Evaluation of porous carbon substrates as catalyst supports for the cathode of direct methanol fuel cells
Ashley D.
Moore
,
Stuart M.
Holmes
and
Edward P. L.
Roberts
*
School of Chemical Engineering and Analytical Science, The University of Manchester, Oxford Road, Manchester, M13 9PL, United Kingdom. E-mail: edward.roberts@manchester.ac.uk; Fax: +44 (0) 161 306 9321; Tel: +44 (0) 161 306 9320
First published on 22nd December 2011
Abstract
This paper presents an investigation into the effect of the cathode catalyst substrate morphology on the performance of supported catalysts for direct methanol fuel cells (DMFC). Two porous carbons were synthesized by a sacrificial templating method, one using disordered, macroporous silica diatomaceous earth, the other using ordered, mesoporous SBA-15. The Pt particles deposited onto the synthesized mesoporous substrate and commercial Vulcan XC-72 were shown to have fairly uniform dispersion, with particle size in the range 2–3 nm, and high catalyst surface areas. In situ single cell DMFC testing showed that the Pt catalyst supported on carbon based on diatomaceous earth (Pt/C–Celatom) exhibited the lowest cell impedance, which may be attributed to the large pores, which facilitate oxygen transport, and to the low porosity, which enhances protonic and electronic conductivity of the catalyst layer. In DMFC polarization tests, the Pt/C–Celatom also exhibited the highest power output of the tested catalysts, with a peak power density 28% better than the current commercial standard, Vulcan XC-72.
1. Introduction
In recent years, polymer electrolyte fuel cells (PEMFCs) have garnered attention as alternate power sources for both portable and stationary applications thanks to their low operating temperature and low emissions at the point of use. Direct methanol fuel cells (DMFC) in particular have been considered for use in portable and vehicular technologies, due to their high specific energy densities when compared to competing battery technologies.1 Despite the multitude of research done on PEMFCs and DMFCs, the commercial applications of these technologies are still limited. One of the major barriers to fuel cell commercialization is the cost of the membrane electrode assembly (MEA), which is dominated by the cost of the precious metal catalyst required on the electrode. Commonly, platinum particles are distributed onto a high surface area conductive support, usually carbon black, which can be used to increase the available catalyst surface area, thereby increasing the catalytic activity and decreasing the necessary loading. Not only do the size and dispersion of the catalyst particles affect the catalytic performance, but the structure of the catalyst support can affect both the catalytic activity and the transport processes within the MEA.2–4There has been a great deal of research devoted to improving fuel cell performance by changing the microstructure of the catalyst support, which needs to be optimized in order to maximize the efficiency of multiphase transport. Materials with three-dimensional interconnected structure have demonstrated improved transport characteristics in fuel cell conditions.5,6 These materials are preferred over two-dimensional structures, as they tend to be more tolerant to pore blockage.7–9
Microporous materials (pore sizes < 2 nm diameter) are desirable for many applications, due to their high specific surface areas and large pore volumes. There are major drawbacks in using microporous materials, namely the slow mass transport of molecules due to the small pores, lack of accessibility of electrolyte and reduced catalyst utilization.7,10 Macroporous materials (pore sizes > 50 nm diameter), on the other hand, offer excellent mass transport characteristics, but relatively smaller surface areas and, often, high electrical resistance.11 Mesoporous carbons (2 nm < pore sizes > 50 nm) have shown many desirable characteristics for fuel cell use, as they allow access to the electrolyte and reagents while also exhibiting large surface areas and conductivities.12 Vulcan XC-72 is a commonly used material due to its high availability and low cost.13 Vulcan is also the most conductive of commercially available furnace blacks, exhibits high chemical stability in the fuel cell environment, and has a relatively large surface area.14,15 It is made of near-spherical particles of graphite, typically 30–60 nm in diameter.16 The porosity of the carbon comes about by the three dimensional agglomeration of the small particles into branched chains and clusters of around 250 nm. Particle size and distribution determine the surface area and porosity of the material. Smaller pores in the size distribution can be attributed to spaces between primary particles within the agglomerate, while the larger are due to the distance between agglomerates.2
A range of different materials have been synthesized and evaluated for fuel cell performance, such as carbon nanotubes and nanofibers,17carbon gels,18 conductive diamonds,11 templated ordered carbons,19carbon nanohorns and nanocoils, and mesocarbon microbeads.20 There are many different carbon materials that can be used as support materials in DMFC, and many parameters to consider when choosing one for fuel cell catalysis. Ideally, the support material would be cost-effective for scale-up, requiring no expensive reagents or equipment for synthesis. From this perspective, templated carbons have an advantage over other materials.
Commonly used templates often require expensive chemical reagents and, most importantly, time for synthesis and characterization before the carbon material can be made. Diatomaceous earth is an abundant naturally occurring mineral that can be milled, consisting mostly of mineralized diatoms with hollow, cylindrical silica structure of similar size.21 Examples of SEM images showing the structure of the mineralised diatom material used in this study (Celatom® FW-80 supplied by Eagle Picher) are shown in Fig. 1. The average length of each particle was 20 μm with an outer diameter of 10–15 μm, inner diameter 4–6.5 μm, and an array of pores 0.5–0.75 μm in diameter in the silica wall.
 | ||
| Fig. 1 SEM images of particles of the diatomaceous earth Celatom® FW-80 (Eagle Picher) as supplied, showing dimensions of individual structures. | ||
Diatomaceous earth is an abundant, low-cost silica that can be used as a template for carbon synthesis, without requiring template synthesis or characterization.22 In this study, a macroporous carbon templated from diatomaceous earth was evaluated for DMFC use, and the effect of the cathode catalyst substrate morphology was investigated when compared to a regular mesoporous templated carbon structure (CMK-3) and the commercial standard, Vulcan XC-72.
2. Experimental
2.1 Preparation of carbon substrates
The mesoporous silica template SBA-15 was synthesized according to the method reported by Zhao et al.23 In this procedure, tetraethylorthosilicate (TEOS 98%, Aldrich) was used as the silica source, and triblock copolymer Pluronic P123 (EO20PO70EO20, Aldrich) was used to create the surfactant template, and the reaction was carried out under acidic condition using hydrochloric acid (HCl 37%, Sigma Aldrich). The molar composition of the mixture was 1 TEOS![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.018 P123:16.33 HCl:183 H2O.24
:0.018 P123:16.33 HCl:183 H2O.24
Typically, the reagents were mixed in a flask and stirred, and then the mixture was heated at 45 °C for 24 h, then at 100 °C for a further 24 h. The sample was allowed to cool and the solid product was then filtered, without washing, and dried at 45 °C. The material was then calcined under static air at 550 °C for 5 h in order to decompose the template. The end product was characterized by powder X-ray diffraction (XRD, Philips Analytical X-Pert X-ray Diffractometer).
The CMK-3 mesoporous carbon was replicated from the template synthesized as above using a technique developed by Ryoo et al.,24viasucrose impregnation and decomposition. Briefly, the SBA-15 silica template was mixed in water with sucrose as a carbon precursor and H2SO4. The sucrose infiltration was done incrementally in two steps by incipient wetness, drying at 100 °C for 6 h and at 160 °C for six hours in between steps in order to fill the pores of the SBA-15 completely.
A macroporous carbon was synthesized in a similar manner, using diatomaceous earth (Celatom® FW-80, Eagle-Picher) as a silica template, according to the procedure described by Holmes et al.22 The pore volume of the Celatom was filled incrementally with three infiltrations, in order to optimize the carbon structure and to prevent overfilling of the pores. The carbonaceous inverse of the silica template is denoted as C–Celatom.
In each case, the silica–sucrose mixture was carbonized by pyrolysis under nitrogen gas at 900 °C for four hours. After pyrolysis, the resulting carbon–silica composite was refluxed with 1 M NaOH in 1:1 ethanol:water solution at 90 °C for 24 h, in order to dissolve the silica template completely. The carbon sample was filtered and washed with a hot solution of 1:1 EtOH:H2O. The template-free carbon was then dried at 45 °C overnight.
Following the silica removal step, the resulting carbonaceous Celatom (C–Celatom) was characterized by visual inspection of the material by scanning electron microscopy (SEM, FEI Quanta 200 Environmental) following silica template removal. The result was a macroporous carbon with surface area around 300 m2 g−1. The structure of the CMK-3 was confirmed by XRD.
2.2 Preparation of Pt/C catalysts and determination of Pt loading
Pt deposition was carried out by identical procedures on three different carbon substrates, using a colloidal sodium alkoxide reduction method.25 Three electrocatalysts were prepared: Pt/C–Celatom, Pt/CMK-3 and Pt/Vulcan XC-72 were obtained by deposition onto the macroporous carbon templated from Celatom, the mesoporous carbon CMK-3, and microporous carbon black Vulcan XC-72 (Fuel Cell Store), respectively. Samples were examined by transmission electron microscopy (TEM, FEI TECNAI G2) to determine the particle size on CMK-3 and Vulcan XC-72, as the structure of the C–Celatom was too thick to allow for TEM imaging. The average Pt particle size and estimated catalyst surface area was estimated from XRD.In each case, the amount of Pt precursor was calculated to obtain a catalyst with a final loading of 20 wt% of Pt. In order to determine the final Pt loading on the prepared catalyst, samples were digested by aqua regia and diluted in a 3.7% HCl matrix. The Pt concentration in the digested catalyst solution was determined by inductively coupled plasma optical emission spectroscopy (ICP-OES, Perkin-Elmer Optima 5300 ICP-AES). The dissolution of Pt from any carbon remaining after digestion was confirmed by EDAX.26
2.3 Electrochemical characterization
Electrochemical measurements were carried out using an Autolab PGSTAT30 Potentiostat/Galvanostat with a three-electrode cell, using a platinum rod and Ag/AgCl as counter and reference electrodes, respectively. The working electrode was a 0.5 mm diameter glassy carbon electrode (Pine Instruments), and was prepared by depositing dilute Pt/C ink onto the electrode surface using a micropipette and drying at 80 °C for 1 h in a vacuum oven. The electrochemically active surface areas (ESA) of the catalyst samples were determined by cyclic voltammetry (CV) performed in N2-purged 1M H2SO4 solution. The potential was swept in the range of 0 to 1.5 V versusstandard hydrogen electrode (SHE), at scan rates of 20, 50, and 100 mV s−1. The activity of the synthesized catalysts for the oxygen reduction reaction (ORR) was determined by linear sweep voltammetry in air-saturated 1M H2SO4, from 1.0 to 0 V versusSHE at a scan rate of 1 mV s−1.2.4 Single cell DMFC testing
Cathode catalyst inks were prepared by dispersing the prepared Pt/C catalysts in acetone with Nafion® ionomer solution (5 wt% dispersion, Aldrich). Anode catalyst ink was prepared by dispersing commercial 60 wt% Pt:Ru on Vulcan XC-72 (E-TEK) in acetone with Nafion® solution. A loading of 1 mg Pt cm−2 was sprayed onto each electrode and followed by a coating of 1 mg cm−2 of Nafion® solution. All prepared cathodes were identical in Nafion® content and platinum loading. The MEA was fabricated by hot-pressing the fabricated electrodes on either side of a pre-treated Nafion® 117 membrane.
Electrochemical impedance spectroscopy (EIS) was carried out using an Autolab PGSTAT30 Potentiostat/Galvanostat at 50 °C using a frequency range from 50 kHz to 10 mHz under galvanostatic cell control. EIS, as well as polarization results, were obtained under real cell conditions, with 5 mL min−1 of 1M methanol flow at the anode and 1 L min−1 of air flow at the cathode. Polarization data was obtained at a cell temperature of 90 °C.
3. Results and discussion
3.1 Scanning electron microscopy (SEM) observations
Fig. 2 shows SEM images of the C-Celatom, showing the structure obtained from the templating of the diatomaceous earth. Based on the internal structure of the Celatom particles, different carbon structures can be obtained, from simple cylindrical particles, to more complex torus shapes. These particles are 10–20 μm long, with diameters of 5–10 μm. One notable detail is that the surfaces of the C-Celatom particles have small protrusions that arise from the pores in the Celatom silica walls. These increase the roughness of the carbon and the overall surface area.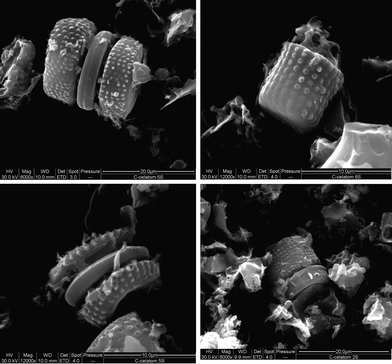 | ||
| Fig. 2 SEM images of the C–Celatom obtained from the templating procedure | ||
Table 1 shows the specific surface areas of the investigated carbon substrates and their pore characteristics. Vulcan XC-72 has a broad pore size distribution including both micro- (< 2 nm) and mesopores (2–40 nm). CMK-3 on the other hand has a narrow pore size distribution in the mesoporous range. The BET surface areas obtained for CMK-3 is consistent with the value of 1500 reported in the literature.28 C–Celatom has negligible mesoporosity and has macroporous characteristics.
| Vulcan XC-72a | CMK-3 | C–Celatom | |
|---|---|---|---|
| a N2 adsorption data (BET) for Vulcan XC-72 obtained from Liu and Craeger.27 b Pore volume and diameter of C–Celatom obtained using mercury porosimetry. | |||
| BET surface area (m2 g−1) | 237 | 1320 | 312 |
| Total pore volume (cm3 g−1) | 0.62 | 1.05 | 0.36b |
| Mean pore diameter | 10.4 nm | 3.2 nm | 2.5 μmb |
| Pore characteristic | Micro/meso | Meso | Macro |
3.2 Pt nanoparticle characterization on fabricated Pt/C
XRD patterns were obtained for each of the synthesized catalysts and, using the Scherrer equation on data from the (220) peak of the platinum fcc crystal lattice, the average particle size of the Pt deposited on each substrate was estimated.29 These average particle sizes calculated from the XRD data are shown in Table 2. The average particle sizes from TEM were 2.1 nm and 3.1 nm for Pt/Vulcan XC-72 and CMK-3, respectively, which agree well with the results obtained from XRD analysis. The catalyst specific surface area, S, was estimated assuming uniform spherical particles as follows: | (1) |
TEM was also used to compare the distribution and shape of the particles on the substrate surfaces. Fig. 3 shows TEM images of the Pt/CMK-3 and Pt/Vulcan XC-72, respectively. The TEM images show that spherical particles of Pt, 2–3 nm in diameter, were deposited onto the substrates, and that the dispersion was fairly uniform over the substrate surface. The small particle sizes, high catalyst specific surface areas, and even dispersion indicate that the fabricated Pt/C catalysts were suitable for fuel cell testing.
 | ||
| Fig. 3 TEM images of a) Pt/CMK-3 and b) Pt/Vulcan XC-72 at two different magnifications. | ||
Cyclic voltammograms (CVs), shown in Fig. 4, were obtained at room temperature in N2-purged 1M H2SO4 solution. The electrochemically active surface area was determined from the area under the hydrogen desorption region of each voltammogram, the scan rate, the platinum loading and the charge for H2 adsorption on a smooth platinum surface (210 μC cm−2Pt).31 The calculated electrochemically active specific surface areas are shown in Table 3.
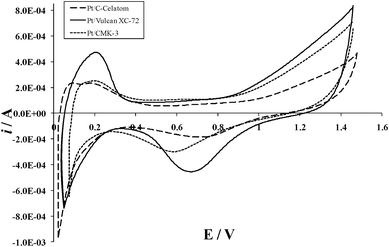 | ||
| Fig. 4 Cyclic voltammograms of synthesized catalysts at a scan rate of 20 mV s−1. | ||
The platinum utilization ratio was determined from the ratio of the active specific surface area, determined by CV, to the catalyst specific surface area, estimated from XRD (Table 2), and is also reported in Table 3. These results show a significant discrepancy between the total and active surface areas for the Nafion® impregnated samples, with a surface area activity loss of 75–85%. This loss may be attributed to inhibited access of the Nafion® electrolyte to the catalyst surface, pore blockage from particle agglomeration in the bulk, and the inaccessibility of protons to the platinum surfaces between the platinum crystallite and the carbon substrate.32 Inaccuracy in the catalyst specific surface area measurement (estimated from XRD data) could also contribute to the low utilization values. The method of calculation was based on the assumption of a homogenous distribution of spherical catalyst particles, with accurate average diameter. Any significant agglomeration, differently shaped particles or wide particle size distribution can have a considerable effect on the specific surface area, and thus on the utilization.
3.3 DMFC Performance of Pt/C catalysts
The electrocatalytic performance of the cathode in an operating fuel cell depends on many factors, not only the electrochemical surface area of the catalyst. Factors which will affect the performance in the fuel cell will include:•the oxygen concentration distribution in the cathode (which is associated with mass transport effects and depends on the substrate structure, porosity etc);
•the potential distribution in the catalyst layer (which will be effected by the substrate conductivity);
•and the distribution and depolarisation effects of methanol cross-over (which again will be influenced by mass transport effects associated with the substrate).
The performance of the catalysts was evaluated in situ using a single cell DMFC. The impedance effects of the fabricated MEAs were examined by EIS. Fig. 5 shows the Nyquist impedance plots obtained for the three MEAs prepared using the Pt/C–Celatom, Pt/CMK-3 and Pt/Vulcan XC-72 cathode electrocatalysts at a cell temperature of 50 °C and an operating current density of 15 mA cm−2. As the same anode and membrane material were used in each case, the differences in impedance behavior should only be due to the differences in the cathode catalyst substrate morphology. There is a large effect of substrate morphology on both real and imaginary impedance, evidenced by the large differences in the impedance curves. The ohmic and total resistances of the MEAs obtained from the impedance data are shown in Table 4.
 | ||
| Fig. 5 Nyquist plots for the MEAs used in this study at 50 °C and 15 mA cm−2. | ||
| R Ω (Ω) | R C (Ω) | OCV (V) | Peak power (mW cm−2) | |
|---|---|---|---|---|
| Pt/C–Celatom | 0.013 | 0.109 | 0.565 | 35.5 |
| Pt/CMK-3 | 0.013 | 0.461 | 0.603 | 29.0 |
| Pt/Vulcan XC-72 | 0.017 | 0.357 | 0.641 | 27.8 |
The ohmic resistances of the MEAs (RΩ) were determined from the high frequency intercepts of the impedance curves with the real (Z′) axis, where the phase shift is equal to zero.33 The total resistance of the MEA, which includes the anodic and cathodic charge transfer and mass transport resistances, was determined as the difference between the high and low frequency Z′-intercepts on the Nyquist plot.34
Based on the impedance spectroscopy data, it can be seen that the C–Celatom MEA exhibits the lowest cell resistance by a significant margin. The low frequency arc is associated with the cathodic mass transfer resistance, which is small for both Pt/C–Celatom and Pt/Vulcan XC-72, but much larger for the Pt/CMK-3. The differences in impedance might be attributed to the differences in porosity of the catalyst substrates. The effective protonic and electronic conductivity of a catalyst layer decreases quickly with increasing substrate porosity.35 Overall, the C–Celatom demonstrates the lowest substrate porosity,22 followed by Vulcan XC-72,35 and then CMK-3.10 The low porosity and the large pore size, which facilitates reagent transport in the catalyst layer and accelerates the ORR, explain the low impedance of the Pt/C–Celatom.
DMFC polarization and power density curves of the MEAs with Pt/C–Celatom, Pt/CMK-3, and Pt/Vulcan XC-72 as cathode catalysts are shown in Fig. 6. The open circuit voltages and peak power densities achieved are shown in Table 4. The power densities obtained for Pt/Vulcan XC-72 are typical for DMFC operating under similar conditions.36
 | ||
| Fig. 6 Power density curves for different electrocatalyst samples, at 90 °C with flow rates of 5 mL min−1 1M MeOH and 1 L min−1 air. | ||
Despite the low open circuit performance, the highest power density, 35.5 mW cm−2, was demonstrated for the Pt/C–Celatom MEA. Conversely, the Pt/CMK-3 exhibits the highest open-circuit performance, but the lowest peak power density, at 27.8 mW cm−2.
Inspection of the results presented in Fig. 6 show that the largest differences in performance are evident in the ‘ohmic’ region of the polarization curve. Comparison of the linear portion of the graphs shows that the Pt/C–Celatom exhibits the lowest cell resistance in this region, followed by the Pt/Vulcan XC-72, and the Pt/CMK-3 has the highest resistance by a noticeable margin. This result is consistent with the impedance results presented in Fig. 5 and Table 4.
The differences in the performance of the MEAs using the three different catalyst support materials may be due to a number of complex interactions. One possibility is that the low porosity C–Celatom support may reduce the effect of methanol crossover by reducing methanol transport and increasing the pressure gradient in the catalyst layer.37 However, the higher OCV observed with the Vulcan XC-72 and CMK-3 catalyst supports indicates lower methanol crossover at open circuit. This effect is lost at increasing cell polarization probably due to the ion and oxygen transport processes in the cathode catalyst layer.38 The differences in DMFC performance are thus most likely to be influenced by differences in the oxygen and ion transport in the cathode catalyst layer, brought about by changes in the structure of the triple phase boundary with the different support materials.
4. Conclusions
In this paper an investigation of the effect of the porous cathode catalyst substrate morphology on catalytic activity and DMFC performance has been discussed. Two templated carbons were synthesized—C–Celatom, a macroporous carbon based on diatomaceous earth, and CMK-3, an ordered mesoporous carbon negative of SBA-15. Electrocatalysts were synthesized from these templated substrates, as well as a commercial substrate, by a colloidal platinum deposition technique. Fabricated catalysts exhibited even dispersions of small, uniformly sized Pt particles with high surface areas.In situ DMFC single cell testing showed that the cathode substrate morphology has a significant effect on the both the EIS and polarization performance under normal cell conditions. The Pt/C–Celatom exhibited the lowest ohmic and overall cell impedances, followed by Pt/Vulcan XC-72. This was most likely due to the differences in porosity of the tested substrate materials. Under the conditions studied, the macroporous C–Celatom exhibited the best cathode substrate structure, both in terms of impedance and power density performance. This indicates the viability of Pt/C–Celatom as a low-cost, high activity material for DMFC catalysis.
Acknowledgements
The financial support from the North American Foundation for the University Of Manchester (NAFUM) is gratefully acknowledged.References
- S. K. Kamarudin, F. Achmad and W. R. W. Daud, Int. J. Hydrogen Energy, 2009, 34, 6902–6916 CrossRef CAS.
- M. Uchida, Y. Aoyama, N. Eda and A. Ohta, J. Electrochem. Soc., 1996, 143, 2245–2252 CrossRef CAS.
- M. Uchida, Y. Aoyama, M. Tanabe, N. Yanagihara, N. Eda and A. Ohta, J. Electrochem. Soc., 1995, 142, 2572–2576 CrossRef CAS.
- E. Auer, A. Freund, J. Pietsch and T. Tacke, Appl. Catal., A, 1998, 173, 259–271 CrossRef CAS.
- S. H. Joo, S. J. Choi, I. Oh, J. Kwak, Z. Li, O. Terasaki and R. Ryoo, Nature, 2001, 412, 169–172 CrossRef CAS.
- Z.-Y. Yuan and B.-L. Su, J. Mater. Chem., 2006, 16, 663 RSC.
- C. Liang, Z. Li and S. Dai, Angew. Chem., Int. Ed., 2008, 47, 3696–3717 CrossRef CAS.
- B. Fang, J. H. Kim, M. Kim and J.-S. Yu, Chem. Mater., 2009, 21, 789–796 CrossRef CAS.
- R. Ryoo, Nat. Chem., 2009, 1, 105–106 CrossRef CAS.
- V. Rao, P. A. Simonov, E. R. Savinova, G. V. Plaksin, S. V. Cherepanova, G. N. Kryukova and U. Stimming, J. Power Sources, 2005, 145, 178–187 CrossRef CAS.
- Y. Shao, J. Liu, Y. Wang and Y. Lin, J. Mater. Chem., 2009, 19, 46–59 RSC.
- L. F. Giraldo, B. L. Lopez, L. Perez, S. Urrego, L. Sierra and M. Mesa, Macromol. Symp., 2007, 258, 129–141 CrossRef CAS.
- E. Antolini, Appl. Catal., A, 2009, 88, 1–24 CrossRef CAS.
- Z. Qu, W. Huang, M. Cheng and X. Bao, J. Phys. Chem. B, 2005, 109, 15842–15848 CrossRef CAS.
- G. Hoogers, Fuel Cell Technology Handbook, CRC Press LLC, Boca Raton, 2003 Search PubMed.
- J. Bayer and S. Ergun, Carbon, 1967, 5, 107–108 CrossRef CAS.
- P. Serp, M. Corrias and P. Kalck, Appl. Catal., A, 2003, 253, 337–358 CrossRef CAS.
- J. Fricke and T. Tillotson, Thin Solid Films, 1997, 297, 212–223 CrossRef CAS.
- R. Ryoo, S. H. Joo, M. Kruk and M. Jaroniec, Adv. Mater., 2001, 13, 677 CrossRef CAS.
- S. Tang, G. Sun, J. Qi, S. Sun, J. Guo, Q. Xin and G. M. Haarberg, Chin. J. Catal., 2010, 31, 12–17 CrossRef CAS.
- EP Minerals, Celatom® Diatomaceous Earth Products: General Classification, http://www.epminerals.com/product_certifications/Celatom%20Diatomaceous%20Earth%20General%20Classification.pdf, 2011.
- S. M. Holmes, B. E. Graniel-Garcia, P. Foran, P. Hill, E. P. L. Roberts, B. H. Sakakini and J. M. Newton, Chem. Commun., 2006, 2662–2663 RSC.
- D. Zhao, Q. Huo, J. Feng, B. F. Chemlka and G. D. Stuckey, J. Am. Chem. Soc., 1998, 120, 6024–6036 CrossRef CAS.
- R. Ryoo, S. Joo and S. Jun, J. Phys. Chem. B, 1999, 103, 7743–7746 CrossRef CAS.
- United States Pat., 7557057, 2009 Search PubMed.
- D. Mirabile Gattia, M. V. Antisari, L. Giorgi, R. Marazzi, E. Piscopiello, A. Montone, S. Bellitto, S. Licoccia and E. Traversa, J. Power Sources, 2009, 194, 243–251 CrossRef CAS.
- B. Liu and S. Craeger, J. Power Sources, 2010, 195, 1812–1820 CrossRef CAS.
- R. Ryoo, S. H. Joo, S. Jun, T. Tsubakiyama and O. Terasaki, in Studies in Surface Science and Catalysis, Elsevier, 2001, vol. Volume 135, pp. 150–150 Search PubMed.
- V. K. Pecharsky and P. Y. Vavalij, Fundamentals of powder diffraction and structural characterization of materials, Springer Science + Business Media, Inc., New York, 2003 Search PubMed.
- P. Stonehart, J. Appl. Electrochem., 1992, 22, 995–1001 CrossRef CAS.
- J. Wu, X. Z. Yuan, H. Wang, M. Blanco, J. J. Martin and J. Zhang, Int. J. Hydrogen Energy, 2008, 33, 1735–1746 CrossRef CAS.
- G. Tamizhmani, J. P. Dodelet and D. Guay, J. Electrochem. Soc., 1996, 143, 18–23 CrossRef CAS.
- M. A. Danzer and E. P. Hofer, J. Power Sources, 2009, 190, 25–33 CrossRef CAS.
- F. Liu, B. Yi, D. Xing, J. Yu, Z. Hou and Y. Fu, J. Power Sources, 2003, 124, 81–89 CrossRef CAS.
- C. Y. Du, P. F. Shi, X. Q. Cheng and G. P. Yin, Electrochem. Commun., 2004, 6, 435–440 CrossRef CAS.
- Chan Lim, R. G. Allen and K. Scott, J. Power Sources, 2006, 162, 11–18 CrossRef.
- D. Ye, X. Zhu, Q. Liao, J. Li and Q. Fu, J. Power Sources, 2009, 192, 502–514 CrossRef CAS.
- T. V. Reshetenko, H.-T. Kim, H. Lee, M. Jang and H.-J. Kweon, J. Power Sources, 2006, 160, 925–932 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |