Ugi-4CR/aza-Wittig sequence.12 Very recently our group has also developed a protocol for the synthesis of substituted oxadiazoles through a copper catalyzed imine bond activation/C–O bond formation.13 It is noteworthy to mention here that in the latter two protocols the oxadiazole derivatives are devoid of a 2-amino functionality.
Although numerous procedures are enumerated in literature for the synthesis of oxadiazoles, most of them suffer a major set back from the standpoint of environmental concerns as they involve direct or indirect use of strong alkaline or acidic conditions, toxic and corrosive reagents, and emission of odoriferous thiol as a byproduct which are undesirable in pharmaceuticals. Besides expensive reagents and catalysts, elevated temperatures and longer reaction times are also some of the associated drawbacks. In addition, a number of methods seem to have limited substrate scope, resulting in the stoichiometric formation of intractable byproducts which impedes their generality. Thus a more sustainable synthesis for their synthesis is of prime importance.
In recent years, molecular iodine has been identified as an effective reagent to carry out various organic transformations owing to its inexpensive, ready availability and eco-friendly nature.14 Our interest to explore the thiophilic properties of hypervalent iodine and molecular iodine toward relevant synthetic transformations15 has led us to envisage a tandem one-pot synthesis of [1,3,4]-oxadiazol-2-yl-amines. Our anticipated tandem synthetic sequence involves the in situ generation of isothiocyanate from the dithiocarbamate salt using molecular iodine. This would then form thiosemicarbazide upon treatment with acyl hydrazide in the same pot and upon reaction with further equivalent of iodine would undergo cyclodesulfurisation to render the corresponding 2-amino-[1,3,4]-oxadiazole.
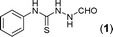
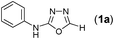
Results and discussion
In one of our recent achievements15f we have seen that the dithiocarbamate salt upon treatment with iodine and inorganic base such as K2CO3 or Na2CO3 in a biphasic medium of ethyl acetate and water afforded the corresponding isothiocyanate. Taking cues from this observation, an initial experiment was performed using freshly prepared phenyl dithiocarbamate salt (1 equiv.) in ethylacetate/water medium, to which was sequentially added K2CO3 (3 equiv.) and iodine (1.1 equiv.) pinchwise over a period of 10 min. A complete consumption of salt to isothiocyanate was observed (judged by TLC) along with the precipitation of colloidal sulfur. To the same reaction mixture formic acid hydrazide (1 equiv.) was added and the formation of thiosemicarbazide (1) was detected in the reaction mixture. Addition of a further 1.1 equiv. of iodine and stirring the reaction mixture for 0.5 h resulted in formation of the product (1a) to our delight in a good yield of 77% isolated yield (Table 1, entry 1). It is worth mentioning here that further investigations toward the optimization of the reaction conditions with respect to the appropriate solvent systems suggested that the EtOH/water system was equally effective in promoting the reaction (Table 1, entry 2). Other solvent systems such as acetonitrile/water (Table 1, entry 3) or THF/water (Table 1, entry 4) or simple organic solvents (Table 1, entry 5–8) either failed or gave poorer yields of the product. This substantial decrease in yield could be attributed to the inefficient formation of isothiocyanate or insolubility of base in them. Thus, after subsequent trials we figured out that EtOAc/water was most suitable for this transformation to meet out with the work up procedures in the EtOH/water system. Among the inorganic bases, Na2CO3 gave slightly poorer yield while (Table 1, entry 9) K2CO3 and Cs2CO3 (Table 1, entry 10) were found to be almost equally effective but from cost consideration K2CO3 was used. Since we were focusing on the use of milder inorganic carbonates, organic bases were not tried for these reactions.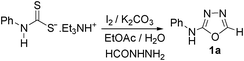
| Entry | Solvent(s) | Base (3 equiv.) | Yield (%)b |
|---|---|---|---|
| a Reactions were monitored by TLC. b Isolated yield. | |||
| 1 | EtOAc/H2O (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) :1) |
K2CO3 | 77 |
| 2 | EtOH/H2O (3:1) |
K2CO3 | 75 |
| 3 | CH3CN/H2O (3:1) |
K2CO3 | 52 |
| 4 | THF/H2O (3:1) |
K2CO3 | 40 |
| 5 | EtOAc | K2CO3 | 10 |
| 6 | EtOH | K2CO3 | 15 |
| 7 | CH3CN | K2CO3 | 8 |
| 8 | THF | K2CO3 | 5 |
| 9 | EtOAc/H2O (3:1) |
Na2CO3 | 63 |
| 10 | EtOAc/H2O (3:1) |
Cs2CO3 | 74 |
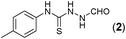
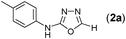
Thus having a standardized protocol in hand the method was subsequently applied to various in situ generated thiosemicarbazides to examine the generality and scope of the reaction. It was gratifying to observe that all these reactions proceeded smoothly affording the corresponding 2-amino-[1,3,4]-oxadiazole in good to excellent yields (Table 2). Various in situ generated thiosemicarbazides (2, 3, 4, and 5) bearing electron-donating groups all gave satisfactory yields of the respective [1,3,4]-oxadiazol-2-yl-amines (2a), (3a), (4a), and (5a). Structure of the product (4a) has been confirmed by crystal X-ray crystallography as shown in (Fig. 2).
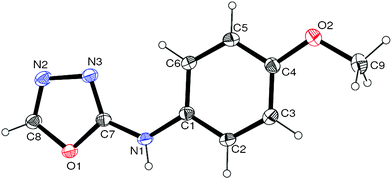 | ||
| Fig. 2 ORTEP view of compound (4a). | ||
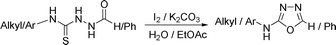
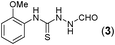
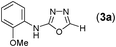
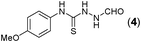
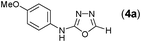
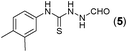
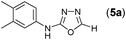
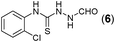
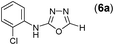
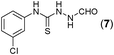
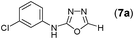
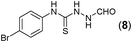
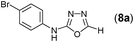
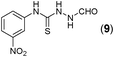
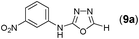
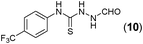
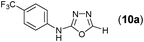
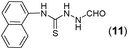
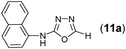
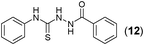
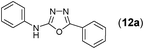
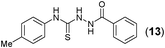
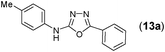
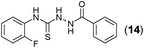
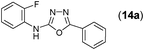
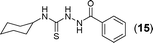
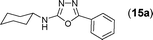
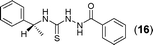
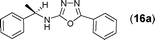
This methodology was also equally successful for substrates (6, 7, and 8) bearing moderately electron-withdrawing groups (–Cl and –Br) and for substrates (9 and 10) having strong electron-withdrawing groups, giving corresponding oxadiazoles as shown in Table 2. 1-Naphthyl thiosemicarbazide (11) derived from the corresponding dithiocarbamate salt yielded oxadiazole (11a) in decent yield under the present condition. In addition to formic acid hydrazide (NH2NHCHO), benzhydrazide (NH2NH2COPh) can also be used. Thus the in situ generated thiosemicarbazides (12, 13 and 14) yielded the corresponding N,5-diphenyl-1,3,4-oxadiazole-2-amines, (12a), (13a), and (14a) respectively, in modest yields. Aliphatic thiosemicarbazide (15) and chiral benzylthiosemicarbazide (16) gave their corresponding oxadiazoles (15a) and (16a) respectively as shown in Table 2 in fairly good yields. However in case of aliphatic thiosemicarbazides an excess of K2CO3 (5 equiv.) had to be used to effect the transformation.
Our recent study on the formation of isothiocyanate from dithiocarbamate salt and carbodiimide from 1,3-disubstituted thiourea,15f,g led us to propose the following reaction mechanism for the formation of 2-amino-[1,3,4]-oxadiazoles. Initially this one-pot strategy isothiocyanate (A) can be obtained from dithiocarbamate salt and molecular iodine following our green strategy.15f Thiosemicarbazide formation (B) can take place in the same pot (Scheme 1) by adding formichydrazide (NH2NHCHO) to the biphasic reaction mixture containing isothiocyanate (A). Subsequent desulfurization using iodine in the presence of base should lead to the formation of a carbodiimide type intermediate (D) via intermediate (C)15g to which an intramolecular attack of thioamidic oxygen would generate intermediate (E). Aromatization of the intermediate (E) would yield the target [1,3,4]-oxadiazol-2-yl-amines (F) (Scheme 1).
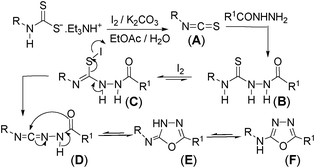 | ||
| Scheme 1 Proposed mechanism for the formation of 1,3,4-oxadiazol-2-yl-amine. | ||
Conclusion
In conclusion we have developed a tandem one-pot method for the synthesis of 3-amino-1,3,4-oxadiazoles which have been achieved starting from dithiocarbamate salts. The in situ generated thiosemicarbazides undergo cyclodesulfurization mediated by iodine to give 2-amino-1,3,4-oxadiazoles. This method gives easy access to the preparation of pharmaceutically important heterocyclic scaffold and is compatible with a wide range of functional groups. In comparison to the reported methods, our method perhaps provides the simplest and most convenient approach and thus has potential industrial application.General procedure
To a freshly prepared dithiocarbamate salt of aniline (540 mg, 2 mmol) in a biphasic medium of water/ethyl acetate (1:2, 10 mL) containing K2CO3 (828 mg, 6 mmol) was added iodine (554.4 mg, 2.2 mmol) pinchwise over a period of ten minutes whilst stirring. Instant precipitation of colloidal sulfur could be observed with the disappearance of iodine color in the medium. After stirring for further 20 min complete conversion of the dithiocarbamate salt to isothiocyanate was observed (by TLC). Formichydrazide (NH2NHCHO) (120 mg, 2 mmol) was added to the above reaction mixture. Formation of thiosemicarbazide (1) having lower Rf was detected in the reaction mixture. Further 2.2 mmol of iodine (554.4 mg, 2.2 mmol) was added portion wise over a period of 10 min to the thiosemicarbazide and stirring continued for 0.5 h resulted in the complete disappearance of thiosemicarbazide (1) to yield a product having higher Rf compared to (1). After filtering out colloidal sulphur, the reaction mixture was treated with a 5% hypo solution (5 mL). It was then further extracted with ethyl acetate (2 × 10 mL) and the combined ethyl acetate layer was washed with water (1 × 5 mL), dried over anhydrous Na2SO4 and concentrated under reduced pressure. The product was purified over a column of silica gel and eluted with (3:1, hexane/ethyl acetate) to give 1a (248 mg, 77% yield).
Acknowledgements
BKP acknowledges the support of this research by the Department of Science and Technology (DST) (SR/S1/OC-79/2009), New Delhi, and the Council of Scientific and Industrial Research (CSIR) (01(2270)/08/EMR-II). SG, SKR, TG and NK thank CSIR for fellowships. Thanks are due to Arghya Basu and Sandeep Dey for crystallographic help. Thanks are due to Central Instruments Facility (CIF) IIT Guwahati for NMR spectra and DST-FIST for XRD facility.References
- Z. Jin, Nat. Prod. Rep., 2003, 20, 584 RSC.
- (a) J. Thomas, Ger. Offen., 1974, 2403357 Search PubMed ; Chem. Abstr. 1974, 81, 136153; (b) H. L. Yale and K. Losse, J. Med. Chem., 1966, 9, 478 CrossRef CAS; (c) D. Ghirian, I. Schwatz and I. Simiti, Farmacia, 1974, 22, 141 Search PubMed; (d) S. J. Dolman, F. Gosselin, P. D. O'Shea and I. W. Davies, J. Org. Chem., 2006, 71, 9548 CrossRef CAS.
- (a) X. Zheng, Z. Li, Y. Wang, W. Chen, Q. Huang, C. Liu and G. Song, J. Fluorine Chem., 2003, 123, 163 CrossRef CAS; (b) X. J. Zou, L. H. Lai, G. Y. Jin and Z. X. Zhang, J. Agric. Food Chem., 2002, 50, 3757 CrossRef CAS.
- (a) M. J. Fray, K. Cooper, M. J. Parry, K. Richardson and J. Steele, J. Med. Chem., 1995, 38, 3514 CrossRef CAS; (b) G. W. Adelstein, C. H. Yen, E. Z. Dajani and R. G. Bianchi, J. Med. Chem., 1976, 19, 1221 CrossRef CAS.
- (a) Y. Ducharme, M. Blouin, C. Brideau, A. Châteauneuf, Y. Gareau, E. L. Grimm, H. Juteau, S. Laliberté, B. MacKay, F. Massé, M. Ouellet, M. Salem, A. Styhler and R. W. Friesen, ACS Med. Chem. Lett., 2010, 1, 170 CrossRef CAS; (b) P. C. Theime, A. Franke, D. Denke, H. D. Lehmann and J. Gries, Ger. Offen., 1981, 3006351 Search PubMed; (c) R. Schlecker and P. C. Thieme, Tetrahedron, 1988, 44, 3289 CrossRef CAS; (d) X. Ouyang, E. L. Piatnitski, V. Pattaropong, X. Chen, H. -Y. He, A. S. Kiselyov, A. Velankar, J. Kawakami, M. Labelle, L. Smith, J. Lohman, S. P. Lee, A. Malikzay, J. Fleming, J. Gerlak, Y. Wang, R. L. Rosler, K. Zhou, S. Mitelman, M. Camara, D. Surguladze, J. F. Doody and M. C. Tuma, Bioorg. Med. Chem. Lett., 2006, 16, 1191 CrossRef CAS; (e) I. Hirao, Nippon Kagaku Zassi, 1967, 88, 574 CrossRef CAS; (f) M. Ogata, H. Atobe, H. Kushida and K. Yamamoto, J. Antibiot., 1971, 24, 443 CrossRef CAS.
- (a) U. Mitschke and P. Bäuerle, J. Mater. Chem., 2000, 10, 1471 RSC; (b) G. S. He, L.-S. Tan, Q. Zheng and P. N. Prasad, Chem. Rev., 2008, 108, 1245 CrossRef CAS; (c) E. Schinzel, T. Martini, W. Spatzier and H. Probst, Ger. Offen., Chem. Abstr., 1983, Search PubMed98, 199850; (d) N. K. Chudgar, S. N. Shah and R. A. Vora, Mol. Cryst. Liq. Cryst., 1989, 172, 51 CrossRef CAS; (e) B. Verheyde and W. Dehaen, J. Org. Chem., 2001, 66, 4062 CrossRef CAS.
- (a) C. K. Reddy, P. C. S. Reddy, P. S. N. Reddy, C. V. Ratnam and V. M. Reddy, Ind. J. Het. Chem., 1997, 7, 17 CAS; (b) G. S. Poindexter, M. A. Bruce, J. G. Breitenbucher, M. A. Higgins, S. -Y. Sit, J. I. Romine, S. W. Marin, S. A. Ward, R. T. McGovern, W. Clarke, J. Russell and I. Antal-Zimanyi, Bioorg. Med. Chem., 2004, 12, 507 CrossRef CAS; (c) M. Demina, G. Sarapulova, A. Borisova, L. Larina and A. Medvedeva, Russ. J. Org. Chem., 2003, 10, 1522 CrossRef; (d) A. -S. S. Hamad and A. I. Hashem, J. Heterocycl. Chem., 2002, 39, 1325 CrossRef CAS; (e) M. Al-Talib, H. Tashtoush and N. Odeh, Synth. Commun., 1990, 20, 1811 CrossRef CAS; (f) S. Sharma, V. K. Srivastava and A. Kumar, Eur. J. Med. Chem., 2002, 37, 689 CrossRef CAS; (g) S. Sharma, V. K. Srivastava and A. Kumar, Ind. J. Chem., 2002, 41B, 2647 CAS; (h) M. Tyagi and A. Kumar, Orient. J. Chem., 2002, 18, 125 CAS; (i) S. E. Wolkenberg and D. L. Boger, J. Org. Chem., 2002, 67, 7361 CrossRef CAS.
- (a) J. Dumciute, V. Martynaitis, W. Holzer, S. Mangelinckx, N. DeKimpe and A. Sackus, Tetrahedron, 2006, 62, 3309 CrossRef CAS; (b) C. H. Lee, H. I. Cho and K.-J. Lee, Bull. Korean Chem. Soc., 2001, 22, 1153 CAS; (c) C. G. Levins and Z. -K. Wan, Org. Lett., 2008, 10, 1755 CrossRef CAS; (d) R. Appel, Angew. Chem., Int. Ed. Engl., 1975, 14, 801 CrossRef; (e) C. T. Brain, J. M. Paul, Y. Loong and P. J. Oakley, Tetrahedron Lett., 1999, 40, 3275 CrossRef CAS; (f) C. T. Brain and S. A. Brunton, Synlett, 2001, 3, 382 Search PubMed.
- (a) S. A. Rostom, M. A. Shalaby and M. A. El-Demellawy, Eur. J. Med. Chem., 2003, 38, 959 CrossRef CAS; (b) X. Wang, Z. Li, B. Wei and J. Yang, Synth. Commun., 2002, 32, 1097 CrossRef CAS; (c) R. S. Sharma and S. C. Bahel, J. Indian Chem. Soc., 1982, LIX, 877 Search PubMed.
- (a) M. Amir, M. S. Y. Khan and M. S. Zaman, Ind. J. Chem. Sec. B, 2004, 43B, 2189 CAS; (b) S. P. Hiremath, J. S. Biradir and S. M. Kudari, J. Ind. Chem. Soc., 1984, 61, 74 CAS; (c) S. Shelka, G. Mhaske, S. Gadakh and C. Gill, Bioorg. Med. Chem. Lett., 2010, 20, 7200 CrossRef; (d) J. Y. Hwang, H.-S. Choi, D.-H. Lee and Y.-D. Gong, J. Comb. Chem., 2005, 7, 816 CrossRef CAS.
- (a) R. Severinsen, J. P. Kilbrun and J. F. Lau, Tetrahedron, 2005, 61, 5565 CrossRef CAS; (b) E. L. P. Chekler, H. M. Elokdah and J. Butera, Tetrahedron Lett., 2008, 49, 6709 CrossRef; (c) M. M. Ahmed, O. M. Aboulwafa and O. Kader, Monatsh. Chem., 1989, 120, 571 CrossRef; (d) O. M. Aboulwafa, A. Omar and M. E. Mohsen, Sulfur Lett., 1992, 14, 181 CAS; (e) F. T. Coppo, K. A. Evans, T. L. Graybill and G. Burton, Tetrahedron Lett., 2004, 45, 3257 CrossRef CAS; (f) F. Fulop, E. Semega, G. Dombi and G. Bernath, J. Heterocycl. Chem., 1990, 27, 951 CrossRef CAS; (g) N. Okajima and Y. Okada, J. Heterocycl. Chem., 1991, 28, 177 CrossRef CAS; (h) S. G. Kucukguzel, E. E. Oruc, S. Rollas, F. Sahin and A. Ozbek, Eur. J. Med. Chem., 2002, 37, 197 CrossRef CAS.
- A. Ramazani and A. Rezaei, Org. Lett., 2010, 12, 2852 CrossRef CAS.
- S. Guin, T. Ghosh, S. K. Rout, A. Banerjee and B. K. Patel, Org. Lett., 2011, 13, 5976 CrossRef CAS.
- (a) J. S. Yadav, B. V. S. Reddy, K. Premalatha and T. Swamy, Tetrahedron Lett., 2005, 46, 2687 CrossRef CAS; (b) L.-Y. Zeng and C. Cai, J. Comb. Chem., 2010, 12, 35 CrossRef CAS; (c) L.-Y. Zeng and C. Cai, Org. Biomol. Chem., 2010, 8, 4803 RSC; (d) X. S. Wang, Q. Li, J. R. Wu, Y. L. Li, C. S. Yao and S. J. Tu, Synthesis, 2008, 1902 CrossRef CAS; (e) X. S. Wang, Q. Li, C. S. Yao and S. J. Tu, Eur. J. Org. Chem., 2008, 3513 CrossRef CAS; (f) X. S. Wang, J. Zhou, M. Y. Yin, K. Yang and S. J. Tu, J. Comb. Chem., 2010, 12, 266 CrossRef CAS; (g) N. Fei, Q. Hou, S. Wang, H. Wang and Z.-J. Yao, Org. Biomol. Chem., 2010, 8, 4096 RSC; (h) D. Yue and R. C. Larock, Org. Lett., 2004, 6, 1037 CrossRef CAS; (i) D. Yue, T. Yao and R. C. Larock, J. Org. Chem., 2006, 71, 62 CrossRef CAS; (j) X. Zhang, M. A. Campo, T. Yao and R. C. Larock, Org. Lett., 2005, 7, 763 CrossRef CAS; (k) D. Yue, N. Della Ca and R. C. Larock, J. Org. Chem., 2006, 71, 3381 CrossRef CAS; (l) J.-G. Kim and D. O. Jang, Synlett, 2010, 2093 CAS; (m) J. S. Yadav, B. V. S. Reddy, K. S. Shankar and S. T. Swamy, Tetrahedron Lett., 2010, 51, 46 CrossRef CAS; (n) X. Lin, X. Dai, Z. Mao and Y. Wang, Tetrahedron, 2009, 65, 9233 CrossRef CAS; (o) J. Jaratjaroonphong, S. Sathalalai, P. Techa-sauvapak and V. Reutrakul, Tetrahedron Lett., 2009, 50, 6012 CrossRef CAS; (p) M. Jereb, D. Vrazica and M. Zupan, Tetrahedron Lett., 2009, 50, 2347 CrossRef CAS; (q) B. Jiang, C. Li, S. J. Tu and F. Shi, J. Comb. Chem., 2010, 12, 482 CrossRef CAS.
- (a) C. B. Singh, H. Ghosh, S. Murru and B. K. Patel, J. Org. Chem., 2008, 73, 2924 CrossRef CAS; (b) H. Ghosh, R. Yella, J. Nath and B. K. Patel, Eur. J. Org. Chem., 2008, 6189 CrossRef CAS; (c) H. Ghosh, R. Yella, A. R. Ali, S. K. Sahoo and B. K. Patel, Tetrahedron Lett., 2009, 50, 2407 CrossRef CAS; (d) J. Nath, H. Ghosh, R. Yella and B. K. Patel, Eur. J. Org. Chem., 2009, 1849 CrossRef CAS; (e) J. Nath, B. K. Patel, L. Jamir, U. B. Singh and K. V. V. Satyanarayana, Green Chem., 2009, 11, 1503 RSC; (f) J. Nath, L. Jamir and B. K. Patel, Green Chem. Lett. Rev., 2011, 4, 1 CrossRef CAS; (g) A. R. Ali, H. Ghosh and B. K. Patel, Tetrahedron Lett., 2010, 51, 1019 CrossRef CAS; (h) R. Yella, N. Khatun, S. K. Rout and B. K. Patel, Org. Biomol. Chem., 2011, 9, 3235 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra and HRMS spectra. CCDC reference numbers 824583. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2ra00044j |
| This journal is © The Royal Society of Chemistry 2012 |