Role of temperature in [3+2]-cycloaddition of isoselenocyanates with oxiranes using BF3·Et2O†
Mani
Sengoden
and
Tharmalingam
Punniyamurthy
*
Department of Chemistry, Indian Institute of Technology Guwahati, Guwahati, 781039, India. E-mail: tpunni@iitg.ernet.in; Fax: +91 0361 2690762; Tel: +91 0361 2582309
First published on 27th January 2012
Abstract
The [3+2]-cycloaddition reaction of isoselenocyanates with oxiranes using BF3·Et2O is temperature dependent affording generally either substituted 2-imino-1,3-oxaselenolanes (−5 °C) or 1,3-oxazolidinones (40 °C) in short reaction times with high yields.
The [3+2]-cycloaddition reaction of heterocumulene with oxirane provides a powerful tool for the construction of five-membered heterocycles that are important in synthetic organic chemistry as well as in biological and medicinal sciences.1,2 Among them, the reactions of carbodiimide, isocyanate and isothiocyanate with oxirane have been studied considerably.3–5 In contrast, the reaction of the analogue isoselenocyanate6 with oxirane is yet to be investigated, which could lead to a straightforward route for the construction of polysubstituted 1,3-oxaselenolanes.7 In a continuation of our studies on carbon–heteroatom bond formation,8 we report herein a new temperature dependent selective synthesis of substituted 2-imino-1,3-oxaselenolanes (−5 °C) and 1,3-oxazolidinones (40 °C) from isoselenocyanates and oxiranes using BF3·Et2O with excellent regioselectivity. The method is efficient and affords a straightforward route for the construction of the target heterocyclic compounds in short reaction times. To the best of our knowledge, this is the first example of the temperature dependent selective cycloaddition reaction of heterocumulene with oxiranes as well as for the formation of 1,3-oxazolidinones from isoselenocyanates and oxiranes.
First, optimization of the reaction conditions was carried out with phenyl isoselenocyanate 1a and styrene oxide 2a as the model substrates using different Lewis acids and solvents at varied temperatures (Table 1). As anticipated, the protocol was effective and interestingly, afforded selectively either Z-phenyl-2-imino-1,3-oxaselenolane 3a (−5 °C) or 3,4-diphenyloxazolidin-2-one 4a (40 °C) as the sole product, depending on the temperature, with excellent regioselectivity. Recrystallization of the compound 4a in hexane gave crystals whose structure was confirmed by single crystal X-ray analysis (Fig. 1). Among the Lewis acids screened, BF3·Et2O, SnCl4, InCl3 and FeCl3, all were active, and BF3·Et2O yielded the best results. CH2Cl2 was found to be the solvent of choice, while toluene and CH3CN exhibited moderate to good results. In contrast, THF and 1,4-dioxane yielded inferior results. Using 10 mol% of BF3·Et2O the target molecules could be obtained in quantitative yield. The reaction at −5 °C gave complete conversion with 1.5 equiv. of styrene oxide, while the process at 40 °C required 2 equiv. of styrene oxide to give the best results. Control experiments for both processes confirmed that no reaction was observed without the Lewis acid.

| Entry | Catalyst | Solvent | Time (h) | Conv. (%)a | |
|---|---|---|---|---|---|
| 3a b | 4a c | ||||
| a Determined by 400 MHz 1H NMR. b Isoselenocyanate 1a (0.5 mmol), oxirane 2a (0.75 mmol) and catalyst (10 mol%) were stirred at −5 °C in solvent (1 mL) under a N2 balloon. c Reactions were carried out with 2 equiv. of oxirane 2a at 40 °C. d BF3·Et2O (5 mol%) was used. e 1 equiv. of oxirane 2a was used. f Catalyst was not used. n.d. = not detected. | |||||
| 1 | BF3·Et2O | CH2Cl2 | 0.5 | 98 | 92 |
| 2 | SnCl4 | CH2Cl2 | 3 | 60 | 53 |
| 3 | Cu(OTf)2 | CH2Cl2 | 8 | n.d. | — |
| 4 | Sc(OTf)3 | CH2Cl2 | 18 | 70 | 68 |
| 5 | InCl3 | CH2Cl2 | 3 | 65 | 52 |
| 6 | FeCl3 | CH2Cl2 | 3 | <5 | — |
| 7 | BF3·Et2O | CHCl3 | 0.5 | 90 | 85 |
| 8 | BF3·Et2O | THF | 1 | <3 | — |
| 9 | BF3·Et2O | Toluene | 1 | 55 | 41 |
| 10 | BF3·Et2O | CH3CN | 1 | 90 | 79 |
| 11 | BF3·Et2O | 1,4-Dioxane | 1 | <3 | — |
| 12d | BF3·Et2O | CH2Cl2 | 0.5 | 78 | 71 |
| 13e | BF3·Et2O | CH2Cl2 | 0.5 | 80 | — |
| 14f | — | CH2Cl2 | 24 | n.d. | n.d. |
 | ||
| Fig. 1 ORTEP diagram of (Z)-4-iodo-N-(4-phenyl-1,3-oxaselenolan-2-ylidene)benzenamine 3f and 3,4-diphenyloxazolidin-2-one 4a. Thermal ellipsoids are drawn at a 40% probability level. Hydrogen atoms have been omitted for clarity. | ||
With optimized conditions in hand, the scope of the procedure was next pursued. The reactions of a series of aryl and cyclohexyl isoselenocyanates 1b–l with oxirane 2a were examined (Table 2). Aryl isoselenocyanates having electron donating as well as -withdrawing substituents on the aromatic ring readily proceeded in the reactions to give the corresponding target heterocycles in good to high yield. For example, aryl isoselenocyanates 1b–h having 2-Cl, 3-F, 4-Cl, 4-F, 4-I, 4-OMe and 4-Me substituents on the aromatic ring proceeded in the cycloaddition reactions with 2a to give the heterocyclic compounds 3b–h in 42–92% yield (−5 °C) and 4b–h in 61–84% yield (40 °C). Compound 3f in hexane gave crystals whose structure was confirmed by X-ray analysis (Fig. 1).

| Entry | Isoselenocyanate | R1 | Product (yield, %)c | |
|---|---|---|---|---|
| −5 °Ca | 40 °Cb | |||
| a Reaction conditions: isoselenocyanate 1a–l (0. 5 mmol), oxirane 2a (0.75 mmol) and BF3·Et2O (10 mol%) were stirred in CH2Cl2 at −5 °C for 0.5 h under N2. b Reactions were performed at 40 °C for 0.5 h under N2. c Isolated yield. n.d. = not detected. | ||||
| 1 | 1a | Ph | 3a (86) | 4a (73) |
| 2 | 1b | 2-ClC6H4 | 3b (42) | 4b (72) |
| 3 | 1c | 3-FC6H4 | 3c (76) | 4c (61) |
| 4 | 1d | 4-ClC6H4 | 3d (90) | 4d (75) |
| 5 | 1e | 4-FC6H4 | 3e (92) | 4e (65) |
| 6 | 1f | 4-lC6H4 | 3f (85) | 4f (84) |
| 7 | 1g | 4-MeOC6H4 | 3g (88) | 4g (68) |
| 8 | 1h | 4-MeC6H4 | 3h (78) | 4h (70) |
| 9 | 1i | 4-NO2C6H4 | 3i (n.d.) | 4i (n.d.) |
| 10 | 1j | 3,4-DiMeC6H3 | 3j (83) | 4j (78) |
| 11 | 1k | 1-Naphthyl | 3k (87) | 4k (71) |
| 12 | 1l | Cyclohexyl | 3l (90) | 3l (90) |
Under these conditions, aryl isoselenocyanate 1i with a 4-NO2 substituent underwent no cycloaddition reaction. However, the disubstituted aryl isoselenocyanate 1j with 3,4-dimethyl groups underwent reaction with 2a to provide the heterocycles 3i and 4i in 83% and 78% yield, respectively. Similarly, 1-naphthyl isoselenocyanate 1k proceeded in reactions with 2a to afford the heterocycles 3k and 4k in 87% and 71% yield respectively. In contrast, cyclohexyl isoselenocyanate 1l, at both the reaction temperatures (−5 and 40 °C), gave 3l as the sole product.
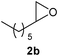
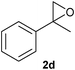
Finally, the reactions of substituted oxiranes 2b–f with phenyl isoselenocyanate 1a were studied (Table 3). As above, the reaction was efficient and the product formation was dependent on the nature of the oxirane. For example, n-octene-1,2-oxide 2b and epichlorohydrin 2c proceeded in reactions with 1a to give the heterocyclic compounds 3m–n in 79–86% yield (−5 °C) and 4l–m in 71–73% yield (40 °C). In contrast, the reactions of α-methylstyrene oxide 2d, 4-acetoxystyrene oxide 2e and cyclohexene oxide 2f with 1a at both the reaction temperatures (−5 and 40 °C) afforded selectively heterocycles 3o–q in 62–76% yield. Among them, the compound 3q afforded single crystals in hexane whose structure was determined by X-ray analysis (see ESI†). These results suggest that the reactions of isoselenocyanates with oxiranes are regioselective and the product formation depends on the reaction temperature and nature of the oxirane.











To reveal the mechanism, compound 3h was reacted with 1.1 equiv. of 2a and the heterocycle 4h, styrene and Se were obtained in 100% conversion along with a trace of phenyl acetaldehyde9 (Scheme 1). This result suggests that the oxirane 2 first undergoes reaction with isoselenocyanate 1 in the presence of the Lewis acid to give the heterocycle 3 (at −5 °C) that could further react with oxirane 2 at 40 °C to give the heterocycle 4 along with alkene and Se. Thus, the reaction of the Lewis acid with oxirane 2 can lead to the formation of a secondary carbocation that could undergo reaction with the Se of the isoselenocyanate to afford the intermediate a (Scheme 2). The cyclization of a could lead to the formation of the heterocycle 3. At 40 °C, the compound 3 may undergo further reaction with oxirane 2 to give the intermediate4bb which could give the heterocycle 4, alkene and Se.
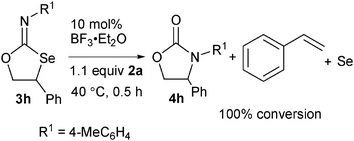 | ||
| Scheme 1 | ||
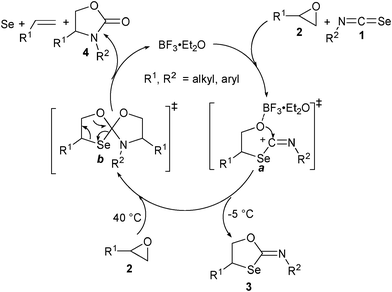 | ||
| Scheme 2 Proposed catalytic cycle. | ||
In summary, the [3+2]-cycloaddition reaction of isoselenocyanate with oxirane has been developed. The process is efficient, regioselective, temperature dependent and affords a straightforward route for the synthesis of 2-imino-1,3-oxaselenolanes and 2-oxazolidinones in short reaction times.
Acknowledgements
We thank the Department of Science and Technology, New Delhi, and the Council of Scientific and Industrial Research, New Delhi, for financial support.References
- (a) H. Ulrich, Cycloaddition Reactions of Heterocumulenes, Academic Press, New York, 1967 Search PubMed; (b) M. E. Dyen and D. Swern, Chem. Rev., 1967, 67, 197 CrossRef CAS; (c) S. Ozaki, Chem. Rev., 1972, 72, 457 CrossRef; (d) C. Larksarp and H. Alper, J. Am. Chem. Soc., 1997, 119, 3709 CrossRef CAS.
- For examples, see: (a) E. J. Brnardic, M. E. Fraley, R. M. Garbaccio, M. E. Layton, J. M. Sanders, C. Culberson, M. A. Jacobsen, B. C. Magliaro, P. H. Huston, J. A. O’Brien, S. L. Huszar, J. M. Uslaner, K. L. Fillgrove, C. Tang, Y. Kuo and S. M. Sur, Bioorg. Med. Chem. Lett., 2010, 20, 3129 CrossRef CAS; (b) J. A. Demaray, J. E. Thuener, M. N. Dawson and S. J. Sucheck, Bioorg. Med. Chem. Lett., 2008, 18, 4868 CrossRef CAS.
- I. Shibata, A. Baba, H. Iwasaki and H. Matsuda, J. Org. Chem., 1986, 51, 2177 CrossRef CAS.
- For examples, see: (a) B. M. Trost and R. Hurnaus, Tetrahedron Lett., 1989, 30, 3893 CrossRef CAS; (b) K. Yano, N. Amishiro, A. Baba and H. Matsuda, Bull. Chem. Soc. Jpn., 1991, 64, 2661 CrossRef CAS; (c) B. M. Trost and A. R. Sudhakar, J. Am. Chem. Soc., 1987, 109, 3792 CrossRef CAS; (d) C.-Q. Sun and D. H. Rich, Tetrahedron Lett., 1988, 29, 5205 CrossRef CAS.
- (a) Y. Xie, X. Chen and W. Su, J. Chem. Res., 2009, 129 CrossRef; (b) J.-Y. Wu, Z.-B. Luo, L.-X. Dai and X.-L. Hou, J. Org. Chem., 2008, 73, 9137 CrossRef CAS.
- (a) A. K. Sharma, A. Sharma, D. Desai, S. V. Madhunapantula, S. J. Huh, G. P. Robertson and S. Amin, J. Med. Chem., 2008, 51, 7820 CrossRef CAS; (b) G. W. Gokel, R. P. Widera and W. P. Weber, Org. Synth., 1988, Coll. 6, p. 232 Search PubMed; (c) J. Zakrzewski and M. Krawczyk, Phosphorus, Sulfur Silicon Relat. Elem., 2009, 184, 1880 CrossRef CAS; (d) G. L. Sommen, A. Linden and H. Heimgartner, Eur. J. Org. Chem., 2005, 3128 CrossRef CAS; (e) D. R. Garud, M. Koketsu and H. Ishihara, Molecules, 2007, 12, 504 CrossRef CAS.
- (a) D. R. Garud, M. Makimura, H. Ando, H. Ishihara and M. Koketsu, Tetrahedron Lett., 2007, 48, 7764 CrossRef CAS; (b) J. Castilla, I. Marin, M. I. Matheu, Y. Diaz and S. Castillon, J. Org. Chem., 2010, 75, 514 CrossRef CAS.
- (a) M. M. Guru, M. A. Ali and T. Punniyamurthy, Org. Lett., 2011, 13, 1194 CrossRef CAS; (b) P. Saha, M. A. Ali, P. Ghosh and T. Punniyamurthy, Org. Biomol. Chem., 2010, 8, 5692 RSC; (c) P. Saha, T. Ramana, N. Purkait, M. A. Ali, R. Paul and T. Punniyamurthy, J. Org. Chem., 2009, 74, 8719 CrossRef CAS.
- (a) H. O. House, J. Am. Chem. Soc., 1955, 77, 3070 CrossRef CAS; (b) S. M. Naqvi, J. P. Horwitz and R. Filler, J. Am. Chem. Soc., 1957, 79, 6283 CrossRef CAS; (c) K. Maruoka, N. Murase, R. Bureau, T. Ooi and H. Yamamoto, Tetrahedron, 1994, 50, 3663 CrossRef CAS; (d) R. Sudha, K. M. Narasimhan, V. G. Saraswathy and S. Sankararaman, J. Org. Chem., 1996, 61, 1877 CrossRef CAS; (e) B. C. Ranu and U. Jana, J. Org. Chem., 1998, 59, 8212 CrossRef; (f) K. A. Bhatia, K. J. Eash, N. M. Leonard, M. C. Oswald and R. S. Mohan, Tetrahedron Lett., 2001, 42, 8129 CrossRef CAS; (g) I. Karame, M. L. Tommasino and M. Lemaire, Tetrahedron Lett., 2003, 44, 7687 CrossRef CAS; (h) A. Procopio, R. Dalpozzo, A. De Nino, M. Nardi, G. Sindona and A. Tagarelli, Synlett, 2004, 2633 CrossRef CAS; (i) K. Suda, K. Baba, S. Nakajima and T. Takanami, Tetrahedron Lett., 1999, 40, 7243 CrossRef CAS; (j) K. Suda, K. Baba, S. Nakajima and T. Takanami, Chem. Commun., 2002, 2570 RSC; (k) T. Takanami, R. Hirabe, M. Ueno, F. Hino and K. Suda, Chem. Lett., 1996, 1031 CrossRef CAS; (l) E. Erturk, M. Gollu and A. S. Demir, Tetrahedron, 2010, 66, 2373 CrossRef CAS; (m) M. W. C. Robinson, K. S. Pillinger and A. E. Graham, Tetrahedron Lett., 2006, 47, 5919 CrossRef CAS; (n) M. W. C. Robinson, K. S. Pillinger and A. E. Graham, Tetrahedron Lett., 2010, 66, 8377 CAS; (o) M. W. C. Robinson, A. M. Davies, R. Buckle, I. Mabbet, S. H. Taylor and A. E. Graham, Org. Biomol. Chem., 2009, 7, 2559 RSC; (p) K. Maruoka, T. Ooi and H. Yamamoto, J. Am. Chem. Soc., 1989, 111, 6431 CrossRef CAS; (q) K. Ishihara, N. Hanaki and H. Yamamoto, Synlett, 1995, 721 CrossRef CAS; (r) K. Suda, T. Kikkawa, S. Nakajima and T. Takanami, J. Am. Chem. Soc., 2004, 126, 9554 CrossRef CAS; (s) K. Suda, S. Nakajima, Y. Satoh and T. Takanami, Chem. Commun., 2009, 1255 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and NMR spectra (1H and 13C). CCDC 843126 (3q), 843127 (4a) and 843128 (3f). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2ra00042c |
| This journal is © The Royal Society of Chemistry 2012 |