A facile method for estimating viable bacterial cells in solution based on “subtractive-aggregation” of gold nanoparticles†
Manab Deb
Adhikari
a,
Biswa Ranjan
Panda
b,
Umakanth
Vudumula
a,
Arun
Chattopadhyay
*bc and
Aiyagari
Ramesh
*a
aDepartment of Biotechnology, Indian Institute of Technology Guwahati, Guwahati 781039, India. E-mail: aramesh@iitg.ernet.in; Fax: +91 361 2582249; Tel: +91 361 2582205
bDepartment of Chemistry, Indian Institute of Technology Guwahati, Guwahati 781039, India. E-mail: arun@iitg.ernet.in; Fax: + 91 361 2582349; Tel: +91 3612582304
cCentre for Nanotechnology, Indian Institute of Technology Guwahati, Guwahati 781039, India
First published on 22nd December 2011
Abstract
In this study we report a new method for sensitive estimation of bacterial cells in solution based on a systematic decrease in the degree of poly-L-lysine (PLL)-mediated aggregation of gold nanoparticles (Au NPs). The quantification of cells was done by interacting bacterial cell suspensions with PLL and recovering the unbound PLL fraction which was subsequently used to initiate aggregation of Au NPs. A progressive decline in nanoparticle aggregation evidenced by a reduced broadening of the surface plasmon resonance (SPR) peak of Au NPs revealed a strong correlation with increasing bacterial cell numbers. Using this method we could readily estimate both Gram-negative strains of Escherichia coliMTCC 433, Enterobacter aerogenesMTCC 2822 and Pseudomonas aeruginosaMTCC 2488 as well as Gram-positive bacteria such as Bacillus cereusMTCC 1305, Staphylococcus aureusMTCC 740 and Listeria monocytogenes Scott A. The limit of detection was as low as 10 colony forming unit (CFU) of bacterial cells. The method was specific for viable cells as heat-killed bacterial cells failed to reveal the characteristic decline in the broadening of the SPR peak of Au NPs. We also demonstrate that a centrifugal filter device could be used for rapid estimation of bacterial cells in solution based on the same principle used herein. The application potential of the method was further validated in an antimicrobial susceptibility test, wherein estimation of viable cell numbers of target bacteria Leuconostoc mesenteroidesNRRL B640 following treatment with nisin, a membrane permeabilizing cationic peptide, was on par with conventional plating. The present method of bacterial cell estimation offers a distinct advantage over other conventional methods in terms of ease of operation, rapidity, high sensitivity and quantitative detection of viable cells.
1 Introduction
There is an ever increasing demand to develop rapid, sensitive and reliable methods of estimating bacterial cell numbers especially in the context of clinical diagnosis, food safety, and environmental monitoring. This assumes even greater significance in the case of pathogenic bacteria as the presence of even a single viable cell may imply grave health concerns. Conventional microbiological methods for estimating bacterial cells rely on plating in a specific growth media. Essentially, the process lacks sensitivity, is long-drawn-out and hence undesirable especially when rapid diagnosis is paramount. In recent times, powerful methods such as polymerase chain reaction (PCR) and immunoassays have been developed to achieve high sensitivity and specificity.1–4 However, the selection of appropriate target genes and surface antigens can be tricky and overall the methods are invasive, costly and time-consuming. Alternative new generation detection methods such as magneto-sensors, electrochemical impedance spectroscopy and quartz crystal microbalance biosensor have emerged, which offer fast detection, great sensitivity and specificity.5–7 However, the relatively high degree of sophistication in the instrumentation and the user expertise required for implementing these tools may have to be addressed prior to their routine application.The emergence of nanotechnology in recent years has ushered in great promise in the field of diagnostics and therapeutics. Nanoparticles (NPs) represent a particularly innovative regime, displaying unique size-dependent properties. Profound examples of the application of nanoscale materials in biology and medicine are rife in literature.8,9 Amongst metal NPs, Au NPs have been particularly interesting in terms of a wide array of biological applications.10–12 Depending on their size, shape, degree of aggregation, and local environment, Au NPs exhibit a characteristic surface plasmon resonance (SPR), which can be exploited as a bio-responsive probe.13,14,15Nanoparticle-based detection systems, with special emphasis on probing proteins, nucleic acids, and pathogens have come of age.16,17 Significant strides have also been made with regard to detection of bacterial cells by implementing nanotechnology-based tools. For example, magnetic glyco-nanoparticles have been used to specifically detect E. coli with appreciable speed and precision and with a detection limit of 104cells/mL.18 Rapid and specific detection of Mycobacterium avium spp. paratuberculosis was accomplished on the basis of magnetic relaxation with antibody-functionalized superparamagnetic iron oxide nanoparticles, wherein detection of the pathogen was demonstrated in a complex matrix like milk and blood.19 In another study, magnetic nanoparticles functionalized with antibodies were used in conjunction with infra-red fingerprinting to detect E. coli O157:H7 and S. typhimurium in food matrices.20
While specific detection of pathogenic bacteria is critical and draws immense attention, functionalization of NPs with antibodies for specific and efficient target capture can be quite challenging. Hence there is a constant impetus to develop broad-affinity based methods which can circumvent bottlenecks in conjugation chemistry on one hand and provide a platform based on a universal principle which could facilitate detection or estimation of cell numbers of a range of bacterial strains. In this direction vancomycin functionalized nanoparticles have been used as effective small molecule probe to detect various Gram-positive and Gram-negative bacteria.21 Magnetic iron oxide NPs coated with titania have been employed as affinity probe to target bacteria under microwave heating. The concept was that metal oxide-coated magnetic NPs would interact with bacteria possessing lipopolysaccharide (LPS) via metal-phosphate chelation, followed by MALDI-MS analysis which could identify potential biomarker ions for pathogens such as Escherichia coli O157:H7, Shigella sonnei, Pseudomonas aeruginosa and Klebsiella pneumoniae.22
The bacterial cell surface is known to be negatively charged and this hallmark is attributed to the presence of either teichoic acid in Gram-positive bacteria or the outer membrane lipopolysaccharide (LPS) in Gram-negative bacteria.23,24 In the context of developing a detection system which is versatile and universally applicable to a wide group of bacterial strains, the anionic charge characteristics of bacterial cell surface can be envisaged as an attractive option to mediate electrostatic interactions with an oppositely charged sensor probe. In a previous study our research group has demonstrated the potential application of a fluorescent gold nanoparticle-polythiophene nanocomposite in estimating various bacterial strains. The quantification was based on loss of the fluorescence intensity of the positively charged nanocomposite upon interaction with negatively charged bacterial cell surface.25 Rapid and discriminatory identification of bacteria has been demonstrated using an array of Au NP-conjugated polymer constructs. The basis was the displacement of anionic conjugated polymer from cationic NPs by negatively charged bacterial surfaces.26 In a recent study, glyco-dendronized poly-L-lysine (PLL) has been used to bind to cell-surface carbohydrate receptors and detect E. coli by microscopic analyses.27
In the present investigation we demonstrate a rapid and sensitive method of estimating bacterial cells in solution for which the fundamental premise was to use the polycationic PLL and set off aggregation of negatively charged citrate-stabilized Au NPs. PLL being polycationic has a propensity to interact electrostatically with bacterial cells. The strategy for estimation of bacterial cell numbers was to initiate bacteria–PLL interactions, separate unbound PLL fraction and measure the extent of aggregation of Au NPs by the unbound fraction of PLL. We refer this phenomenon as “subtractive-aggregation” wherein the extent of Au NP aggregation by unbound PLL provides a measure of bacterial cell numbers, in the absence of the interfering bacterial cells. The significant features of the proposed method were the estimation of cell numbers in solution for a wide variety of bacterial strains with a lower detection limit of 10 CFU, and that the method could account for viable cell numbers in solution. Additionally, the potential of the method is demonstrated by implementing a centrifugal filter device which augmented rapid and easy estimation of bacterial cells. We have also validated the utility of the subtractive-aggregation method in an antimicrobial susceptibility test wherein the viable cell count obtained for target bacteria following treatment with a potent antimicrobial agent by both conventional plating as well as the present method were highly comparable.
2 Experimental
2.1 Chemicals and growth media
HAuCl4 (17 wt% solution of HAuCl4 in dilute HCl; 99.99%), poly-L-lysine (PLL) hydrochloride (molecular weight ranging from 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–30000), propidium iodide (PI) and nisin were obtained from Sigma-Aldrich Chemicals, USA. Nutrient broth (NB), Brain-Heart Infusion (BHI) broth and deMan, Rogosa and Sharpe (MRS) broth were purchased from HiMedia, Mumbai, India. Trisodium citrate 2-hydrate was procured from Merck Chemicals, Mumbai, India.
000–30000), propidium iodide (PI) and nisin were obtained from Sigma-Aldrich Chemicals, USA. Nutrient broth (NB), Brain-Heart Infusion (BHI) broth and deMan, Rogosa and Sharpe (MRS) broth were purchased from HiMedia, Mumbai, India. Trisodium citrate 2-hydrate was procured from Merck Chemicals, Mumbai, India.
2.2 Bacterial strains
The bacterial strains used in the present investigation comprised of Gram-positive strains of Staphylococcus aureusMTCC 740, Listeria monocytogenes Scott A, Bacillus cereusMTCC 1305, Leuconostoc mesenteroidesNRRL B640 and Gram-negative strains of Escherichia coliMTCC 433, Pseudomonas aeruginosaMTCC 2488 and Enterobacter aerogenesMTCC 2822. S. aureusMTCC 740 and L. monocytogenes Scott A were propagated in BHI broth at 37 °C and 180 rpm for 12 h, whereas B. cereusMTCC 1305, E. coliMTCC 433, P. aeruginosaMTCC 2488 and E. aerogenesMTCC 2822 were grown in NB growth medium at 37 °C and 180 rpm for 12 h. L. mesenteroidesNRRL B640 was grown in MRS broth at 37 °C under static conditions.2.3 Preparation of citrate-stabilized gold nanoparticle
Synthesis of gold nanoparticle (Au NP) was accomplished by following the procedure of citrate reduction of HAuCl4.14 Following synthesis a 1.0 mL portion of the Au NP solution was taken in a cuvette and the UV-vis extinction spectrum was recorded (Cary 300, Varian, USA). An aliquot of the synthesized Au NP sample was also subjected to transmission electron microscopy (TEM) analysis.2.4 Interaction of poly-L-lysine and citrate-stabilized Au NPs
A stock solution of PLL was prepared in MilliQ grade water at a concentration of 10−4% (w/v). In a series of microcentrifuge tubes, 1.5 mL portions of citrate-stabilized Au NP solution (0.25 nM) was taken to which various volumes of PLL (starting from 10 μL to 200 μL) was added. The addition of PLL to Au NP solution was pursued drop wise to achieve adequate mixing. The final volume of the mixture was adjusted to 1.9 mL with MilliQ grade water so as to achieve an approximate concentration gradient of PLL in the range of 5.2 × 10−7, 10.5 × 10−7, 21 × 10−7, 31.5 × 10−7, 42 × 10−7, 52 × 10−7, 63 × 10−7, 73.6 × 10−7, 84 × 10−7, 95 × 10−7 and 105 × 10−7% w/v. The control sample consisted of 1.5 mL portion of citrate-stabilized Au NP solution and 0.4 mL of MilliQ grade water. Following incubation at room temperature for 15, 30 and 60 min respectively, UV-vis extinction spectrum of the samples were recorded at a wavelength set from 400–800 nm. The area under the extinction spectrum was estimated by using OriginPro 7.0 (OriginLab Corporation, MA, USA). All measurements were taken from three independent experimental samples. Aliquots of the sample from the series were also subjected to TEM analysis.2.5 PLL-bacterial cell interaction studies
These experiments were conducted with bacterial cultures of E. coliMTCC 433 and L. monocytogenes Scott A. The objective was to ascertain the effect of concentration and interaction time of PLL with bacterial cells. Bacterial cells were harvested from a 1.0 mL aliquot of an overnight grown culture broth by centrifugation at 8,000 rpm for 3 min. The cells were washed twice with phosphate-buffered saline (PBS) to remove residual media components and then resuspended in 0.2 mL PBS in separate tubes to achieve a cell number of 102, 104 and 106 CFU. Various volumes of PLL stock solution were added separately to 0.2 mL of bacterial cells and the volume was made upto 0.4 mL to achieve a final PLL concentration range of 0.25 × 10−5, 0.5 × 10−5, 1.0 × 10−5, 3.0 × 10−5, 3.5 × 10−5, 4.0 × 10−5 and 5.0 × 10−5% w/v. Hence the bacterial cell concentrations in the samples were 102, 104 and 106 CFU per 0.4 mL. The samples were incubated at room temperature under mild shaking conditions for 15, 30 and 60 min. Subsequently the bacterial cells were separated by centrifugation at 8000 rpm for 3 min. The recovered supernatant which was essentially unbound PLL (nearly 0.4 mL) was added drop wise to 1.5 mL citrate-stabilized Au NP. The bacterial cells bound with PLL were resuspended in 0.4 mL PBS and added separately to a 1.5 mL aliquot of citrate-stabilized Au NP. Both these samples were incubated at room temperature for 30 min. UV-vis extinction spectrum of all the samples were recorded for three independent experimental samples and the area under each extinction spectrum was estimated. Additional control experiments were also performed to study the effect of the order of addition of the three interacting components namely bacterial cells, PLL and Au NPs on the broadening of the extinction spectrum of Au NPs. The description of these experiments is mentioned in ESI†.
2.6 Effect of PLL on bacterial cell viability
A 0.2 mL aliquot of PLL was added to varying cell numbers of E. coliMTCC 433 and L. monocytogenes Scott A suspended in 0.2 mL PBS and incubated at room temperature for 30 min and 60 min under mild shaking conditions. Subsequently the samples were plated to determine the viable cell numbers. A control sample consisting of untreated bacterial cells was also included. Cell viability was also assessed by inoculating both PLL-treated and untreated samples into culture media and monitoring the growth of the cells.2.7 Estimation of bacterial cell numbers in solution
For estimation of cell numbers, Gram-positive bacterial strains of Staphylococcus aureusMTCC 740, Listeria monocytogenes Scott A, Bacillus cereusMTCC 1305 and Gram-negative strains of Escherichia coliMTCC 433, Pseudomonas aeruginosaMTCC 2488 and Enterobacter aerogenesMTCC 2822 were selected. Bacterial cells were harvested from a 1.0 mL aliquot of an overnight grown culture broth by centrifugation at 8000 rpm for 3 min. The cells were washed twice with PBS to remove residual media components and then resuspended in 0.2 mL PBS to achieve a logarithmic series of cell number in the range of 1.0–107 CFU. A 0.2 mL portion of PLL solution was added separately to the series of tubes with varying cell number to achieve a final PLL concentration of 3.5 × 10−5% w/v in 0.4 mL. The bacterial cell concentrations in the samples ranged from 1.0–107 CFU per 0.4 mL. The samples were incubated at room temperature under mild shaking conditions for 30 min. Subsequently the bacterial cells were separated by centrifugation at 8000 rpm for 3 min. The recovered supernatant which consisted of unbound PLL was added to 1.5 mL of citrate-stabilized Au NP solution and incubated at room temperature for 30 min. Subsequently UV-vis extinction spectrum of all the samples were recorded as mentioned before and the area under each extinction spectrum was estimated. The control samples consisted of (i) bacterial solution or PLL alone (both were volume adjusted to 0.4 mL) added to 1.5 mL of Au NP solution and (ii) volume adjusted Au NP solution alone. The areas under UV-vis extinction spectrum were considered for calculating the difference between the areas due to maximum aggregation of Au NP by PLL alone (AaggMax) and aggregation of Au NP by unbound PLL (AaggUnbound-PLL) for every sample of the series ranging from 1.0–107 CFU. The difference in the areas gave a measure of subtractive-aggregation (ΔAagg) and a plot between ΔAagg and bacterial cell number was constructed. A two-way analysis of variance (ANOVA) followed by all pair wise multiple comparison (Holm-Sidak method) was performed using Sigma Plot.
2.8 Estimation of cell numbers in centrifugal device
A logarithmic series of cell numbers of E. coliMTCC 433 and L. monocytogenes Scott A were taken in the range of 1.0–107 CFU in 0.2 mL PBS and interacted with 0.2 mL PLL (final concentration of 3.5 × 10−5% w/v in 0.4 mL) in a microcentrifuge tube. Following 30 min of interaction, the contents of the tube were added to Costar Spin-X centrifuge tube (Sigma-Aldrich, USA) having a 0.22 μm cellulose acetate membrane filter and spun immediately at 8000 rpm for 1 min. The bacterial cells with bound PLL were trapped in the filter whereas the unbound PLL fraction (nearly 0.4 mL) was collected as flow through in the collection tube. As mentioned earlier, 1.5 mL Au NP solution was then added to the unbound PLL fraction and incubated for 30 min. Aggregation of Au NP was pursued by recording the UV-visible extinction spectrum of the samples and determining the area under each spectrum. As mentioned earlier, the areas were considered for obtaining a measure of subtractive-aggregation (ΔAagg) and a plot between ΔAagg and bacterial cell number was constructed.
2.9 Interaction with heat-killed bacterial cells
In this set of experiments, varying cell numbers of bacterial cells (102, 104 and 106 CFU) were taken in separate tubes and subjected to heat treatment at 55 °C, 65 °C, 85 °C for 30 min each and autoclaving at 121 °C and 15 lb/in2 for 20 min. To verify bacterial cell death and membrane damage following various heat treatments, aliquots of the samples were (a) subjected to plating to determine cell numbers, (b) re-inoculated in fresh growth media to ascertain the effect of heat treatment on cell viability and (c) stained with propidium iodide (PI) to determine outer membrane damage in bacterial cells (refer to ESI† for a description of PI staining method). Subsequently, 0.2 mL of PLL solution was added separately to 102, 104 and 106 CFU heat- treated bacterial cells (in 0.2 mL PBS) to achieve a final PLL concentration of 3.5 × 10−5% w/v in 0.4 mL. The samples were incubated at room temperature under mild shaking condition for 30 min. The bacterial cells were then separated by centrifugation at 8000 rpm for 3 min and the supernatant consisting of unbound PLL was added to 1.5 mL of Au NP solution and incubated at room temperature for 30 min. UV-vis extinction spectrum of all the samples were recorded as mentioned before. The control sample consisted of either heat treated bacterial solution or PLL alone (both were volume adjusted to 0.4 mL) added to 1.5 mL of Au NP solution.
2.10 Application studies
The potential of the proposed bacterial cell estimation method as a tool to determine viable cell numbers was determined following exposure of the cells to a potent antimicrobial agent. Nisin was chosen as the model antimicrobial agent and a stock solution of the same was prepared at a concentration of 10000 IU/mL in 0.75% NaCl (pH 5.3). Leuconostoc mesenteroidesNRRL B640 was used as target bacteria to test the activity of nisin. Initially a dose-dependent test for nisin activity against L. mesenteroidesNRRL B640 was performed by an agar well diffusion assay (refer to ESI†). For testing antimicrobial activity of nisin in solution, 1.0 mL of bacterial cells corresponding to 106 CFU of L. mesenteroidesNRRL B640 were taken in PBS to which nisin solution was added at various concentrations (100, 200, 400 and 800 IU/mL) and the cells were incubated for 12 h. Following incubation, the cells were separated by centrifugation, washed twice with PBS, serially diluted and plated to determine viable cell number. The control sample consisted of untreated bacterial cells alone.
In a parallel set of sample, 1.0 mL aliquot of the cells of L. mesenteroidesNRRL B640 was treated with nisin in a similar manner and the bacterial cells were harvested by centrifugation and resuspended in 0.2 mL of PBS. To this, 0.2 mL of PLL solution was added and the samples were incubated at room temperature for 30 min under mild shaking conditions. Subsequently, samples were centrifuged and 1.5 mL of citrate-stabilized Au NP solution was added to the supernatant and incubated further for 30 min at room temperature. UV-vis extinction spectrum of the samples was recorded and area under each spectrum was calculated. A calibration plot between subtractive-aggregation areas (ΔAagg) and cell numbers of L. mesenteroidesNRRL B640 was constructed. In the case of nisin treated samples, this calibration plot was used to calculate cell numbers for L. mesenteroidesNRRL B640 following treatment with the antimicrobial peptide.
3 Results and discussion
The primary aim of the present work was to develop a fast, convenient and sensitive method of estimating bacterial cell numbers in solution. An important criterion in our quest was to adopt a strategy that is based on a broad principle so as to permit estimation of a wide range of bacterial cells. In this regard the characteristics of the bacterial cell surface provided us the lead. It is well established that the bacterial cell surface is negatively charged at near neutral pH owing to the presence of teichoic acid in Gram-positive bacteria and lipopolysaccharide (LPS) in Gram-negative bacteria.23,24 We conceived that a positively charged polymeric species like PLL would readily interact with the bacterial cell surface based on electrostatic interaction. PLL would also interact with negatively charged citrate-stabilized Au NPs leading to change in extinction quantitatively, which would enable estimation of the concentration of PLL. If PLL led to aggregation of Au NPs like the ones reported with other proteins,14,15 then a quantitative estimation would be possible based on the broadening of the extinction spectrum. Further, if PLL is taken in stoichiometric excess, some fraction of the polymer would be bound to bacterial cells while the unbound fraction could then be separated and used for initiating aggregation of negatively charged citrate-stabilized Au NPs. It was also reasonable to fathom that increasing cell concentration in solution would result in enhanced sequestration of the positively charged PLL by bacterial cells, leading to reduced amount of the polymer as unbound fraction and consequently the degree of aggregation of Au NP would also decrease correspondingly. Hence a measure of the extent of aggregation of Au NPs by unbound PLL can be used as a way to estimate bacterial cell numbers and this formed the basis of our endeavour.3.1 PLL-mediated aggregation of Au NPs
Citrate-stabilized Au NPs used in the present investigation exhibited a prominent extinction peak at 520 nm, which is the characteristic SPR band of Au NPs (ESI†, Fig. S1a). TEM analysis revealed that Au NPs were well dispersed and spherical in shape (ESI†, inset of Fig. S1a). The average particle size as determined using ImageJ software (http://rsb.info.nih.gov/ij) was observed to be 12.0 ± 2.0 nm (ESI†, Fig. S1b). Initially, experiments were performed to determine the effect of increasing concentration of PLL on the degree of aggregation of Au NPs. Interaction of PLL and Au NPs in solution was pursued for 15, 30 and 60 min following which UV-vis spectral measurements were taken. Fig. 1a corresponds to measurements conducted after 30 min of Au NP and PLL interaction. It is clear from the figure that an increase in the concentration of PLL resulted in a distinct broadening of the extinction spectra of Au NPs. It is also noteworthy that there was only marginal broadening beyond a PLL concentration of 84 × 10−7% w/v probably indicating saturation. It can also be observed that there was no shift of the absorbance maximum in the samples, although at higher concentrations of PLL a hump appeared at higher wavelength, which is a well-known phenomenon, associated with localized SPR spectra of Au NPs. The effect of increasing concentration of PLL was also manifested as a change in the color of Au NP solution from ruby red to deep violet (inset of Fig. 1a). When the area under the UV-vis extinction spectrum was calculated, a systematic increase was observed as a function of PLL concentration and saturation was observed at PLL concentrations of 95 × 10−7 and 105 × 10−7% w/v (Fig. 1b). In order to understand the effect of incubation time of PLL and Au NPs on spectral broadening, measurements were also taken after 15 min and 60 min interaction of PLL and Au NP (refer to ESI†, Fig. S2). The extent of spectral broadening and the corresponding area under extinction spectrum observed in case of samples following 15 min incubation period of PLL and Au NP (ESI†, Fig. S2a and S2c) was less than that for 30 min (Fig. 1a and 1b) and 60 min (ESI†, Fig. S2b and S2d) interaction time, respectively. With regard to linear response of Au NP aggregation as a function of PLL concentration it was observed that the best response was obtained for 30 min interaction time (R2 value of 0.97) followed by 60 min and 15 min interaction time (R2 value of 0.966 and 0.95, respectively). Based on the results obtained in these experiments spectral measurements were taken after 30 min incubation of PLL with Au NPs for all subsequent experiments.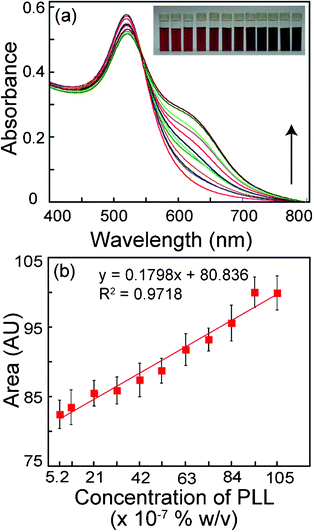 | ||
| Fig. 1 (a) UV-vis spectrum of citrate-stabilized Au NPs with increasing concentration of poly-L-lysine (PLL). Spectral measurements were performed following 30 min of addition of PLL solution to Au NPs. The lowest trace (red color) is for the control sample consisting of Au NP alone. The subsequent traces were obtained with increasing concentration of PLL as indicated by arrow. Inset shows photographs of vials of Au NP solution in the presence of increasing concentration of PLL (left to right). (b) Area under the UV-vis spectrum in (a) versus the corresponding PLL concentrations. | ||
Poly-L-lysine hydrochloride (molecular weight ranging from 15000–30000) provides a large number of positively charged residues, which can interact electrostatically with negatively charged citrate-stabilized Au NPs, resulting in a concentration-dependent aggregation of Au NPs. To ascertain this, we performed TEM analysis of as synthesized Au NPs as well as Au NPs in presence of various concentrations of PLL. It is clear from Fig. 2a that citrate-stabilized Au NPs were spherical in shape and well dispersed. However, upon addition of increasing concentration of PLL, a progressive increase in Au NP aggregation was manifested (Fig. 2b–d). At lower concentrations of PLL (21 × 10−7% w/v), discrete clusters of aggregated Au NPs were observed (Fig. 2b) along with a few dispersed particles. With further increase in PLL concentration to 42 × 10−7% w/v, the size of the clusters increased (Fig. 2c). At a PLL concentration as high as 84 × 10−7% w/v, majority of the particles were involved in the formation of aggregates. The sizes of such aggregates were distinctly larger than at lower concentrations of PLL (Fig. 2d). Higher magnification TEM images for select samples of PLL-aggregated Au NPs are shown in ESI† (Fig. S3). TEM experiments clearly suggest a progressive increase in the aggregation of Au NPs with increasing PLL concentration and this probably explains the broadening of UV-vis spectra observed in the samples, following interaction of PLL and Au NPs.
 | ||
| Fig. 2 TEM images of (a) as synthesized citrate-stabilized Au NPs, and (b–d) Au NPs in the presence of increasing concentration of PLL (21 × 10−7, 42 × 10−7, and 84 × 10−7% w/v). Scale bar is 50 nm for all the images. | ||
3.2 PLL-bacterial cell interaction studies
A systematic increase in Au NP aggregation as a function of PLL concentration motivated us to explore the possibility of studying PLL–bacterial cell interactions and device a method of bacterial cell estimation. In this context, the concentration of PLL and its interaction time with bacterial cells are critical parameters which are likely to influence the final estimation of cell numbers. To address these issues experiments were performed with Gram-negative E. coliMTCC 433 and Gram-positive L. monocytogenes Scott A. A higher concentration of PLL (0.25–5.0 × 10−5%w/v) was used to interact with bacterial cells in solution, compared to earlier experiments where Au NP aggregation was pursued at different concentrations of PLL, so as to ensure recovery of sufficient unbound PLL fraction. Essentially the observation is that Au NP aggregation increased as a function of PLL concentration for both E.coliMTCC 433 as well as L. monocytogenes Scott A, as evident from the broadening of UV-vis extinction spectrum (ESI†, Fig. S4 and S5) and corresponding increase in the area under the curve (Fig. 3). This increase in area was observed for all cell numbers used in the experiments (102, 104 and 106 CFU) as well as the individual interaction times (15, 30 and 60 min). With increasing PLL concentrations, the fraction of unbound PLL recovered following separation of PLL-bound bacterial cells shows a corresponding increase and thus accounts for a systematic increase in Au NP aggregation and area under extinction spectrum. In case of E.coliMTCC 433, at a PLL concentration of 3.5 × 10−5% w/v and 30 min interaction time (Fig. 3c), difference in the areas under the curve correlated with differences in cell numbers (91.52, 88.8 and 84.2 AU, corresponding to 102, 104 and 106 CFU, respectively). This trend was also consistent in case of L. monocytogenes Scott A (Fig. 3d). It is also noteworthy that the calculated areas under extinction spectrum corresponded to the linear range of response of Au NP aggregation as a function of PLL concentration (refer to Fig. 1b). The differences in the areas with respect to varying cell numbers were again observed to reduce in magnitude when the initial PLL concentration was enhanced to 4.0 × 10−5 and 5.0 × 10−5% w/v. This probably indicates a saturation effect. Quantitative differences in the area under extinction spectrum amongst samples representing various cell numbers (102, 104 and 106 CFU) were prominently manifested in case of 30 and 60 min of interaction between 3.5 × 10−5% w/v PLL and bacterial cells. However, the corresponding difference in area for 15 min interaction time between bacterial cells and PLL was only marginal. An extended interaction time between PLL and bacterial cells (30 and 60 min) resulted in quantitative sequestration of PLL from solution as a function of bacterial cell numbers and the degree of aggregation of Au NP by unbound PLL was inversely correlated with bacterial cell numbers. This fact is evident from the TEM analysis shown in ESI† (Fig. S6), wherein both the number as well as sizes of Au NP aggregates appear to be larger for the sample with 102 CFU in comparison to the sample with 104 CFU of E.coliMTCC 433. Based on the results obtained, it is clear that an initial interaction time of 30 min between 3.5 × 10−5% w/v PLL and bacterial cells can be pursued to achieve quantitative cell estimation in the shortest time.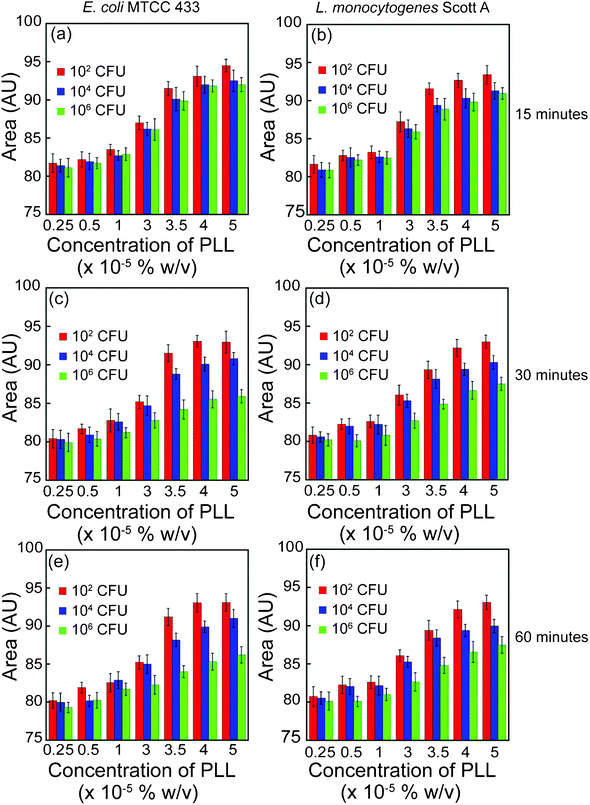 | ||
| Fig. 3 Area under UV-vis extinction spectrum for PLL interaction with varying bacterial cell number. PLL concentrations indicated in the plots represent the initial concentrations used for interaction with bacterial cells. | ||
Bacterial cell numbers could not be estimated by following aggregation of Au NPs by cell-bound PLL as the UV-vis extinction spectrum coincided with the control sample (Au NP alone), indicating lack of Au NP aggregation (ESI†, Fig. S7). Our next endeavour was to study the effect of the sequence of addition of the three interacting components namely bacterial cells, PLL and Au NPs (refer to control experiments in ESI†). When varying concentrations of bacterial cells, PLL and Au NPs were incubated together for 30 min, bulk of the PLL was involved in causing rapid aggregation of Au NPs in solution and hence the UV-vis extinction spectrum (ESI†, Fig. S8a–b) and areas under extinction spectrum for E.coli and L. monocytogenes failed to reflect differences in cell numbers (ESI†, Fig. S9a–b). When PLL and Au NPs were initially incubated, considerable aggregation of Au NPs resulted and a hump was observed at a higher wavelength in the extinction spectrum (ESI†, Fig. S8c–d). Subsequent addition of E.coliMTCC 433 and L. monocytogenes Scott A failed to disrupt the PLL-Au NP complex, and thus the areas under extinction spectrum did not reflect quantitative differences in cell numbers (ESI†, Fig. S9c–d). Further when varying concentrations of bacterial cells were initially interacted with Au NPs, binding of Au NPs by bacterial cells was minimal as both the cell surface as well as citrate-stabilized Au NPs possesses negative charges. Thus when the unbound Au NPs were recovered and incubated with PLL, the extent of spectral broadening for Au NPs revealed no significant difference for varying cell numbers (ESI†, Fig. S8e–f and Fig. S9e–f). Based on the control experiments it was thus established that quantitative estimation of bacterial cell numbers in solution could be readily pursued by initially interacting PLL and bacterial cells followed by recovery of unbound PLL and measuring the extent of aggregation of Au NPs by the unbound PLL fraction.
3.3 Effect of PLL on cell viability
If the present method of estimating bacterial cells is to be pursued by interacting cells with PLL, it was pertinent to investigate the effect of PLL on bacterial cell viability as there are reports suggesting that PLL affects cell viability.28,29 The essential observation from our experiments was that the viability of bacterial cells was not affected. For instance, when approximately 6.6 × 106 CFU E.coliMTCC 433 per 0.4 mL sample volume was interacted with 3.5 × 10−5% w/v PLL, the viable cell count obtained after 30 min and 60 min of interaction was determined to be 6.5 × 106 CFU. Similar observations were seen for L. monocytogenes Scott A. Even at lower cell numbers in the range of 104 and 102 CFU, bacterial cell viability was unaffected by PLL. Further when PLL-interacted cells were re-inoculated into fresh growth media and incubated overnight, high media turbidity resulted indicating cell growth. It is worthwhile to mention that the concentration of PLL (3.5 × 10−5% w/v) used for initial interaction with bacteria is way below the minimum inhibitory concentration (MIC) reported for various bacterial strains.28,29 The fact that PLL did not affect cell viability renders the present method non-destructive to the samples. This is an additional advantage since PLL-bound bacterial cells can be plated to determine viable cell count and compared with the cell number estimations obtained by the Au NP aggregation method in order to validate the accuracy and reliability of the latter.3.4 Estimation of cell numbers of various bacterial strains by subtractive-aggregation
Two key issues which we were interested to address while developing a method of bacterial cell estimation were (a) the wide scale applicability of the method for various types of bacterial strains including both Gram-positive and Gram-negative bacteria and (b) the sensitivity of the method. To pursue these goals we chose various Gram-positive and Gram-negative pathogenic bacteria and varied their cell numbers over a range of 1.0–107 CFU. A significant observation was that the extent of broadening of the extinction spectrum of Au NPs (ESI†, Fig. S10) and the area under extinction spectrum (Fig. 4) revealed a systematic decrease in samples with increasing bacterial cell numbers. Interestingly this phenomenon was unequivocally observed for all the bacterial strains tested. The linear relationship between area under UV-vis extinction spectrum and cell numbers (Fig. 4) for all the tested bacterial strains is also substantiated by the equations indicated in Table S1 (refer to ESI†). In case of bacterial cells the presence of either teichoic acid in Gram-positive bacteria or lipopolysaccharide in Gram-negative bacteria confers a negative charge to bacterial cell surface.23,24 The large number of positive charge residues on PLL ensures more or less equal propensity of interaction with negatively charged cell surface of Gram-positive and Gram-negative bacteria. This in turn results in equal proportion of unbound PLL recovered in all the samples, which explains the nearly equivalent levels of estimation across various bacterial strains. The differences observed in the broadening of UV-vis extinction spectrum and corresponding area under the spectrum could possibly be due to differences in charge characteristics of the cell surface as well as cell surface area which is likely to be seen as a strain level difference. In other words, the differences in area under curve observed for different bacterial strains reflect their innate nature of difference which could be captured in this method. However, the linearity in the relationship for all the strains indicates a general nature of interaction between bacteria and PLL. It may be mentioned that the decrease in area under extinction spectrum from 1.0 to 10 CFU was nearly to the same extent as from 10 to 100 CFU. The plausible explanation is that at lower cell concentrations, PLL is in stoichiometric excess in solution and the difference in the amount of unbound PLL recovered when cell concentration increases from 1.0 to 10 CFU as well as 10 to 100 CFU is only minimal. Hence, similar extent of decrease in area under extinction spectrum is observed. However, as cell numbers increase beyond 100 CFU, quantitative sequestration of PLL takes place as a function of increasing cell number and the extent of decrease in area under extinction spectrum becomes more pronounced. It may be mentioned that the area under UV-vis extinction spectrum obtained for the lowest limit of detection i.e. for 10 CFU was close to 92.0 AU for E.coliMTCC 433 and was within the linear range of response of Au NP aggregation as a function of PLL concentration as evident from Fig. 1b. This fact was also applicable to all other bacterial strains.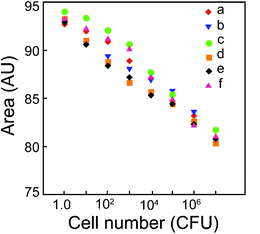 | ||
| Fig. 4 Area under UV-vis extinction spectrum as a function of bacterial cell numbers. (a) Escherichia coliMTCC 433, (b) Listeria monocytogenes Scott A, (c) Staphylococcus aureusMTCC 740, (d) Enterobacter aerogenesMTCC 2822, (e) Bacillus cereusMTCC 1305 and (f) Pseudomonas aeruginosaMTCC 2488. The area represents mean value of three independent measurements. | ||
The present method of bacterial cell estimation provided some interesting mechanistic facet which enabled sensitive estimation of bacterial cells in solution. In the absence of any bacterial cells, maximum Au NP aggregation by PLL was observed. Subsequently, when increasing concentration of bacterial cells in solution were used as a bait to sequester PLL, there was a progressive decline in Au NP aggregation. Essentially, bacterial cells effectively entrap PLL onto their surface by electrostatic interactions leading to a decrease in the availability of PLL in solution. Consequently, when Au NP is interacted with the unbound PLL fraction there is a systematic decrease in Au NP aggregation, which could account quantitatively for bacterial cell numbers. We refer this decline in Au NP aggregation which accounted for increasing bacterial cell numbers as “subtractive-aggregation” and Fig. 5 is an explicit pictorial representation of the phenomenon.
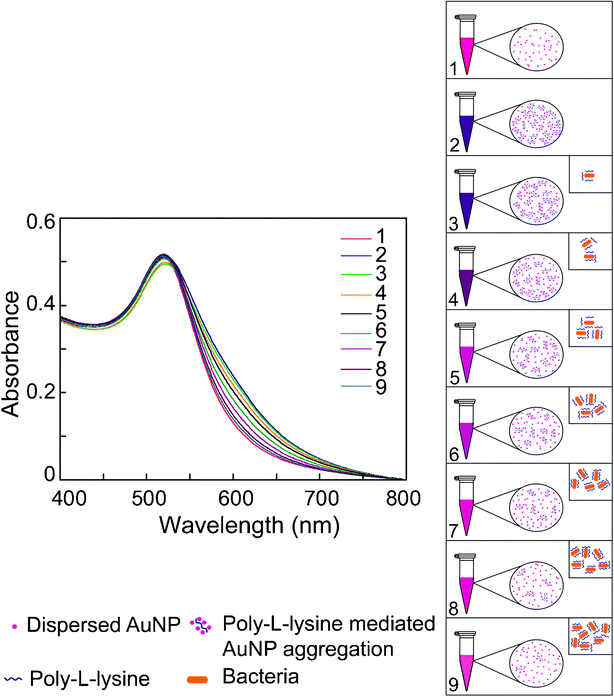 | ||
| Fig. 5 Pictorial representation of subtractive-aggregation as a measure of bacterial cell numbers. Traces 1: Au NP alone (control sample); 2: Maximum broadening of UV-vis extinction spectrum of Au NP in presence of 3.5 × 10−5% w/v PLL; 3–9: Progressive decline in broadening of UV-vis extinction spectrum of Au NP for samples with increasing bacterial cells. Panel on the right side depicting systematic decrease in Au NP aggregation by unbound PLL which is also manifested as a gradient of color change in Au NP solution in microcentrifuge tubes. Inset indicates incremental sequestration of PLL by bacterial cells as a function of cell number. | ||
We may state the bacterial cell-PLL interaction in solution as follows:
| [B] + [PLL] = [B-PLL] + [PLL]Free | (1) |
| [Δagg] = [aggMax] - [aggUnbound-PLL] | (2) |
| [ΔAagg] = [AaggMax] - [AaggUnbound-PLL] | (3) |
It is clear from Fig. 6 that cell numbers in the range of 101–107 CFU per 0.4 mL sample volume could be readily estimated on the basis of ΔAagg values. In the case of 1.0 CFU, ΔAagg values for the bacterial strains were very low. A two-way analysis of variance (ANOVA) followed by all pair wise multiple comparison (Holm-Sidak method) revealed that with regard to difference in ΔAagg values among the bacterial strains across every cell number (CFU), the values obtained for Enterobacter aerogenesMTCC 2822 versus Escherichia coliMTCC 433 as well as Bacillus cereusMTCC 1305 versus Escherichia coliMTCC 433 were statistically significant (P value <0.005). The relationship between subtractive-aggregation area and bacterial cell number is represented by linear equations (refer to ESI†, Table S2).
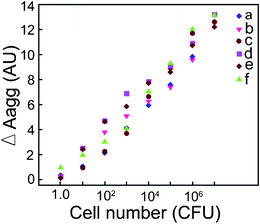 | ||
| Fig. 6 Plot for subtractive-aggregation (ΔAagg) versus bacterial cell numbers. (a) Escherichia coliMTCC 433, (b) Listeria monocytogenes Scott A, (c) Staphylococcus aureusMTCC 740, (d) Enterobacter aerogenesMTCC 2822, (e) Bacillus cereusMTCC 1305 and (f) Pseudomonas aeruginosaMTCC 2488. | ||
3.5 Estimation in centrifugal device
Our next aim was to explore the possibility of using a centrifugal filter device to separate bacterial cells following initial interaction with PLL. We conceived that the use of such a device which enables bacteria to be entrapped in the filter and unbound PLL to be recovered as flow through would cut down the number of steps involved as well as reduce the overall estimation time. Initial interaction of bacterial cells (ranging from 1.0–107 CFU) with PLL was conducted in a separate microcentrifuge tube so as to avoid any non-specific binding of cells or PLL to the filter device of the centrifugal tube. Subsequently, unbound PLL fraction recovered as flow through from the centrifugal device was used for initiating aggregation of Au NPs. With increase in cell numbers the extent of the spectral broadening diminished progressively (ESI†, Fig. S11a–b). This strong correlation between cell number and spectral broadening is explicit in the area under extinction spectrum indicated in Fig. S11c. The magnitude of the areas obtained for E.coliMTCC 433 and L. monocytogenes Scott A was comparable with that obtained earlier (Fig. 4). Quantitative estimations for maximum Au NP aggregation (AaggMax) as well as Au NP aggregation by unbound PLL (AaggUnbound-PLL) were also obtained and subtractive-aggregation area from Fig. S11d indicated that cell numbers in the range of 101–107 CFU could be readily estimated.The results obtained with the centrifugal device have encouraging implications. The separation of bacterial cells following initial interaction with PLL and recovery of the unbound PLL fraction as flow through is accomplished in a single step and thus the duration of the process of estimating cell numbers is reduced. There is ample scope of developing a rapid cell estimation kit based on the leads obtained. It would be interesting to explore the use of an appropriate membrane filter in the centrifugal device, which shows minimal non-specific binding to either bacterial cells or PLL so that even the initial PLL-bacteria interaction can be pursued in the same tube.
3.6 Estimation of viable versus non-viable cells
For any cell estimation method, determination of viable cell count is a significant point of contention which needs to be addressed. In order to test the potential of the present method of cell estimation to distinguish viable vis-a-vis non-viable cells, experiments were performed with cells subjected to various heat treatment. When 102 CFU of E. coliMTCC 433 were subjected to 55 °C for 30 min, the cells failed to survive the heat treatment as determined by plating, re-inoculation and subtractive-aggregation method (ESI†, Table S3). When higher cell numbers (106 and 104 CFU) of E. coliMTCC 433 were subjected to the same condition, enumeration of cells which survived the heat treatment could be performed. Interestingly, the cell numbers estimated by conventional plating and subtractive-aggregation method nearly coincided (refer to ESI†, Table S3), indicating reliability of the developed method in estimating viable cell numbers. Similar results were also obtained for L. monocytogenes Scott A (data not shown). For all other conditions of heat treatment, cell numbers could not be estimated indicating cell death, which was verified by lack of growth in plating as well as re-inoculation experiments (ESI†, Table S3). The UV-vis extinction spectrum obtained by interacting heat-killed cells (autoclaving) of E. coliMTCC 433 and L. monocytogenes Scott A with PLL followed by measuring Au NP aggregation by unbound PLL is indicated in Fig. 7. It was interesting to observe that for both bacteria, non-viable heat-killed cells failed to generate any change in the extent of Au NP aggregation and this trend was unequivocally observed for all cell numbers. It was also observed that cells subjected to heat treatment at 65 °C and 80 °C responded in a similar manner. We had also observed that in the absence of PLL, heat-killed bacterial cells alone had no effect on the characteristic extinction spectra of Au NPs. To account for the lack of change in the extent of Au NP aggregation in case of heat-killed non-viable cells, it may be construed that PLL fails to bind to heat-killed cells. As a consequence, bulk of the PLL is recovered as unbound fraction and hence the aggregation observed in case of heat-killed cells is more or less on par with that observed for PLL alone. Heat treatment at higher temperature presumably leads to outer membrane damage in bacterial cells. In that case, it is anticipated that the charge characteristics of such cells is likely to be lost. This would hinder the initial binding of PLL to bacterial cells, which is primarily driven by electrostatic interaction. To verify our hypothesis we stained heat-treated bacterial cells with propidium iodide (PI) which is an indicator of outer membrane damage. Viable cells with intact outer membrane are known to be refractory to PI uptake whereas membrane compromised cells are expected to readily incorporate PI, which upon intercalation with cellular DNA exhibits enhanced fluorescence.30 Evidence for outer membrane damage in heat-killed (autoclaved) cells is clearly observed in Fig. S12 (ESI†) wherein cells of E. coliMTCC 433 exhibited intense red fluorescence. Discrimination of viable and non-viable cells has been achieved earlier using fluorescent probes based on cell viability markers such as membrane potential, intracellular esterase activity and membrane integrity.31,32,33,34 These probes have been used alone or in combination to decipher live and dead cells quantitatively. Although the probes render considerable sensitivity and differentiating ability, they are used in conjunction with specialized techniques such as flow cytometry, which requires careful standardization and interpretation of results. In comparison, the present method of estimating viable cells is essentially label free, rapid and relatively simple in mode of operation.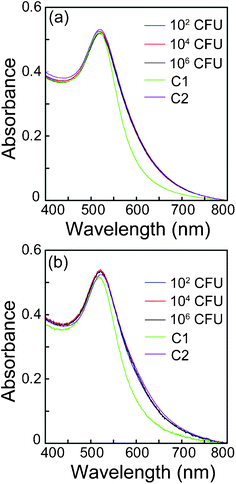 | ||
| Fig. 7 UV-vis extinction spectrum of Au NP in the presence of PLL following initial interaction of PLL with heat-killed bacteria. (a) E. coliMTCC 433, (b) L. monocytogenes Scott A. C1:Au NP alone (volume adjusted) and C2: Au NP interacted with 3.5 × 10−5% w/v PLL. | ||
3.7 Estimation of antimicrobial susceptibility by subtractive-aggregation
It was encouraging to observe that the present method of bacterial cell estimation clearly provided a measure of viable cell numbers on the basis of subtractive-aggregation of Au NPs. This motivated us to set up an experiment wherein the application potential of the method of cell estimation could be tested to determine viable bacterial cell numbers following exposure to an antimicrobial agent. We chose nisin as a model antimicrobial peptide and studied the effect of different concentrations of the peptide on the viability of the target strain Leuconostoc mesenteroidesNRRL B640. Nisin is a lantibiotic and a widely used food preservative which is known to act on the membrane and disrupt membrane potential of target bacterial strains.35,36 Preliminary experiments based on agar well diffusion assay clearly revealed that L. mesenteroidesNRRL B640 was susceptible to nisin, as evident from the zone of growth inhibition (ESI†, Fig. S13). Following treatment with varying levels of nisin, viable cell numbers of L. mesenteroidesNRRL B640 ascertained by both plating method as well as Au NP subtractive-aggregation was compared. The results of the experiment are indicated in Fig. 8. It is quite evident from Fig. 8a that the trend of Au NP aggregation by PLL as well as the same in the presence of varying cell numbers of L. mesenteroidesNRRL B640 as manifested by the broadening of extinction spectrum of Au NPs revealed a pattern similar to other bacterial strains shown in Fig. S10 (ESI†). Likewise the area under each UV-vis extinction spectrum obtained for L. mesenteroidesNRRL B640 decreased with increasing cell numbers (Fig. 8b). When subtractive-aggregation areas (ΔAagg) were calculated from the extinction spectra and plotted against cell numbers, a linear function was evident which revealed a strong correlation with cell numbers (Fig. 8c) as observed earlier for other bacterial strains (Fig. 6). Treatment of L. mesenteroidesNRRL B640 with increasing concentration of nisin resulted in a progressive decline in the viability of the cells. For instance conventional plating revealed that following exposure to a nisin concentration of 100, 200 and 400 IU/mL for 12 h, the viable cell count of the target strain reduced to 5.12, 4.63 and 3.20 log10 CFU per mL, respectively, as compared to the untreated cells wherein the cell number was nearly 6.0 log10 CFU per mL. At the highest nisin concentration of 800 IU/mL there was a dramatic decrease in viability of the cells resulting in a cell count as low as 2.3 log10 CFU per mL (Fig. 8d). When the cell numbers were estimated by the subtractive-aggregation method, the results nearly coincided with that of the plating method (Fig. 8d). By using the equation indicated in Fig. 8c, the cell numbers calculated by the subtractive-aggregation method amounted to 5.25, 4.59, 3.42 and 2.25 log10 CFU per mL, when cells were treated with 100, 200, 400 and 800 IU/mL nisin, respectively. Upon exposure to an antimicrobial agent like nisin it may be assumed that both viable as well as nisin-affected non-viable cells would be present in the sample. The latter would be high in number with increasing concentration of nisin. It is worthwhile to mention that the present method of viable cell estimation by Au NP subtractive-aggregation provided an accurate estimation of viable cells even in the midst of nisin- affected non-viable target cells present in the same sample. This feature renders the method on par with conventional plating for estimating viable cell numbers. However, the rapidity of the present method provides an edge over plating which is usually arduous and protracted. We have clearly demonstrated the application potential of the subtractive-aggregation method of viable bacterial cell estimation to test the potency of a model antimicrobial agent. This method could well be extended to screen other antimicrobial agents and candidate drugs in future.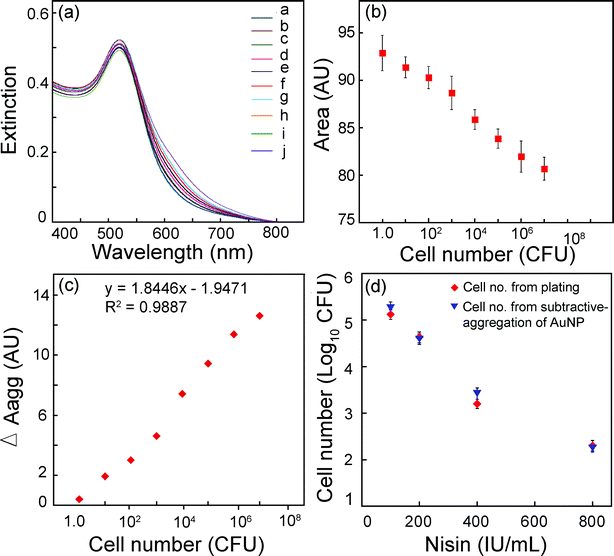 | ||
| Fig. 8 (a) UV-vis extinction spectrum for estimation of cell numbers for Leuconostoc mesenteroidesNRRL B640 by Au NP aggregation. Traces a–h correspond to decreasing cell numbers (107–1.0 CFU), trace i: Au NP alone (volume adjusted) and trace j: Au NP interacted with 3.5 × 10−5% w/v PLL. (b) Area under UV-vis extinction spectrum as a function of bacterial cell numbers. (c) Plot for subtractive-aggregation (ΔAagg) versus bacterial cell numbers. (d) Estimation of cell numbers for L. mesenteroidesNRRL B640 to determine antimicrobial susceptibility to nisin treatment. Cell number from subtractive-aggregation of Au NP was determined by calculating ΔAagg for nisin treated samples and using the equation shown in Panel c. | ||
4. Conclusions
In the present study, we have demonstrated the judicious use of a polycationic species PLL in conjunction with citrate-stabilized Au NPs and developed a convenient tool based on Au NP aggregation for rapid estimation of bacterial cells in solution. The significant achievements of the method is that it is applicable to a wide range of bacterial strains, offers high sensitivity with a detection limit of 10 CFU and can estimate viable cell numbers with a high degree of accuracy. The other benefits are that the method is operationally simple, devoid of any conjugation chemistry and is essentially non-destructive. The significance of the method is enhanced owing to the distinct possibility of translating the concept from laboratory bench to the development of a miniaturized centrifugal device for bacterial cell estimation. The reliability of the method in estimating viable cell numbers in solution vindicates its application potential in the domain of pathogen detection, screening of antimicrobial compounds and microbiological quality control. For practical applications, it would be important to assess the feasibility and promise of the method with complex samples. Development of appropriate strategies for sample preparation and sequestration of bacterial cells from complex matrix could significantly expand the application potential of the developed method.Acknowledgements
We thank the Department of Biotechnology (BT/01/NE/PS/08) and Department of Science and Technology, Ministry of Science and Technology, Government of India for research grants. We also thank Central Instruments Facility, IIT Guwahati for their valuable help. M.D.A. acknowledges IIT Guwahati, for a research fellowship.References
- N. Mancini, S. Carletti, N. Ghidoli, P. Cicehro, R. Burioni and M. Clementi, Clin. Microbiol. Rev., 2010, 23, 25–251 CrossRef.
- A. Lauri and P. O. Mariani, Genes Nutr., 2009, 4, 1–12 CrossRef CAS.
- T. Geng, M. T. Morgan and A. K. Bhunia, Appl. Environ. Microbiol., 2004, 70, 6138–6146 CrossRef CAS.
- O. Tokarskyy and D. L. Marshall, Food Microbiol., 2008, 25, 1–12 CrossRef CAS.
- Q. Lu, H. Lin, S. Ge, S. Luo, Q. Cai and C. A. Grimes, Anal. Chem., 2009, 81, 5846–5850 CrossRef CAS.
- R. Maalouf, C. Fournier-Wirth, J. Coste, H. Chebib, Y. Sailkali, O. Vittori, A. Errachid, C. Jean-Pierre, M. Claude and N. Jaffrezic-Renault, Anal. Chem., 2007, 79, 4879–4886 CrossRef CAS.
- Z. Shen, M. Huang, C. Xiao, Y. Zhang, X. Zeng and P. G. Wang, Anal. Chem., 2007, 79, 2312–2319 CrossRef CAS.
- S. S. Agasti, S. Rana, M. Park, C. K. Kim, C. You and V. M. Rotello, Adv. Drug Delivery Rev., 2010, 62, 316–328 CrossRef CAS.
- M. De, P. S. Ghosh and V. M. Rotello, Adv. Mater., 2008, 20, 4225–4241 CrossRef CAS.
- R. A. Sperling, P. R. Gil, F. Zhang, M. Zanella and W. J. Parak, Chem. Soc. Rev., 2008, 37, 1896–1908 RSC.
- C. J. Murphy, A. M. Gole, J. W. Stone, P. N. Sisco, A. M. Alkilany, E. C. Goldsmith and S. C. Baxter, Acc. Chem. Res., 2008, 41, 1721–1730 CrossRef CAS.
- C. M. Cobley, J. Chen, E. C. Cho, L. V. Wang and Y. Xia, Chem. Soc. Rev., 2011, 40, 44–56 RSC.
- J. Deka, A. Paul, A. Ramesh and A. Chattopadhyay, Langmuir, 2008, 24, 9945–9951 CrossRef CAS.
- J. Deka, A. Paul and A. Chattopadhyay, J. Phys. Chem. C, 2009, 113, 6936–6947 CAS.
- J. Deka, A. Paul and A. Chattopadhyay, Nanoscale, 2010, 2, 1405–1412 RSC.
- F. Xia, X. Zuo, R. Yang, Y. Xiao, D. Kang, A. Vallee-Belisle, X. Gong, J. D. Yuen, B. B. Y. Hsu, A. J. Heeger and K. W. Plaxco, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 10837–10841 CrossRef CAS.
- C. Fournier-Wirth and J. Coste, Biologicals, 2010, 38, 9–13 CrossRef.
- K. El-Boubbou, C. Gruden and X. Huang, J. Am. Chem. Soc., 2007, 129, 13392–13393 CrossRef CAS.
- C. Kaittanis, S. A. Naser and J. M. Perez, Nano Lett., 2007, 7, 380–383 CrossRef CAS.
- S. P. Ravindranath, L. J. Mauer, C. Deb-Roy and J. Irudayaraj, Anal. Chem., 2009, 81, 2840–2846 CrossRef CAS.
- A. J. Kell, G. Stewart, S. Ryan, R. Peytavi, M. Boissinot, A. Huletsky, M. G. Bergeron and B. Simard, ACS Nano, 2008, 2, 1777–1788 CrossRef CAS.
- W. J. Chen, P. J. Tsai and Y. C. Chen, Anal. Chem., 2008, 80, 9612–9621 CrossRef CAS.
- V. Berry, A. Gole, S. Kundu, C. J. Murphy and R. F. Saraf, J. Am. Chem. Soc., 2005, 127, 17600–17601 CrossRef CAS.
- W. W. Wilson, M. M. Wade, S. C. Holman and F. R. Champlin, J. Microbiol. Methods, 2001, 43, 153–164 CrossRef CAS.
- B. R. Panda, A. K. Singh, A. Ramesh and A. Chattopadhyay, Langmuir, 2008, 24, 11995–12000 CrossRef CAS.
- R. L. Phillips, O. R. Miranda, C. C. You, V. M. Rotello and U. H. F. Bunz, Angew. Chem., Int. Ed., 2008, 47, 2590–2594 CrossRef CAS.
- P. Laurino, R. Kikkeri, N. Azzouz and P. H. Seeberger, Nano Lett., 2011, 11, 73–78 CrossRef CAS.
- T. Yoshida and T. Nagasawa, Appl. Microbiol. Biotechnol., 2003, 62, 21–26 CrossRef CAS.
- M. Conte, F. Aliberti, L. Fucci and M. Piscopo, World J. Microbiol. Biotechnol., 2007, 23, 1679–1683 CrossRef CAS.
- R. Virto, P. Manas, I. Alvarez, S. Condon and J. Raso, Appl. Environ. Microbiol., 2005, 71, 5022–5028 CrossRef CAS.
- K. B. Amor, P. Breeuwer, P. Verbaarschot, F. M. Rombouts, A. D. L. Akkermans, W. M. De Vos and T. Abee, Appl. Environ. Microbiol., 2002, 68, 5209–5216 CrossRef.
- K. Papadimitriou, H. Pratsinis, G. Nebe-von-Caron, D. Kletsas and E. Tsakalidou, Int. J. Food Microbiol., 2006, 111, 197–205 CrossRef CAS.
- D. Hoefel, W. L. Groobya, P. T. Monisa, S. Andrews and C. P. Saint, J. Microbiol. Methods, 2003, 52, 379–388 CrossRef CAS.
- C. J. Bunthof, K. Bloemen, P. Breeuwer, F. M. Rombouts and T. Abee, Appl. Environ. Microbiol., 2001, 67, 2326–2335 CrossRef CAS.
- P. D. Cotter, C. Hill and R. P. Ross, Nat. Rev. Microbiol., 2005, 3, 777–788 CrossRef CAS.
- T. Zendo, F. Yoneyama and K. Sonomoto, Appl. Microbiol. Biotechnol., 2010, 88, 1–9 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: Description of control experiments, propidium iodide staining of heat-killed cells, agar well diffusion assay for nisin activity, additional UV-vis extinction spectrum, TEM analysis, equations for area under UV-vis extinction spectrum (AU) versuscell number (CFU), equations for calculating cell numbers from subtractive-aggregation, data for centrifugal device-based cell estimation, estimation of viable cell numbers for heat-treated cells, propidium iodide stained heat-killed cells of E.coli, Dose-dependent agar well diffusion assay for nisin activity against L. mesenteroides. See DOI: 10.1039/c1ra01023a/ |
| This journal is © The Royal Society of Chemistry 2012 |