Printable thin film supercapacitors utilizing single crystal cobalt hydroxide nanosheets
Xiaobo
Ji
b,
Philip M.
Hallam
a,
Said M.
Houssein
a,
Rashid
Kadara
a,
Leiming
Lang
c and
Craig E.
Banks
*a
aSchool of Chemistry and the Environment, Division of Chemistry and Environmental Science, Manchester Metropolitan University, John Dalton Building, Chester St., Manchester, M1 5GD, UK. E-mail: c.banks@mmu.ac.uk; Fax: +44(0)1612476831; Tel: +44(0)1612471196 Web: www.craigbanksresearch.com
bCollege of Chemistry and Chemical Engineering and State Key Laboratory for Powder Metallurgy, Central South University, Changsha, 410083, China
cBiochemical and Environmental Engineering College, Nanjing Xiaozhuang University, Nanjing, 211171
First published on 19th December 2011
Abstract
Cobalt hydroxide (β-Co(OH)2) thin film supercapacitors are developed for the first time via screen printing and their potential application towards energy storage (as an electrode material within a supercapacitor) explored. The highly uniform, porous nanostructures were synthesized via a novel, hydrothermal, template-free approach. The nanostructures consist of amassing, arbitrarily layered, interconnecting nanosheets, resulting in a flower-like structure with many fissures. These nanostructures were fabricated into flexible thin film supercapacitors via screen printing for the first time. The β-Co(OH)2 asymmetric supercapacitor is found to deliver a maximum specific capacitance of ∼170 F g−1 at a current density of 0.5 μA in an aqueous electrolyte solution (3M KOH) retaining 99.69% of its maximum capacity over 600 cycles. Given the ability to mass produce such supercapacitors via screen printing, where readily, synthetically fabricated nanostructures can be incorporated, the application of this approach will likely be widely implemented.
1. Introduction
The rapidly growing market in portable electronic devices, electric vehicles and the ever worsening global warming issues call for not only urgent development of clean alternative energies and emission control of global warming gases, but also more environmentally friendly, high-performance energy-storage systems.1,2 Supercapacitors, also known as electrochemical capacitors or ultracapacitors,3,4 are electronic components that can be rapidly charged and discharged and relied upon to store energy reliably for long periods, offering transient but extremely high powers, and are probably the most important next generation energy storage device.5 Emerging as an ideal model, they have been touted as a solution to the mismatch between the fast growth in power required by devices and the inability of batteries in various applications which require transient but high/peak power pulses for the time-dependent usage.6The materials mostly used as electrochemical double layer capacitors (EDLC) are based on carboneous materials such as activated carbon, carbide-derived carbon, graphene and carbon nanotubes7–15 which have a large surface area for the charge to reside. Capacitors in the alternative class, called pseudocapacitors, utilise transition metal oxides or electrically conducting polymers as electrode materials.16 The storage mechanism in this case is the undergoing of fast surface faradaic reactions while the amount of charge held is proportional to the voltage.16 While supercapacitors benefit from the highest known power capability (10 kW kg−1), high current capability, ease of maintenance, and long-term cycling stability (> 106 cycles),6,16 their moderate energy density for the widespread deployment in everyday technology as clean and renewable energy media are still far away to meet the performances and cost requirements, in particular providing power boosts to start the engine or to assist acceleration.
To make capacitors commercially viable and become extensively used, the energy that they can store needs to increase significantly and hence to attain supercapacitors with sufficient energy for higher requirements in the future still remains very challenging. Extensive efforts have been devoted to improve the specific capacitance of supercapacitors by introducing pseudocapacitive metal oxides. Compared with the EDLC-based capacitors, pseudocapacitors based on transition metal oxide electrodes such as RuO2,5MnO2,17,18CoOx,19 and NiO20 are available for charge storage through faradaic or redox-type reactions because they can produce much higher capacitances than double-layer carbonaceous materials. Impressively, cobalt oxide is very attractive in view of its outstanding pseudocapacitive behaviour (theoretical capacitance, 3460 F g−1),21 practical availability, environmental compatibility and lower cost when compared to the state-of-the-art supercapacitor material RuO2. Despite its importance, efforts are still needed to further improve the practical use of cobalt materials in supercapacitors.
A key methodology to boost the specific capacitance of such electroactive cobalt-based materials is their morphological and/or chemical composition design at the nanometre-scale, because high surface-to-volume ratio with suitable pore sizes is desirable for the penetration of electrolytes and reactants into the whole electrode matrix, and can promote the electric double-layer capacitances and accommodate a large amount of superficial electroactive species to participate in faradaic redox reactions. Thus, considerable effort has been done to improve the electrochemical performances of β-Co(OH)2 by developing effective strategies to synthesize β-Co(OH)2 nanostructures particularly with more controllable and uniform sizes.
Also it should be noted that supercapacitors can be made flexible with sheet-like structures, which are light-weight. The fabrication method of the supercapacitors should be inexpensive and high-throughput, just like batteries. Various methods exist but are costly and expensive and not appropriate to be used to make millions of supercapacitors. State-of-the-art flexible thin-film supercapacitors based on sprayed networks of single-walled carbon nanotubes (SWCNTs)22 were fabricated, showing the performances of the devices with very high energy (6 Wh kg−1). The results underline the potential of printable thin film supercapacitors. Despite this scientific advance towards semi-printable/production supercapacitors, there have been limited reports using printing processes for fabricating flexible supercapacitors; to the best of our knowledge there have been no reports of using true screen printing despite its inherent advantages.22,23
Novel electrochemical designs can be readily fabricated via screen printing by designing and producing screens which derive the geometry and thickness of the screen printed material, usually graphite but can be applied to other novel materials.22,23 In this present paper we utilize screen printing as a new methodology to fabricating flexible and reproducible supercapacitors with high surface areas and minimal wasted space with respect to geometric arrangement and in terms of multiple printing. The advantage of this fabrication approach is that it can be readily scaled up and is economical which produces reproducible surface shapes and thicknesses, something which is lacking in this area. Thus both the resistance and weight of printing electrodes were reduced via eliminating the interface between the charge collectors and working electrodes, leading to higher power density.
We report a novel, hydrothermal, template-free approach towards the fabrication of flower-like β-Co(OH)2 produced from the aggregation of single crystal nanosheets structure, integrated into the working surface of a screen printed electrode. We explore the electrochemistry of the nanostructure, comprised of layered transition β-Co(OH)2, towards understanding how to increase faradaic capacitance. In doing so, we demonstrate enhanced pseudocapacitance, achieved using this unique structure of layered single crystalline β-Co(OH)2, is dependent on the ability to accommodate elevated numbers of electroactive ions and is consequently accessible to fast faradaic reactions.
2. Experimental
2.1 Materials
Cobalt(II) chloride hexahydrate (CoCl2·6H2O), hexamethylenetetramine (HMT), cetyltrimethylammonium bromide (CTAB) and monoethanolamine were purchased from Shanghai Chemical Co. LTD, China. All chemicals used were of analytical grade and were used as received without any further purification.2.2 Synthesis of Co(OH)2
Cobalt(II) chloride (CoCl2, 2 mmol) and HMT (2 mmol) were dissolved into distilled water (20 mL) including CTAB (1 mmol) under vigorous stirring. After the mixture became transparent, monoethanolamine (1 mL) was added and the resulting solution immediately transferred into a Teflon stainless steel autoclave. The autoclave was sealed and maintained at 100 °C for 24 h, and was then allowed to cool to room temperature. The resulting solid was then washed repeatedly with distilled water and finally dried at 80 °C in air. Note that the synthesis reported here is an adaptation from a previous literature report,24 but as will be shown, flower-like structures were observed rather than rod-like.2.3 Characterization
The morphologies were characterized using a scanning electron microscopy (SEM) (EDAX-4800) and transmission electron microscopy (TEM) (Japan JEOL JEM-200CX, transition electron microscope). The high-resolution TEM (HRTEM) images were obtained using a JEOL-2010 TEM at an acceleration voltage of 200 kV. The phase purity of the products was characterized by X-ray power diffraction (XRD), (Shimadzu XD-3A X-ray diffractometer with Cu α radiation, λ = 0.15417 nm). The BET (Brunauer-Emmett-Teller) surface area was measured by ASAP2020 (Micromeritics, United States). The X-ray photoelectron energy spectroscopy (XPS) was recorded on a VG ESCALAB MKII, using an accelerating voltage and working current of 12.5 kV and 20.0 mA, respectively.2.4 Screen printed electrodes
Voltammetric measurements were carried out using a μ-Autolab III (Eco Chemie, The Netherlands) potentiostat/galvanostat and controlled by Autolab GPES software. Screen printed electrodes (SPEs) were fabricated in-house with appropriate stencil designs using a microDEK 1760RS screen-printing machine (DEK, Weymouth, UK). A carbon-graphite ink formulation (C2000802D2, Gwent Electronic Materials Ltd, UK) was first screen printed onto a polyester flexible film (Autostat, 250 μm thickness) defining the carbon contacts, counter and working electrodes. This layer (5–10 microns in depth) was cured in a fan oven at 60 degrees for 30 min. Next a silver/silver chloride reference electrode was included by screen printing Ag/AgCl paste (Gwent Electronic Materials Ltd, UK) on to the plastic substrate. Last a dielectric paste ink (Gwent Electronic Materials Ltd, UK) was printed to cover the connections and define the 9 mm diameter graphite working electrode. After curing at 60 degrees for 30 min the screen printed electrode is ready to use. An example of a flexible screen printed electrode is shown in Fig. 1. The printed carbon material displays a heterogeneous rate constant of 1.7 × 10−3 cm s−1 as determined with potassium ferrocyanide/1 M KCl.23,25
Cobalt hydroxide screen printed electrodes were fabricated as described above with β-Co(OH)2 efficiently mixed into the ink formulation prior to screen printing. Note that in doing so, the rheology of the ink changes such that the careful addition of solvents is needed along with modification of the standard printing parameters. Increasing amounts of cobalt hydroxide were incorporated into the screen printed electrodes over the range of 10, 20 and 30% (MP/MI), where MP is the mass of particulate and MI is the mass of ink formulation used in the printing process. The printing process facilitated the homogenous coverage of the electrode surface by the cobalt hydroxide.
3. Results and discussion
3.1 Characterisation of capacitance material
X-ray diffraction (XRD) analysis (Fig. 2) was used to determine the chemical composition and the crystalline structure of the synthesised material over the range 15°≤ 2 θ ≤ 70° (2θ). The diffraction peaks show excellent conformity with the standard XRD pattern expected for β-Co(OH)2 (JCPDS: 74-1057), confirming the synthesized product is high purity hexagonal brucite-like β-Co(OH)2. The peak intensities of diffraction planes (001), (100) and (011) are significantly larger than those of the other diffraction peaks, not dissimilar to the standard XRD pattern produced by bulk β-Co(OH)2. The morphology and microstructure of the sample was observed by scanning electron microscopy (SEM) and Transmission electron microscopy (TEM). SEM imaging of the β-Co(OH)2 microflower at low magnification (Fig. 3A) demonstrates excellent uniformity throughout, with an average diameter of ∼10 μm. Upon magnification (Fig. 3B), SEM imaging reveals the β-Co(OH)2 microflower is fundamentally an accumulation of individual nanosheets, each displaying a thickness of ∼20 nm. The structure of the porous β-Co(OH)2 microflower was further investigated by TEM and high-resolution TEM (HRTEM). TEM analysis (Fig. 3C) of the β-Co(OH)2 nanosheet petal, indicates that several nanosheets are overlapped together. It further demonstrates that the β-Co(OH)2 microflower is an assembly of the nanosheets. The selected area electron diffraction (SAED) analysis on the nanosheet petal, renders its single crystalline nature and the surface of the nanosheet is the (001) plane of β-Co(OH)2 (inset Fig. 3C). HRTEM imaging of the β-Co(OH)2 nanosheet (Fig. 3D) illustrates the porous character of the nanosheet, typically 2–4 nm in size whilst the inter-planar spacing of 0.276 nm coincide with that expected for a (100) lattice plane. | ||
| Fig. 2 X-ray diffraction (XRD) spectra of the β-Co(OH)2 nanosheets. | ||
 | ||
| Fig. 3 SEM images of as-prepared porous β-Co(OH)2 microflower assembled by nanosheets; C) TEM image and corresponding SAED pattern (inset) of β-Co(OH)2 nanosheets and D) HRTEM image of the nanosheet (magnification of Fig. 3C). | ||
The surface electronic states and the chemical composition of the β-Co(OH)2 were examined by X-ray photoelectron energy spectroscopy (XPS). The survey XPS curves reveal the presence of Co and O, as confirmed by XRD analysis. The binding energies of the Co 2p3/2 and 2p1/2 states are located at 781.3 and 797.4 eV, respectively (Fig. 4A), which agree with the value illustrated in the literature.26 The Co 2p main peaks include a satellite peak on the higher binding-energy side, indicating the Co ion is in a divalent state.25 The binding energy of the O 1s state at 531.0 eV (Fig. 4B) is consistent with values previously reported and confirms the presence of O–H bonds.25,27 The N2 adsorption/desorption isotherm showed a type IV curve with a hysteresis loop with the pore distribution curve exhibiting a peak at approximately 40 nm, demonstrating the porous characteristics of the material. The BET (Brunauer-Emmett-Teller) surface area and single point total pore volume were found to correspond to 26 m2 g−1 and 0.03 cm3 g−1, respectively. The pore size calculated by the BJH (Barrett-Joyner-Halenda) method is ∼40 nm, corresponding to the result illustrated in the inset of Fig. 3.
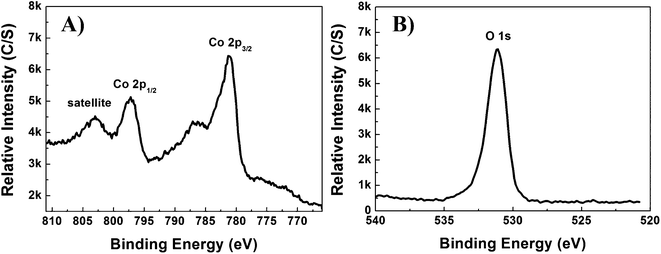 | ||
| Fig. 4 Typical XPS spectra of the β-Co(OH)2 microflower; A) Co 2p and B) O 1s. | ||
In order to gain an insight into the formation mechanism of the β-Co(OH)2 microflower, time-dependent experiments were carried out at 100 °C and the products inspected by SEM. It was found that the reaction time played a crucial role in the formation of flower-like products. The evolution process can be clearly seen from Scheme 1 and Fig. 5. At the beginning of the reaction, nanoparticles of the cobalt-monoethanolamine tertiary complex are primary formed (Fig. 4a), which are sap green in colour. At elevated temperature (up to 90 °C in this experiment), the cobalt-monoethanolamine complex decomposes slowly, resulting in the increase of Co2+ concentration. Meanwhile, OH− concentration increases gradually because of the hydrolysis of both hexamethylenetetramine (HMT, Eqn 1) and monoethanolamine.
 | (1) |
 | ||
| Fig. 5 SEM images of β-Co(OH)2 produced over a time period of: A) 0, B) 2, C) 4, D) 6, E) 12 and F) 24 h. | ||
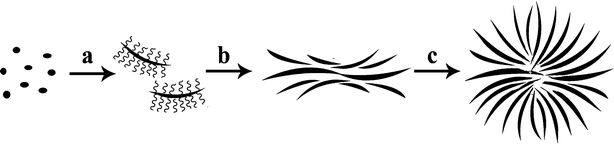 | ||
Scheme 1 The formation process of the β-Co(OH)2 microflower fashioned from individual nanosheets; The conversion of cobalt-monoethanolamine tertiary complex nanoparticles to Co(OH)2 nanosheets affixed with CTAB ( ) (a). The aggregation of Co(OH)2 nanosheets with the help of CTAB (b) and the assembly of the β-Co(OH)2 microflower (c). ) (a). The aggregation of Co(OH)2 nanosheets with the help of CTAB (b) and the assembly of the β-Co(OH)2 microflower (c). | ||
When the reaction continues for 2 h, most of the nanoparticles have been converted to nanosheets accompanied by the colour change of the products from ‘sap green’ to that of ‘wine’, which demonstrates the appearance of β-Co(OH)2 (Fig. 5B). With the increase of reaction time, more and more β-Co(OH)2 nanosheets are obtained (Fig. 5C). Some of these overlap together through layer-by-layer assembly to form flower-like Co(OH)2 with the diameter of 3–5 μm when the reaction time is up to 6 h (Fig. 5D). As the reaction proceeds, large β-Co(OH)2 microflowers with a diameter of 10 μm can been observed in Fig. 5E, which are larger than those of the 6 h reaction, but the structure of the flower-like β-Co(OH)2 is not perfect. The SEM image of the product reacted for 24 h (Fig. 5F) indicating that well-assembled β-Co(OH)2 microflowers can be readily achieved; the morphology of the product will not change if the reaction time is prolonged.
To examine the function of CTAB and monoethanolamine, comparable β-Co(OH)2 nanomaterials with different morphologies were prepared. When HMT is used as the sole supplier of OH− and no monoethanolamine is added, irregular β-Co(OH)2 nanosheets are obtained, rather than flower-like β-Co(OH)2 (Fig. 6A). This indicates that monoethanolamine has significant effects on the morphology of β-Co(OH)2. In this reaction system, cobalt-monoethanolamine tertiary complexes are formed at the beginning of the reaction and release Co2+ slowly with the reaction proceeding, which is favoured for the formation of flower-like β-Co(OH)2. When no CTAB is used, many individual β-Co(OH)2 nanosheets were obtained instead of flower-like β-Co(OH)2 (Fig. 6B). CTAB serves as not only the surfactant for the assembly of the β-Co(OH)2 nanosheets (Scheme 1b and 1c), but also the structure-directing agent for the formation of the mesoporous β-Co(OH)2. Therefore, both CTAB and monoethanolamine cannot be indivisible for the synthesis of β-Co(OH)2 microflowers. Note that the flower-like shapes, i.e. the petals, likely allow for electrolyte ions to penetrate and the large surface area allows energy storage.
 | ||
| Fig. 6 SEM images of β-Co(OH)2 produced under varied reaction conditions: A) without monoethanolamine and B) without CTAB. | ||
3.2 Electrochemical evaluation of the Co(OH)2 microflowers
We first consider the voltammetric performance of the β-Co(OH)2 modified screen printed supercapacitor (20%, (Mp/MI)) in a 3 electrode-cell, with 3 M KOH using a potential range between −0.3 and +0.5 V at an applied scan rate of 0.02 Vs−1 (vs Ag/AgCl). This was considered a suitable test to determine the mechanistic process of the reaction (faradaic and non-faradaic). Fig. 7A displays a predominantly rectangular shaped CV curve, strongly resembling the characteristics of electrochemical double-layer, commonly observed for carbon based materials but also faradaic pseudocapacitance, evident from the oxidation and reduction waves at −0.10, −0.11, 0.12 and 0.3 V (P1, P2, P3 and P4, respectively, Fig. 7A), typical to metal oxides (β-Co(OH)2 in our case). The two redox couples observed in the voltammetric profile are likely attributed to two reaction mechanisms:28Co(OH)2 ⇌ CoO(OH) + H+ + e− (P1/P2) andCoO(OH) ⇌ CoO2 + H+ + e− (P3/P4). Note that in the synthesis CTAB and monoethanolamine are used and we assume that β-Co(OH)2 is adequately washed such that these are not part of the final material and do not contribute electrochemically.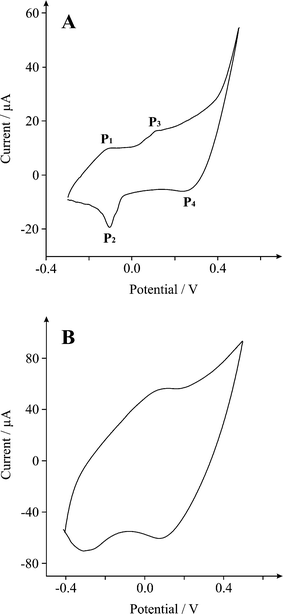 | ||
| Fig. 7 Cyclic voltammograms of a β-Co(OH)2 (20%, (Mp/MI)) modified SPE at scan rate of A) 20 and B) 130 mV s−1 in 3 M KOH. | ||
To determine the optimum coverage of nanomaterial which exhibits the most beneficial electrochemical response, we consequently explored the effect of increasing amounts of cobalt hydroxide (β-Co(OH)2) over the range 10–30% (Mp/MI). The response of the 10% and 20% (Mp/MI) cobalt hydroxide electrodes were initially explored. It was found that the specific capacitance, calculated using galvanostatic charge–discharge method (see below) increased exponentially in relation to the β-Co(OH)2 content. However once a critical mass is reached, in our case 30% (Mp/MI), the electrochemical performance of the electrode drops-off. This suggests that excessive quantities of β-Co(OH)2 amplifying the volume of nanosheets superimposed on the surface of the electrode, decreases porosity, consequently diminishing the ability to store large amounts of charge within the active material. As a result, there is a significant reduction in the specific capacitance of the working electrode. This phenomenon has been observed before when metal oxide modified SPE's have been used in electroanalysis, credited to the reduced number of conductive pathways throughout the electrode.29,30
Galvanostatic charge–discharge was used to directly evaluate the applicability of the cobalt hydroxide screen printed electrodes. A typical charge–discharge plot for the β-Co(OH)2 electrode is depicted in Fig. 8. The hybrid cobalt electrode exhibits good electrochemical capacitance performance, demonstrated by a linear variation of the voltage observed during the charging-discharging process (Fig. 8). The specific capacitance of the electrode can be determined from the charge/discharge test, using with the following equation:1
 | (2) |
 | ||
| Fig. 8 Typical galvanostatic charge–discharge of β-Co(OH)2 modified screen printed electrode in 3M KOH at a discharge current of 5 μA. | ||
Where Cm is the specific capacitance of the capacitor (F g−1), I is the current of the charge–discharge, Δt is the discharging time period in seconds for the potential ΔV, in volts and m is the mass load of the active materials (including the positive and negative electrode). The specific capacitance of the β-Co(OH)2 electrode was calculated to be ∼170 F g−1 at a discharge current of 5 μA. This is amid previous literature values recorded for β-Co(OH)2.31,32,33α-Co(OH)2 nanosheets, fabricated through electrochemical methods, have demonstrated a specific capacitance of up to 860 F g−1.34 However, the electrochemical deposition technique suffers with limitations such as a small area of deposition, severe cleaning subsequent to each deposition and high working cost, thus restricting its application on a large scale. While the α form has higher activity, its hydrotalcite-like phase is metastable and easily undertakes a phase transformation into the more stable brucite-like compounds in strong alkaline media.
A long cycle life constitutes one of the core characteristics sought-after in any energy storage device. The cyclic performance of the hybrid electrode is impressive, retaining 99.69% of its maximum capacity over 600 cycles as shown in Fig. 9, indicating the stability and suitability of the β-Co(OH)2 particles for high-performance energy storage applications. It is apparent from the cycling process, that an activation process is required to attain the optimum energy storage from the cobalt modified electrode (Fig. 10). The thickness of the working electrode area is an important parameter to consider in future studies as increased values (in our case) have exhibited detrimental effects, possibly attributed to a decrease in conductive pathways.
 | ||
| Fig. 9 A plot showing number of charge–discharge cycles vs. specific capacitance for a β-Co(OH)2 modified SPE over 600 cycles in 3M KOH. | ||
 | ||
| Fig. 10 Activation process of the β-Co(OH)2 electrode; galvanostatic charge–discharge cycles 1–50 in 3M KOH. | ||
4. Conclusions
We have reported the first literature report of a supercapacitor fabricated solely by screen printing. This simple methodology, allows for the mass fabrication of inexpensive, reproducible supercapacitors, where potentially any material can be incorporated. In this example we have also reported the novel chemical synthesis of cobalt hydroxideβ-Co(OH)2 and incorporated this into the supercapacitor and explored its electrochemical performance towards energy storage. The β-Co(OH)2 asymmetric supercapacitor delivered a maximum specific capacitance of 170 F g−1 (0.1% RSD), retaining 99.69% of its maximum capacity over 600 scans. The capacitance can likely be improved upon in subsequent studies, through possible manipulation of design, morphology and chemical composition which makes these materials an exciting prospect and should be considered when designing electrochemical devices such as energy storage devices.Acknowledgements
The authors are grateful for the financial supports from the National Natural Science Foundation of China (No. 21050110115), International Joint Project from The Royal Society (No. JP090644), and Hunan Province Foundation of Natural Science (10JJ6026).References
- X. Du, C. Wang, M. Chen, Y. Jiao and J. Wang, J. Phys. Chem. C, 2009, 113, 2643 CrossRef CAS.
- A. S. Arico, P. Bruce, B. Scrosati, J. M. Tarascon and S. W. Van, Nat. Mater., 2005, 4, 366 CrossRef CAS.
- A. G. Pandolfo and A. F. Hollenkamp, J. Power Sources, 2006, 157, 11 CrossRef CAS.
- M. Winter and R. Brodd, Chem. Rev., 2004, 104, 4245 CrossRef CAS.
- C.-C. Hu, K.-H. Chang, M.-C. Lin and Y.-T. Wu, Nano Lett., 2006, 6(12), 2690 CrossRef CAS.
- A. Burke, J. Power Sources, 2000, 91, 37 CrossRef CAS.
- M. Winter and R. Brodd, Chem. Rev., 2004, 104, 4245 CrossRef CAS.
- S. Lipka, IEEE Aerosp. Electron. Syst. Mag., 1997, 12, 27 Search PubMed.
- D. N. Futaba, K. Hata, T. Yamanda, T. Hiraoka, Y. Hayamizu, Y. Kakudate, O. Tanaike, H. Hatori, M. Yumura and S. Iiyima, Nat. Mater., 2006, 5, 987 CrossRef CAS.
- S. Talapatra, S. Kar, S. K. Pal, R. Vajtai, L. Ci, P. Victor, M. M. Shaijumon, S. Kaur, O. Nalamasu and P. M. Ajayan, Nat. Nanotechnol., 2006, 1, 112 CrossRef CAS.
- G. Lota, T. A. Centeno, E. Frackowiak and F. Stoecki, Electrochim. Acta, 2008, 53, 2210 CrossRef CAS.
- K. H. An, W. S. Kim, Y. S. Park, J.-M Moon, D. J. Bae, S. C. Lim, Y. S. Lee and Y. H. Lee, Adv. Funct. Mater., 2001, 11, 387 CrossRef CAS.
- D. Y. Qu, J. Power Sources, 2002, 109, 403 CrossRef CAS.
- M. M. Shaijumon, F. S. Ou, L. J. Ci and P. M. Ajayan, Chem. Commun., 2008, 2373 RSC.
- C. G. Liu, M. Liu, F. Li and H. M. Cheng, Appl. Phys. Lett., 2008, 92, 143108 CrossRef.
- B. E. Conway, Electrochemical super capacitor and technological applications: Kluwer-Plenum Press: New York, 1999 Search PubMed.
- J. H. Jiang and A. Kucernak, Electrochim. Acta, 2002, 41, 2381 Search PubMed.
- B. Djurfors, J. N. Broughton, M. J. Brett and D. G. Ivey, J. Power Sources, 2006, 156, 741 CrossRef CAS.
- C. Lin, J. A. Ritter and B. N. Popov, J. Electrochem. Soc., 1998, 145, 4097 CrossRef CAS.
- J. W. Lang, L. B. Kong, W. J. Wu, Y. C. Luo and L. Kang, Chem. Commun., 2008, 4213 RSC.
- L. Cao, F. Xu, Y. Y. Liang and H. L. Li, Adv. Mater., 2004, 16, 1853 CrossRef CAS.
- M. Kaempgen, C. K. Chan, J. Ma, Y. Cui and G. Gruner, Nano Lett., 2009, 9(5), 1872 CrossRef CAS.
- J. P. Metters, R. O. Kadara and C. E. Banks, Analyst, 2011, 136(6), 1067 RSC.
- C. Yuan, X. Zhang, L. Hou, L. Shen, D. Li, F. Zhang, C. Fan and J. Li, J. Mater. Chem., 2010, 20, 10809 RSC.
- R. O. Kadara, N. Jenkinson and C. E. Banks, Sens. Actuators, B, 2009, 138, 556 CrossRef.
- J.H. Yang, H. Hyodo, K. Kimura and T. Sasaki, Nanotechnology, 2010, 21, 045605 Search PubMed.
- S. Tang, S. Vongehr, Y. Wang, L. Chen and X. Meng, J. Solid State Chem., 2010, 183, 2166 CrossRef CAS.
- Z. Hu, L. Mo, X. Feng, J. Shi, Y. Wang and Y. Xie, Mater. Chem. Phys., 2009, 114, 53 CrossRef CAS.
- N. A. Choudhry, D. K. Kampouris, R. O. Kadara, N. Jenkinson and C. E. Banks, Anal. Methods, 2009, 1, 183 RSC.
- R. O. Kadara, N. Jenkinson and C. E. Banks, Electroanalysis, 2009, 21(22), 2410 CAS.
- E. Hosono, S. Fujihara, I. Homma, M. Ichihara and H. Zhou, J. Power Sources, 2006, 158, 779 CrossRef.
- Z. A. Hu, L. P. Mo, X. J. Feng, J. Shi, Y. X. Wang and Y. L. Xie, Mater. Chem. Phys., 2009, 114, 53 CrossRef CAS.
- J. Zhang, L.-B. Kong, J.-J. Cai, Y.-C. Luo and L. Kang, J. Solid State Electrochem., 2010, 14, 2065 Search PubMed.
- V. Gupta, T. Kusahara, H. Toyama, S. Gupta and N. Miura, Electrochem. Commun., 2007, 9, 2315 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |