Synthesis and reactivity of dialkyldithiophosphate complexes of ruthenium(II)†
Payel
Patel
,
Saira
Naeem
,
Andrew J. P.
White
and
James D. E. T.
Wilton-Ely
*
Department of Chemistry, Imperial College London, South Kensington Campus, London, SW7 2AZ, UK. E-mail: j.wilton-ely@imperial.ac.uk
First published on 2nd December 2011
Abstract
The ruthenium(II) complexes [Ru(CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHR){κ2-S2P(OEt)2}(CO)(PPh3)2] (R = But, C6H4Me-4) are formed on reaction of (NH4)[S2P(OEt)2] with [Ru(CHCHR)Cl(CO)(BTD)(PPh3)2] (BTD = 2,1,3-benzothiadiazole), while the enynyl complex [Ru(C(C
CHR){κ2-S2P(OEt)2}(CO)(PPh3)2] (R = But, C6H4Me-4) are formed on reaction of (NH4)[S2P(OEt)2] with [Ru(CHCHR)Cl(CO)(BTD)(PPh3)2] (BTD = 2,1,3-benzothiadiazole), while the enynyl complex [Ru(C(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CPh)CHPh)Cl(CO)(PPh3)2] loses a phosphine on treatment with the same ligand to yield [Ru(η3-PhCC–CCHPh){κ2-S2P(OEt)2}(CO)(PPh3)]. The stilbenyl complexes [Ru(CPhCHPh)Cl(CA)(PPh3)2] (A = O, S) react with (NH4)[S2P(OEt)2] to provide the dimers [Ru(CPhCHPh){μ–κ1–κ2–S2P(OEt)2}(CA)(PPh3)]2. Treatment of the thiocarbonyl complex with carbon monoxide results in migratory insertion of the vinyl and CS ligands to provide [Ru(η2-SCCPhCHPh){κ2-S2P(OEt)2}(CO)(PPh3)]. On treatment with excess 4-ethynyltoluene, the known compound [RuH{κ2-S2P(OEt)2}(CO)(PPh3)2] undergoes insertion of the alkyne to provide an alternative route to [Ru(CHCHC6H4Me-4){κ2-S2P(OEt)2}(CO)(PPh3)2]. The compounds [Ru(CHCHR){κ2-S2P(OEt)2}(CO)(PPh3)2] (R = n-C4H9, CH2OSi(But)Me2, CO2Me, Fc, CPh2OH, (HO)C6H10) were prepared cleanly by this route using HCCR. On stirring [RuH{κ2-S2P(OEt)2}(CO)(PPh3)2] over a longer period of time with excess 4-ethynyltoluene, the acetylide [Ru(CCC6H4Me–4){κ2-S2P(OEt)2}(CO)(PPh3)2] is generated. Heating [Ru(CHCHC6H4Me-4){κ2-S2P(OEt)2}(CO)(PPh3)2] with excess 3,3-dimethylbut-1-yne in 1,2-dichloroethane led to elimination of the vinyl ligand and formation of the acetylide compound [Ru(CCBut){κ2-S2P(OEt)2}(CO)(PPh3)2]. A small amount of the side product, [RuCl{κ2-S2P(OEt)2}(CO)(PPh3)2], was also formed. This could be obtained directly from the reaction of [RuH{κ2-S2P(OEt)2}(CO)(PPh3)2] with N-chlorosuccinimide. The related mixed-donor ligand, K[OP(S)(OEt)2], reacts with [Ru(CHCHC6H4Me-4)Cl(CO)(BTD)(PPh3)2] to yield [Ru(CHCHC6H4Me-4){κ2-OP(S)(OEt)2}(CO)(PPh3)2]. The molecular structure of [Ru(CHCHCPh2OH){κ2-S2P(OEt)2}(CO)(PPh3)2] was determined crystallographically.
CPh)CHPh)Cl(CO)(PPh3)2] loses a phosphine on treatment with the same ligand to yield [Ru(η3-PhCC–CCHPh){κ2-S2P(OEt)2}(CO)(PPh3)]. The stilbenyl complexes [Ru(CPhCHPh)Cl(CA)(PPh3)2] (A = O, S) react with (NH4)[S2P(OEt)2] to provide the dimers [Ru(CPhCHPh){μ–κ1–κ2–S2P(OEt)2}(CA)(PPh3)]2. Treatment of the thiocarbonyl complex with carbon monoxide results in migratory insertion of the vinyl and CS ligands to provide [Ru(η2-SCCPhCHPh){κ2-S2P(OEt)2}(CO)(PPh3)]. On treatment with excess 4-ethynyltoluene, the known compound [RuH{κ2-S2P(OEt)2}(CO)(PPh3)2] undergoes insertion of the alkyne to provide an alternative route to [Ru(CHCHC6H4Me-4){κ2-S2P(OEt)2}(CO)(PPh3)2]. The compounds [Ru(CHCHR){κ2-S2P(OEt)2}(CO)(PPh3)2] (R = n-C4H9, CH2OSi(But)Me2, CO2Me, Fc, CPh2OH, (HO)C6H10) were prepared cleanly by this route using HCCR. On stirring [RuH{κ2-S2P(OEt)2}(CO)(PPh3)2] over a longer period of time with excess 4-ethynyltoluene, the acetylide [Ru(CCC6H4Me–4){κ2-S2P(OEt)2}(CO)(PPh3)2] is generated. Heating [Ru(CHCHC6H4Me-4){κ2-S2P(OEt)2}(CO)(PPh3)2] with excess 3,3-dimethylbut-1-yne in 1,2-dichloroethane led to elimination of the vinyl ligand and formation of the acetylide compound [Ru(CCBut){κ2-S2P(OEt)2}(CO)(PPh3)2]. A small amount of the side product, [RuCl{κ2-S2P(OEt)2}(CO)(PPh3)2], was also formed. This could be obtained directly from the reaction of [RuH{κ2-S2P(OEt)2}(CO)(PPh3)2] with N-chlorosuccinimide. The related mixed-donor ligand, K[OP(S)(OEt)2], reacts with [Ru(CHCHC6H4Me-4)Cl(CO)(BTD)(PPh3)2] to yield [Ru(CHCHC6H4Me-4){κ2-OP(S)(OEt)2}(CO)(PPh3)2]. The molecular structure of [Ru(CHCHCPh2OH){κ2-S2P(OEt)2}(CO)(PPh3)2] was determined crystallographically.
Introduction
In contrast to dithiocarbamate1a–f and xanthate1c–h complexes of transition metals, which are well documented in the literature, compounds bearing other members of the 1,1-dithio ligand family have remained relatively neglected.1 In an effort to address this oversight, we have recently expanded our interest in dithiocarbamate complexes2 to the dithiocarboxylate1c–e betaine ligand class, NHC·CS2, prepared from N-heterocyclic carbenes (NHCs).3 All these species share the CS2 moiety as the basis of their chelation properties. In an effort to look beyond this class of ligands, we have started to investigate the possibilities afforded by the sulfur-derivatives of dialkylphosphates in the realm of coordination chemistry.Dialkyldithiophosphate species, [S2P(OR)2]−, have been employed in a range of settings with the most significant applications being the use of zinc dialkyldithiophosphates (ZDDP) as antioxidants and oil additives.4 In common with dithiocarbamates, early reports have centred on their use as analytical reagents5 and chelates for extraction purposes.6 They have even been employed to sequester ruthenium fission products from aqueous acid solution.7 In ruthenium chemistry, the coordination of dialkyldithiophosphate ligands has been explored sporadically with examples known for various oxidation states, including Ru(0).8 Unsurprisingly, most examples are of divalent ruthenium,9–13 though a number of Ru(III) compounds have also been reported,14 including homoleptic examples.15 In 2007, tetravalent compounds bearing this ligand were reported for the first time.16
Recently, ruthenium compounds bearing dialkyldithiophosphates, among other 1,1-dithio ligands, have been explored in a surgical setting as nitric oxide scavengers and to modulate metalloproteinase activity.17
Few organometallic ruthenium complexes have been reported bearing dialkyldithiophosphate ligands. Some representative examples are shown in Fig. 1. The first, [RuCl{κ2-S2P(OEt)2}(η6-p-cymene)],9a was obtained by addition of NH4[S2P(OEt)2] to the dimer, [RuCl2(η6-p-cymene)]2. The second originates from reaction of the Grubbs metathesis catalyst, [Ru(CHPh)Cl2(PCy3)2] with two equivalents of KS2P(OEt)2,9b while the third illustrates a bridging mode for the dialkoxydithiophosphate ligand.11
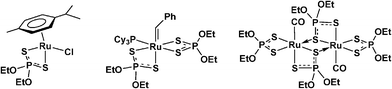 | ||
| Fig. 1 Examples of organometallic ruthenium dialkyldithiophosphate compounds and one in which a bridging mode is adopted. | ||
In this contribution, we report the synthesis and characterization of the first ruthenium vinyl complexes bearing the [S2P(OR)2]− and [S(O)P(OR)2]− ligands. We also show that the resulting compounds demonstrate reactivity which departs significantly from that displayed by the analogous dithiocarbamate and xanthate compounds.
Results and discussion
In 1986 it was demonstrated that alkynes inserted readily into the ruthenium–hydride bond of both [RuHCl(CO)(PPh3)3],18 and [RuHCl(CO)(PiPr3)2]19 to yield the vinyl complexes [Ru(CR1CHR2)Cl(CO)(PR3)2] (R = Ph, iPr). These coordinatively-unsaturated compounds have gone on to become an extremely versatile starting point for ruthenium vinyl chemistry.20–26 Our own interest has centred on the triphenylphosphine compounds [RuHCl(CA)(BTD)(PPh3)2] (A = O,25 S21j), which also insert alkynes into the Ru–H bond to yield [Ru(CR1CHR2)Cl(CA)(BTD)(PPh3)2] (A = O, S). The presence of the labile 2,1,3-benzothiadiazole (BTD) ligand confers both greater crystallinity on the materials and competes successfully with PPh3 to avoid contamination with tris(phosphine) byproducts. A number of comprehensive reviews have covered the fascinating area of ruthenium vinyl chemistry.26
A slight excess of ammonium diethyldithiophosphate, (NH4)[S2P(OEt)2], and [Ru(CHCHC6H4Me-4)Cl(CO)(BTD)(PPh3)2] were dissolved in dichloromethane and stirred for one hour. An immediate colour change was observed from red to yellow, suggesting displacement of the 2,1,3-benzothiadiazole (BTD) chromophore from the metal centre. After work up, the yellow product was analysed by 31P NMR spectroscopy to reveal two singlets at 32.7 and 94.8 ppm. The higher field resonance displayed a different chemical shift to the precursor, while still being typical of a triphenylphosphine ligand bonded to ruthenium. The lower field resonance was attributed to the new diethyldithiophosphate unit on the basis of the resonance of the starting material (112.8 ppm) and the expected deshielding associated with a pentavalent phosphorus centre. The 1H NMR spectrum of the product displayed typical resonances for the vinyl ligand at 7.48 and 5.25 ppm for Hα and Hβ protons (showing mutual JHH coupling of 16.9 Hz), respectively. The lower field resonance also showed coupling (doublet of triplets) to the phosphorus nuclei of the phosphine ligands (JHP = 4.1 Hz) suggesting a mutually trans arrangement for the phosphines. In addition to aromatic resonances for PPh3, the tolyl substituent gave rise to an AB system at 6.17 and 6.82 ppm (JAB = 8.0 Hz) as well as a singlet at 2.22 ppm for the methyl group. Close to this resonance was a feature not found in the vinyl precursor—a triplet at 0.89 ppm (JHH = 7.1 Hz). This resonance (OCCH3) and multiplets at 2.93 and 3.18 ppm (OCH2) were assigned to the ethoxy groups of the coordinated [S2P(OEt)2]− ligand. A triplet at 147.4 ppm (Cα) was observed in the 13C NMR spectrum, again showing coupling to mutually trans phosphorus nuclei, while the dithiophosphate ligand gave rise to two doublet resonances at 61.7 (JPC = 7.4 Hz) and 15.7 (JPC = 8.8 Hz) ppm. The retention of the carbonyl ligand was indicated by a triplet at 205.3 ppm (JPC = 14.9 Hz) in the same spectrum as well as an absorption at 1916 cm−1 in the solid state infrared spectrum. Two intense bands at the lower frequency values of 1015 and 947 cm−1 (not present in the precursor) were attributed to the dithiophosphate ligand. A molecular ion in the mass spectrum (FAB +ve mode) at m/z = 955 and good agreement of elemental analysis with calculated values confirmed the overall formulation (Scheme 1) to be [Ru(CHCHC6H4Me-4){κ2-S2P(OEt)2}(CO)(PPh3)2] (1). A similar reaction ensued between (NH4)[S2P(OEt)2], and [Ru(CHCHBut)Cl(CO)(BTD)(PPh3)2] to form [Ru(CHCHBut){κ2-S2P(OEt)2}(CO)(PPh3)2] (2) as a yellow product in reasonable yield. Spectroscopic data for this complex were similar to those obtained for compound 1 apart from features associated with the vinyl ligand, which gave rise to doublets (JHH = 16.1 Hz) at 4.61 (Hβ) and 6.08 (Hα) ppm in the 1H NMR spectrum and a singlet resonance at 0.36 ppm (tBu).
![Formation of ruthenium dithiophosphate complexes. (i) HCCC6H4Me-4 or HCCBut; (ii) (NH4)[S2P(OEt)2]; (iii) HCCR; (iv) N-chlorosuccinimide. BTD = 2,1,3-benzothiadiazole. R = C6H4Me-4 (1), But (2), n-C4H9 (4), CH2OSi(But)Me2 (5), CO2Me (6), Fc (7), CPh2OH (8), (HO)C6H10 (9).](/image/article/2012/RA/c1ra00973g/c1ra00973g-s1.gif) | ||
| Scheme 1 Formation of ruthenium dithiophosphate complexes. (i) HCCC6H4Me-4 or HCCBut; (ii) (NH4)[S2P(OEt)2]; (iii) HCCR; (iv) N-chlorosuccinimide. BTD = 2,1,3-benzothiadiazole. R = C6H4Me-4 (1), But (2), n-C4H9 (4), CH2OSi(But)Me2 (5), CO2Me (6), Fc (7), CPh2OH (8), (HO)C6H10 (9). | ||
The most closely related dialkyldithiophosphate complex to those presented here is [RuH{κ2-S2P(OEt)2}(CO)(PPh3)2] (3),13a,b formed from [RuHCl(CO)(PPh3)3]. The same product can be obtained in good yield from reaction of [RuHCl(CO)(BTD)(PPh3)2] with (NH4)[S2P(OEt)2] (Scheme 1). Spectroscopic and analytical data for 3 were found to agree well with those reported in the literature.13b Although [RuH{κ2-S2P(OEt)2}(CO)(PPh3)2] is coordinatively saturated, hemilability of the diethyldithiophosphate ligand would afford a vacant coordination site cis to the hydride, allowing coordination of an incoming alkyne and subsequent insertion into the Ru–H bond. Chelates such as dithiocarbamates are normally not sufficiently labile at room temperature to permit this. However, on treatment of [RuH{κ2-S2P(OEt)2}(CO)(PPh3)2] (3) with excess 4-ethynyltoluene, a single product was formed within an hour of stirring in dichloromethane at room temperature. This was identified as compound 1, providing a new route to vinyl complexes supported by the bidentate 1,1-dithio ligand (Scheme 1).
The scope of this synthetic route was explored with a range of terminal alkynes. In particular, the effect of increasing the steric bulk of the substituent was investigated (vide infra). Hex-1-yne reacts with 3 to provide [Ru{CHCH(n-C4H9)}{κ2-S2P(OEt)2}(CO)(PPh3)2] (4) after 40 min. The new vinyl ligand gives rise to resonances at 6.13 and 4.37 ppm (α and β-protons, respectively) in the 1H NMR spectrum as well as multiplets in the region 0.73–1.60 ppm for the n-butyl unit. The same route was employed to form [Ru{CHCHCH2OSi(But)Me2}{κ2-S2P(OEt)2}(CO)(PPh3)2] (5) from 1.5 equivalents of the unusual alkyne, HCCCH2OSi(But)Me2. In addition to typical resonances for the dithiophosphate ligand, 1H NMR analysis revealed a doublet at 6.51 ppm (JHH = 15.8 Hz) for the α-proton, while a multiplet was observed for the β-proton at 4.53 ppm, coupling to the OCH2 resonance at 3.44 (d, JHH = 5.2 Hz) ppm. Singlets were observed for the tertiarybutyl (0.86 ppm) and methyl (0.09 ppm) protons. Methyl propiolate reacted with 3 in a similar way to yield [Ru(CHCHCO2Me){κ2-S2P(OEt)2}(CO)(PPh3)2] (6). An additional metal centre was introduced to the system through the use of ethynylferrocene, which reacted with 3 within 20 min to yield [Ru(CHCHFc){κ2-S2P(OEt)2}(CO)(PPh3)2] (7). The retention of the ferrocenyl group was shown by the resonances at 3.88 (singlet), 3.84 and 3.39 ppm (broadened triplets) in the 1H NMR spectrum along with typical resonances for the α- and β-vinyl protons. In all cases, both phosphine ligands were retained.
This route was also used to prepare the γ-hydroxyvinyl complex [Ru(CHCHCPh2OH){κ2-S2P(OEt)2}(CO)(PPh3)2] (8) from 3 and 1,1-diphenylpropyn-1-ol. The formation of the vinyl ligand was confirmed by 1H NMR spectroscopy, which revealed the presence of a doublet at 5.40 ppm (JHH = 16.4 Hz), assigned to Hβ and a singlet at 0.92 ppm for the hydroxy proton. The resonance for Hα was not located and was presumed to be obscured by the aromatic resonances. Single crystals of the complex were grown by slow diffusion of ethanol into a dichloromethane solution of 8 and the structure determined (See Structural Section and Fig. 2).
![Molecular structure of [Ru(CHCHCPh2OH){κ2-S2P(OEt)2}(CO)(PPh3)2] (8). Selected bond distances (Å) and angles (°): Ru–C(26) 1.835(3), Ru–C(10) 2.081(2), Ru–P(46) 2.3780(6), Ru–P(27) 2.4125(7), Ru–S(3) 2.5238(6), Ru–S(1) 2.5896(6), S(1)–P(2) 1.9808(9), P(2)–O(4) 1.586(2), P(2)–O(7) 1.592(2), P(2)–S(3) 1.9825(9), C(10)–C(11) 1.329(4), P(46)–Ru–P(27) 173.25(2), S(3)–Ru–S(1) 78.28(2), S(1)–P(2)–S(3) 109.07(4), C(11)–C(10)–Ru 127.7(2).](/image/article/2012/RA/c1ra00973g/c1ra00973g-f2.gif) | ||
| Fig. 2 Molecular structure of [Ru(CHCHCPh2OH){κ2-S2P(OEt)2}(CO)(PPh3)2] (8). Selected bond distances (Å) and angles (°): Ru–C(26) 1.835(3), Ru–C(10) 2.081(2), Ru–P(46) 2.3780(6), Ru–P(27) 2.4125(7), Ru–S(3) 2.5238(6), Ru–S(1) 2.5896(6), S(1)–P(2) 1.9808(9), P(2)–O(4) 1.586(2), P(2)–O(7) 1.592(2), P(2)–S(3) 1.9825(9), C(10)–C(11) 1.329(4), P(46)–Ru–P(27) 173.25(2), S(3)–Ru–S(1) 78.28(2), S(1)–P(2)–S(3) 109.07(4), C(11)–C(10)–Ru 127.7(2). | ||
Attempts to obtain crystals of compound 1 from analytically pure (as determined by NMR spectroscopy) samples led on a number of occasions to the isolation of crystals of the hydride (3), for which the structure is known.12b This indicated that the insertion into the Ru–H bond is reversible (β-elimination) over longer periods in solution.
Treatment of [Ru(CHCHCPh2OH)(κ2-S,N-MI)(CO)(PPh3)2] (MI = 1-methylimidazole-2-thiolate) with HBF4 leads to dehydration of the hydroxyvinyl ligand and generation of the vinylcarbene complex, [Ru(CHCHCPh2)(κ2-S,N-MI)(CO)(PPh3)2]BF4.21g Thus, a similar reaction was attempted with compound [Ru(CHCHCPh2OH){κ2-S2P(OEt)2}(CO)(PPh3)2] (8). However, an intractable mixture of products was obtained on treatment of 8 with one equivalent of HBF4, CF3CO2H or p-toluenesulfonic acid, possibly due to reactions taking place at both the dialkyldithiophosphate ligand and the vinyl moiety.
Another γ-hydroxyvinyl complex, [Ru{CHCH(HO)C6H10}{κ2-S2P(OEt)2}(CO)(PPh3)2] (9), was formed on addition of 1-ethynyl-1-cyclohexanol to [RuH{κ2-S2P(OEt)2}(CO)(PPh3)2] (3). In addition to two doublets for the vinyl protons at 6.58 and 4.79 (JHH = 16.4 Hz) ppm, the 1H NMR spectrum displayed multiplets at 1.34, 1.15, 0.92 and 0.78 ppm for the cyclohexyl protons. A singlet at 1.61 ppm was tentatively assigned to the hydroxy proton on the vinyl ligand. Similar resonances to those observed for the other complexes were noted for the ethoxy substituents of the diethyldithiophosphate ligand.
Stirring [RuH{κ2-S2P(OEt)2}(CO)(PPh3)2] (3) in dichloromethane for 3 h (or heating for 10 min in toluene) with excess 4-ethynyltoluene yielded a single new product in low yield after work up. The 1H NMR spectrum of this isolated product revealed retention of the [S2P(OEt)2]− ligand as well as resonances for a tolyl substituent with an AB system at 6.44 and 6.83 (JAB = 7.8 Hz) ppm and a methyl resonance at 2.23 ppm. However, no resonances for a vinyl ligand were evident. The infrared spectrum (solid state) gave rise to a carbonyl absorption at 1936 cm−1 as well as a medium intensity peak at 2105 cm−1. This was assigned to a νCC band for an acetylide ligand bonded to the ruthenium centre and suggested the formulation of [Ru(CCC6H4Me–4){κ2-S2P(OEt)2}(CO)(PPh3)2] (10) for the product (Scheme 1). The carbon nuclei of the acetylide ligand were found to resonate at 115.8 (s, Cβ) and 108.1 (t, Cα, JPC = 21.0 Hz) ppm. Mass spectrometry (FAB, +ve ion) revealed a molecular ion at m/z = 954. Elemental analysis further corroborated the composition of 10. It is likely that the initial reaction between 3 and the excess alkyne forms the vinyl compound (1), before the vinyl ligand is displaced and the acetylide formed. To confirm this 1 was treated with excess HCCC6H4Me-4 and heated at reflux for 10 min in toluene. In addition to the formation of 10, close inspection of the 1H NMR spectrum of the crude reaction product revealed resonances assigned to H2CC(H)C6H4Me-4 at 5.26 (JHH = 10.9 Hz), 5.78 (JHH = 17.6 Hz) and 6.77 (JHH = 17.6, 10.9 Hz) ppm.
In a similar manner, the compound [Ru(CCBut){κ2-S2P(OEt)2}(CO)(PPh3)2] (11) was formed by heating [Ru(CHCHC6H4Me-4){κ2-S2P(OEt)2}(CO)(PPh3)2] (1) with excess HCCBut in tetrahydrofuran or toluene. The presence of the new acetylide ligand was indicated by a singlet resonance integrating to 9 protons at 0.76 ppm in the 1H NMR spectrum of 11.
A plausible mechanism for these reactions involves opening of the diethyldithiophosphate chelate followed by oxidative addition of the alkyne to give a transient Ru(IV) hydrido vinyl acetylide species, which then eliminates the vinyl ligand as the spectroscopically observed alkene (Scheme 2). Although analogous vinyl to acetylide transformations have been observed in the dithiocarbamate compounds [Ru(CHCHC6H4Me-4){κ2-S2CNR2}(CO)(PPh3)2],2f,24l the ease with which they take place in these dialkyldithiophosphate compounds is noteworthy. Of course, it is possible that, rather than hemilability of the 1,1-dithio ligand, a phosphine ligand dissociates to create the vacant site required for the transformations from hydride to vinyl to acetylide. However, following the conversion of [RuH{κ2-S2P(OEt)2}(CO)(PPh3)2] (3) to 1 to 10 by 31P NMR in CD2Cl2 fails to show any free triphenylphosphine during the course of the reaction.
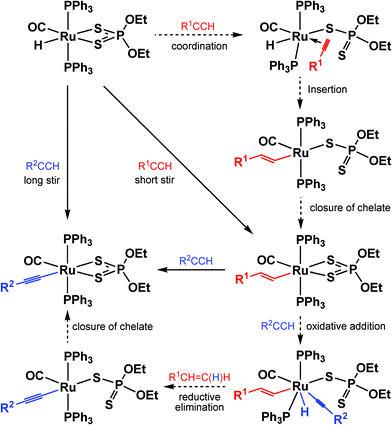 | ||
| Scheme 2 Possible mechanism of formation of vinyl and acetylide diethyldithiophosphate complexes. | ||
Initially, 1,2-dichloroethane was used for the transformation of 3 to 11 with excess HCCBut. In this reaction, a side product was observed in very low yield, which was identified as [RuCl{κ2-S2P(OEt)2}(CO)(PPh3)2] (12) on the basis of spectroscopic and analytical data. Compound 12 can be obtained directly from treatment of 3 with N–chlorosuccinimide.
The only previously reported ruthenium dialkyldithiophosphate complex with a σ-organyl ligand is the acetylide complex, [Ru(CCPh){κ2-S2P(OEt)2}(η6-p-cymene)]. This was obtained in unusual fashion by treatment of [RuCl{κ2-S2P(OEt)2}(η6-p-cymene)] with the molybdenum alkynyl transfer agent, [Mo(CCPh)(η3-allyl)(CO)2(bipy)].9g
The enynyl compound [Ru(C(CCPh)CHPh)Cl(CO)(PPh3)2] reacted readily with (NH4)[S2P(OEt)2] to provide a yellow complex. Analysis of the crude product by 31P NMR revealed significant amounts of free PPh3 and some OPPh3. 1H NMR analysis confirmed the presence of the enynyl ligand (Hβ at 6.39 ppm) and integration of this proton with those in the aromatic region indicated the presence of only one triphenylphosphine ligand. Similar resonances for the coordinated [S2P(OEt)2]− ligand were observed to those found in the spectra of 1—12. The retention of the carbonyl ligand was also confirmed by the solid state infrared spectrum (νCO at 1921 cm−1). The composition was supported by elemental analysis to be [Ru(C(CCPh)CHPh){κ2-S2P(OEt)2}(CO)(PPh3)] (13), as shown in Scheme 3. Three possible structures (Fig. 3) are consistent with the spectroscopic (1H, 31P NMR, IR spectroscopy) and elemental analysis data.
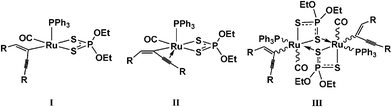 | ||
| Fig. 3 Possible structures for compounds 13, 14 or 15. | ||
![Formation of ruthenium stilbenyl diethyldithiophosphate complexes. A = O, S. (i) (NH4)[S2P(OEt)2]; (ii) CO.](/image/article/2012/RA/c1ra00973g/c1ra00973g-s3.gif) | ||
| Scheme 3 Formation of ruthenium stilbenyl diethyldithiophosphate complexes. A = O, S. (i) (NH4)[S2P(OEt)2]; (ii) CO. | ||
The starting material [Ru(C(CCPh)CHPh)Cl(CO)(PPh3)2] is coordinatively unsaturated,27a and, barring steric factors, it would be unusual for the addition of a bidentate donor, [S2P(OEt)2]−, to yield another 16-electron product (I). Instead, coordinative saturation could be achieved (in I) by interaction of the triple bond with the metal centre as shown in II. This has precedent,27b,c most closely related being the structure of [Ru(η3-PhCC–CCHPh)(CO)2(PPh3)2]PF6.27d If structure II were adopted, resonances for the carbon nuclei of the carbon–carbon triple bond would show coupling to phosphorus through the metal centre. However, in a proton decoupled 13C NMR spectrum of 13, the region in which resonances for these nuclei are typically found was obscured by other features in the aromatic region.
Mass spectrometry measurements (FAB, +ve mode) revealed a molecular ion corresponding to a mononuclear species for 13 (m/z = 780), however, this must be viewed with some caution as fragmentation in the mass spectrometer can easily occur. Hill and co-workers27d have reported that the compound [Ru(η3-PhCC–CCHPh)(CO)2(PPh3)2]PF6 shows no clear ν(CC) absorption in the IR spectrum for the coordinated triple bond. Similarly, no band for an uncoordinated triple bond (typically around 2150 cm−1) was observed in 13. While these data pointed tentatively towards formulation II, the alternative, dimeric structure III could not be ruled out at that stage.
A simple but often overlooked technique for measuring molecular weight is the Signer osmometry method,28 which is based on the fact that the vapour pressure of a solution depends upon the mole fraction of dissolved solute. Using Vaska's complex, [IrCl(CO)(PPh3)2], as a stable reference compound of comparable molecular mass, precise amounts of this reference and 13 were dissolved in dichloromethane and allowed to equilibrate in the apparatus (Supplementary Information†) under a partial vacuum. After two days, the measurements had stabilised to show that the mass calculated from the readings of the apparatus was within 5% of the mass of the monomer, ruling out III. Accordingly, the likely formulation of 13 was taken to be [Ru(η3-PhCC–CCHPh){κ2-S2P(OEt)2}(CO)(PPh3)], as shown in Figure 3 (II) and Scheme 3:
The stilbenyl compound, [Ru(CPhCHPh)Cl(CO)(PPh3)2], was also found to react with (NH4)[S2P(OEt)2] to yield a monophosphine compound. Measurements using the Signer apparatus suggested that, in contrast to 13, this complex was actually a dimer (Fig. 3, III). Indeed, in the absence of an adjacent donor (e.g., a triple bond in 13), it is more difficult to invoke a plausible stabilising interaction provided by the stilbenyl ligand. Accordingly the complex was formulated as a dimer [Ru(CPhCHPh){μ,κ1,κ2-S2P(OEt)2}(CO)(PPh3)]2 (14) (Scheme 3). This assignment is supported by literature precedent in the complex [Ru{κ2-S2P(OEt)2}{μ,κ1,κ2-S2P(OEt)2}(CO)]2, shown in Fig. 1.11
The thiocarbonyl variant of the precursor, [Ru(CPhCHPh)Cl(CS)(PPh3)2] reacted to give dimeric [Ru(CPhCHPh){μ,κ1,κ2-S2P(OEt)2}(CS)(PPh3)]2 (15) under the same conditions (Scheme 3). The main spectroscopic differences between 14 and 15 were the intense νCO (1965 cm−1 in 14) and νCS (1280 cm−1 in 15) absorptions.
We have reported recently that thiocarbonyl vinyl species such as [Ru(CR1CHR2)Cl(CS)(PPh3)2] (R1 = H, Ph; R2 = Ph) react with carbon monoxide by migratory insertion of the thiocarbonyl and vinyl ligands to form the thioacyl species, [Ru(SCCR1CHR2)Cl(CO)(PPh3)2].21j In order to ascertain whether migration could be induced in complexes bearing the diethyldithiophosphate ligand, carbon monoxide was bubbled through a dichloromethane solution of 15. An immediate colour change was observed from yellow to deep red. The product isolated gave rise to two new resonances in the 31P NMR spectrum at 49.7 and 102.7 ppm. A sharp singlet at 7.94 ppm was observed for the Hβ proton in the 1H NMR spectrum, showing no sign of coupling to the triphenylphosphine ligands. This compares well to the value of 7.38 ppm found in [Ru(SCCPhCHPh)Cl(CO)(PPh3)2].21j A new absorption assigned to νCO was observed at 1910 cm−1 and a less intense absorption at 1256 cm−1 was attributed to νCS of the thioacyl ligand. The composition of the product was confirmed by mass spectrometry (molecular ion at m/z = 801), Signer measurements and elemental analysis to be [Ru(η2-SCCPhCHPh){κ2-S2P(OEt)2}(CO)(PPh3)] (16).
Passing carbon monoxide through a dichloromethane solution of the carbonyl analogue (14) did not result in the corresponding acyl complex, but instead gave incomplete conversion to the hydride 3, presumably by β-elimination of diphenylacetylene.
In the context of our work on 1,1-dithio ligands, we have also explored mixed-donor ligands such as Schiff bases,21h1-methylimidazole-2-thiolate21g and IMes·CSNPh (IMes = 1,3-dimesitylimidazol-2-ylidene).3d Thus, it was decided to move beyond symmetrical donor combinations to explore briefly the dialkylthiophosphate ligand, [S(O)P(OEt)2]−. The potassium salt of this ligand reacted readily with [Ru(CHCHC6H4Me-4)Cl(CO)(BTD)(PPh3)2] to yield a yellow product (Scheme 4). In the 31P NMR spectrum, two new resonances were observed at 29.7 and 48.8 ppm, the latter being attributed to the thiophosphate ligand (the phosphorus nucleus in the free ligand resonates at 56.4 ppm). The remaining spectroscopic data were similar to those found for compound 1. The overall formulation was assigned to be [Ru(CHCHC6H4Me-4){κ2-S(O)P(OEt)2}(CO)(PPh3)2] (17).
![Formation of ruthenium diethylthiophosphate complex (17). (i) K[S(O)P(OEt)2]; R = C6H4Me–4, BTD = 2,1,3-benzothiadiazole.](/image/article/2012/RA/c1ra00973g/c1ra00973g-s4.gif) | ||
| Scheme 4 Formation of ruthenium diethylthiophosphate complex (17). (i) K[S(O)P(OEt)2]; R = C6H4Me–4, BTD = 2,1,3-benzothiadiazole. | ||
The exact regiochemistry at the ruthenium centre is difficult to establish unambiguously. The depiction in Scheme 4 is based on the arrangement observed in the vinyl complexes chelated by the 1-methylimidazole-2-thiolate ligand in [Ru(CR1CHR2)(κ2-S,N-MI)(CO)(PPh3)2], with the sulfur donor trans to the vinyl ligand.21g
Structural section
The structure of complex 8 is based on a distorted octahedral arrangement with cis-angles at the metal centre in the range 78.28(2)–100.03(8)°, the smallest of which is the S(3)–Ru–S(1) angle. The S(1)–P(2)–S(3) angle of the diethyldithiophosphate ligand is relatively large at 109.07(4)° compared to 1,1-dithio ligands such as dithiocarbamates and xanthates. The same angle in the structure of [RuH{κ2-S2P(OEt)2}(CO)(PPh3)2] (3) is 108.11(7)°.13b The geometry at the pentavalent phosphorus is tetrahedral with P(2)–S(3) and S(1)–P(2) distances of 1.9825(9) and 1.9808(9) Å, respectively. The steric effect of the ethoxy substituents is apparent in the reactivity observed (ready loss of a phosphine), however, there is little effect on the P(46)–Ru–P(27) angle, which shows only a modest deviation from linearity at 173.25(2)°. The Ru–S(3) and Ru–S(1) distances of 2.5238(6) and 2.5896(6) Å, respectively, are not equal and indicate the superior trans influence of the vinyl ligand, causing an elongation of the Ru–S(1) bond. Compound 8 was formed by insertion of an alkyne into a Ru–H bond, a process which typically occurs regiospecifically to yield the E-isomer. This is reflected in the observed regiochemistry at the double bond of the vinyl ligand in the structure of 8. The C(10)–C(11) distance of 1.329(11) Å is typical for a double bond between carbon atoms.29 Otherwise the structural data associated with the vinyl ligand are unremarkable.Conclusion
Earlier reports of dialkyldithiophosphate ligands suggested very similar coordination chemistry to that displayed by the related 1,1-dithio ligands, dithiocarbamates and xanthates, which have been much more intensively investigated. Although our initial experiments supported this view, subsequent reactions took a markedly different course. This can be traced, in part, to the greater steric impact of the ligand on the coordination environment of the metal (compared to dithiocarbamates or xanthates). This is illustrated by the spontaneous loss of a phosphine when disubstituted vinyl complexes are prepared. The hemilability of coordinated [S2P(OEt)2]− at room temperature is also superior to that observed by other 1,1-dithio chelates. This allows facile insertion of alkynes to form vinyl ligands, displacement of vinyl ligands to form acetylides and coordination of carbon monoxide to induce migratory insertion and thioacyl formation. These properties have considerable potential for exploitation in a catalytic context. Future experiments will include the preparation of a series of dialkyldithiophosphate ligands with differing steric profiles in order to determine the effect of this steric bulk on the reactivity at the metal centre. Investigations focusing on this aspect are currently underway.Experimental section
General Comments. All experiments were carried out under aerobic conditions and the products obtained appear indefinitely stable towards the atmosphere, whether in solution or in the solid state. Solvents were used as received from commercial sources. The complexes [RuHCl(CO)(BTD)(PPh3)2],30 [Ru(CHCHR)Cl(CO)(BTD)(PPh3)2] (R = But, C6H4Me-4) were prepared by literature procedures,25 only using commercially available 2,1,3-benzothiadiazole (BTD) in place of 2,1,3-benzoselenadiazole (BSD). The compounds [Ru(CPhCHPh)Cl(CA)(PPh3)2] (A = O,18 S21j) and [Ru(C(CCPh) CHPh)Cl(CO)(PPh3)2]31 were prepared as described elsewhere. Ammonium diethyldithiophosphate was obtained from Fisher Scientific and potassium diethylthiophosphate was purchased from Sigma-Aldrich. Petroleum ether refers to the fraction boiling in the range 40–60 °C. Electrospray and Fast Atom Bombardment (FAB) mass data were obtained using Micromass LCT Premier and Autospec Q instruments, respectively. Infrared data were obtained using a Perkin-Elmer Spectrum 100 FT-IR spectrometer. Characteristic phosphine-associated infrared data are not reported. Unless otherwise indicated, NMR spectroscopy was performed at 25 °C using Varian Mercury 300 and Bruker AV400 spectrometers. All couplings are in Hertz. Elemental analyses were provided by London Metropolitan University.
[Ru(CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) CHC6H4Me-4){κ2-S2P(OEt)2}(CO)(PPh3)2] (1)
CHC6H4Me-4){κ2-S2P(OEt)2}(CO)(PPh3)2] (1)
a) [Ru(CHCHC6H4Me-4)Cl(CO)(BTD)(PPh3)2] (200 mg, 0.212 mmol) and (NH4)[S2P(OEt)2] (47 mg, 0.233 mmol) were dissolved in dichloromethane (20 mL) and ethanol (10 mL). The reaction mixture was stirred for 1 h. Rotary evaporation to a solvent volume of approximately 10 mL led to precipitation of the yellow product. This was washed with ethanol (10 mL) and petroleum ether (10 mL) and dried. Yield: 147 mg (72%). b) [RuH{κ2-S2P(OEt)2}(CO)(PPh3)2] (40 mg, 0.048 mmol) and HCC6H4Me-4 (11.3 mg, 0.097 mmol) were dissolved in dichloromethane (10 mL). The reaction mixture was stirred for 1 h. Rotary evaporation of solvent led to precipitation of the yellow product, which was washed and dried as above (Yield: 33 mg, 70%). IR: 1916 (νCO), 1514, 1186, 1015, 947, 789, 770, 671 cm−1. NMR (CD2Cl2) 1H: 0.89 (t, 6H, OCCH3, JHH = 7.1 Hz), 2.22 (s, 3H, tolyl–CH3), 2.93, 3.18 (m × 2, 4H, OCH2), 5.25 (d, 1H, Hβ, JHH = 16.9 Hz), 6.17, 6.82 (AB, 4H, C6H4, JAB = 8.0 Hz), 7.34–7.56 (m, 30H, C6H5), 7.48 (dt, 1H, Hα, JHH = 16.9 Hz, JHP = 4.1 Hz) ppm. 13C: 205.3 (t, CO, JPC = 14.9 Hz), 147.4 (t, Cα, JPC = 14.0 Hz), 138.7 (s, tolyl–C1), 135.4 (tv, o/m-PC6H5, JPC = 5.0 Hz), 135.1 (s(br), Cβ), 133.4 (tv, ipso-PC6H5, JPC = 20.6 Hz), 133.4 (s, tolyl–C4), 129.7 (s, p-PC6H5), 126.6 (s, tolyl–C2,6), 127.8 (tv, o/m–PC6H5, JPC = 4.3 Hz), 124.6 (s, tolyl–C2,6), 61.7 (d, OCH2, JPC = 7.4 Hz), 20.9 (d, tolyl–CH3), 15.7 (d, OCH2, JPC = 8.8 Hz) ppm. 31P NMR: 32.7 ppm (s, PPh3), 94.8 (s, S2P) ppm. MS (ES +ve) m/z (abundance): 955(3) [M]+. Calcd. for C50H49O3P3RuS2: C 62.8, H 5.2%. Found: C 59.0, H 4.5%.
[Ru(CHCHBut){κ2-S2P(OEt)2}(CO)(PPh3)2] (2)
[Ru(CHCHBut)Cl(CO)(BTD)(PPh3)2] (100 mg, 0.110 mmol) and (NH4)[S2P(OEt)2] (25 mg, 0.121 mmol) were dissolved in dichloromethane (20 mL) and ethanol (10 mL). The reaction mixture was stirred for 1 h. Rotary evaporation of solvent led to precipitation of an orange solid, which was then dissolved in dichloromethane (5 mL). This was then passed through celite, and treated with ethanol (10 mL). Rotary evaporation to a solvent volume of approximately 10 mL led to precipitation of the orange solid. This was then washed with ethanol (10 mL) and petroleum ether (10 mL) and dried. Yield: 55 mg (47%). IR: 1940 (νCO), 1480, 1433, 1094, 693 cm−1. NMR (CD2Cl2) 1H: 0.36 (s, 9H, But), 0.85 (t, 6H, OCCH3, JHH = 7.0 Hz), 3.01 (m, 4H, OCH2), 4.61 (d, 1H, Hβ, JHH = 16.1 Hz), 6.08 (dt, 1H, Hα, JHH = 16.1 Hz, JHP = 3.6 Hz), 7.32–7.69 (m, 30H, C6H5) ppm. 31P NMR: 31.4 (s, PPh3), 95.5 (s, S2P) ppm. MS (FAB +ve) m/z (abundance): 922(1) [M]+, 660(7) [M − PPh3]+. Calcd. for C47H51O3P3RuS2: C 61.2, H 5.6%. Found: C 61.2, H 5.5%.
[RuH{κ2-S2P(OEt)2}(CO)(PPh3)2] (3)
[RuHCl(CO)(BTD)(PPh3)2] (150 mg, 0.182 mmol) and (NH4)[S2P(OEt)2] (40.6 mg, 0.200 mmol) were dissolved in dichloromethane (20 mL) and ethanol (10 mL). The reaction mixture was stirred for 1 h. Rotary evaporation to a solvent volume of approximately 10 mL led to precipitation of the green product. This was washed with ethanol (10 mL) and petroleum ether (10 mL) and dried. Yield: 106 mg (70%). IR: 1948 ν(Ru–H), 1922 (νCO), 1389, 1185, 1035, 1015, 945, 649 cm−1. NMR (CD2Cl2) 1H: −11.87 (t, 1H, RuH, JHP = 24.0 Hz), 0.87 (t, 6H, OCCH3, JHH = 8.0 Hz), 3.00–3.15 (m, 4H, OCH2), 7.40–7.73 (m, 30H, C6H5) ppm. 31P NMR: 41.8 (s, PPh3), 94.5 (s, S2P) ppm. MS (FAB +ve) m/z (abundance): 839(5) [M]+. Calcd. for C41H41O3P3RuS2: C 58.6, H 4.9%. Found: C 58.7, H 4.8%.[Ru{CHCH(n-C4H9){{κ2-S2P(OEt)2}(CO)(PPh3)2] (4)
[RuH{κ2-S2P(OEt)2}(CO)(PPh3)2] (50 mg, 0.061 mmol) and 1-hexyne (10 mg, 0.122 mmol) were dissolved in dichloromethane (20 mL). The reaction mixture was stirred for 40 min, revealing a colour change from green to yellow. Rotary evaporation of all solvent, and further recrystallisation with dichloromethane (10 mL) and ethanol (10 mL) led to precipitation of the yellow product. This was washed with ethanol (10 mL) and petroleum ether (10 mL) and dried. Yield: 38 mg (68%). IR: 1914 (νCO), 1572, 1387, 1185, 1017, 942, 770, 670 cm−1. NMR (CD2Cl2) 1H: 0.73–1.60 (m, 9H, (CH2)3CH3), 0.88 (t, 6H, OCCH3, JHH = 7.1 Hz), 2.96, 3.14 (m×2, 4H, OCH2), 4.37 (m, 1H, Hβ), 6.13 (dt, 1H, Hα, JHH = 15.9 Hz, JHP unresolved), 7.27–7.68 (m, 30H, C6H5) ppm. 31P NMR: 32.4 (s, PPh3), 95.1 (s, S2P) ppm. MS (FAB +ve) m/z (abundance): 660 (100) [M − PPh3]+. Calcd. for C47H51O3P3RuS2: C 61.2, H 5.6%. Found: C 61.1, H 5.5%.
[Ru{CHCHCH2OSi(But)Me2}{κ2-S2P(OEt)2}(CO)(PPh3)2] (5)
[RuH{κ2-S2P(OEt)2}(CO)(PPh3)2] (50 mg, 0.061 mmol) and HCCCH2OSi(But)Me2 (15 mg, 0.088 mmol) were dissolved in dichloromethane (20 mL). The reaction mixture was stirred for 20 min, leading to a colour change from green to yellow. Rotary evaporation of all solvent, and further recrystallisation with dichloromethane (10 mL) and methanol (10 mL) led to precipitation of the yellow product. This was washed with methanol (10 mL) and petroleum ether (10 mL) and dried. Yield: 33 mg (53%). IR: 1913 (νCO), 1572, 1388, 1249, 1187, 1019, 945, 835, 773, 666 cm−1. NMR (CD2Cl2) 1H: 0.09 (s, 6H, SiMe), 0.86 (s, 9H, SiBut), 0.88 (t, 6H, OCCH3, JHH = 7.1 Hz), 2.96, 3.14 (m×2, 4H, OCH2), 3.44 (d, 2H, OCH2, JHH = 5.2 Hz), 4.53 (m, 1H, Hβ), 6.51 (d, 1H, Hα, JHH = 15.8 Hz), 7.34–7.62 (m×2, 30H, C6H5) ppm. 31P NMR: 32.4 (s, PPh3), 95.1 (s, S2P) ppm. MS (FAB +ve) m/z (abundance): 1009(1) [M]+, 748 (89) [M − PPh3]+. Calcd. for C50H59O4P3RuS2Si: C 59.5, H 5.9%. Found: C 60.5, H 6.0%.
[Ru(CHCHCO2Me){κ2-S2P(OEt)2}(CO)(PPh3)2] (6)
[RuH{κ2-S2P(OEt)2}(CO)(PPh3)2] (50 mg, 0.061 mmol) and HCCCO2Me (8 mg, 0.095 mmol) were dissolved in dichloromethane (20 mL). The reaction mixture was stirred for 40 min. Rotary evaporation of all solvent, and further recrystallisation with dichloromethane (10 mL) and methanol (10 mL) led to precipitation of the olive green product. This was washed with methanol (10 mL) and petroleum ether (10 mL) and dried. Yield: 36 mg (64%). IR: 1923 (νCO), 1674 (νC–O), 1535, 1389, 1247, 1015, 946, 786, 771, 743 cm−1. NMR (CD2Cl2) 1H: 0.88 (t, 6H, OCCH3, JHH = 7.0 Hz), 2.93, 3.12 (m×2, 4H, OCH2), 3.35 (s, 3H, Me), 5.03 (d, 1H, Hβ, JHH = 16.6 Hz), 7.32–7.60 (m, 30H, PC6H5), 9.16 (dt, 1H, Hα, JHH = 16.6 Hz, JHP = 3.2 Hz) ppm. 31P NMR: 31.3 (s, PPh3), 94.5 (s, S2P) ppm. MS (FAB +ve) m/z (abundance): 924(2) [M]+, 739(4) [M − S2P(OEt)2]+, 634 (63) [M − CO − PPh3]+. Calcd. for C45H45O5P3RuS2: C 58.5, H 4.9%. Found: C 58.6, H 5.0%.
[Ru(CHCHFc){κ2-S2P(OEt)2}(CO)(PPh3)2] (7)
[RuH{κ2-S2P(OEt)2}(CO)(PPh3)2] (50 mg, 0.061 mmol) and HCCFc (19 mg, 0.091 mmol) were dissolved in dichloromethane (20 mL). The reaction mixture was stirred for 20 min, revealing a colour change from green to dark yellow. Rotary evaporation of all solvent, and further recrystallisation with dichloromethane (10 mL) and methanol (10 mL) led to precipitation of the yellow product. This was washed with methanol (10 mL) and petroleum ether (10 mL) and dried. Yield: 25 mg (40%). A further crop was obtained by removing all solvent and triturating in diethylether (5 mL). IR: 1917 (νCO), 1560, 1387, 1187, 1016, 945, 781, 663 cm−1. NMR (CD2Cl2) 1H: 0.91 (t, 6H, OCCH3, JHH = 7.1 Hz), 2.95, 3.18 (m×2, 4H, OCH2), 3.39, 3.84 (tv×2, 2×2H, C5H4, JHH = 1.7 Hz), 3.88 (s, 5H, C5H5), 5.06 (d, 1H, Hβ, JHH = 16.4 Hz), 6.93 (dt, 1H, Hα, JHH = 16.4 Hz, JHP = 3.8 Hz), 7.39–7.62 (m, 30H, PC6H5) ppm. 31P NMR: 32.1 (s, PPh3), 94.7 (s, S2P) ppm. MS (FAB +ve) m/z (abundance): 839(7) [M − CO − S2P(OEt)2]+, 788(8) [M − PPh3]+. Calcd. for C53H51FeO3P3RuS2: C 60.6, H 4.9%. Found: C 60.5, H 4.8%.
[Ru(CHCHCPh2OH){κ2-S2P(OEt)2}(CO)(PPh3)2] (8)
[RuH{κ2-S2P(OEt)2}(CO)(PPh3)2] (100 mg, 0.121 mmol) and HCCCPh2OH (25 mg, 0.120 mmol) were dissolved in dichloromethane (20 mL). The reaction mixture was stirred for 90 min, leading to a colour change from green to yellow. Rotary evaporation of all solvent, and further recrystallisation with dichloromethane (10 mL) and ethanol (10 mL) led to precipitation of the yellow product. This was washed with ethanol (10 mL) and petroleum ether (10 mL) and dried. Yield: 95 mg (75%). IR: 1929 (νCO), 1158, 1088, 941, 778 cm−1. NMR (CD2Cl2) 1H: 0.87 (t, 6H, OCCH3, JHH = 7.1 Hz), 0.92 (s, 1H, CPh2OH), 2.86, 3.14 (m×2, 4H, OCH2), 5.40 (d, 1H, Hβ, JHH = 16.4 Hz), 6.81–7.11 (m, 10H, CC6H5), 7.32–7.48 (m, 30H, PC6H5), 6.87 (dt, 1H, Hα, partially obscured) ppm. 31P NMR: 30.8 (s, PPh3), 95.0 (s, S2P) ppm. MS (FAB +ve) m/z (abundance): 1047(1) [M]+, 786(23) [M − PPh3]+. Calcd. for C56H53O4P3RuS2: C 64.2, H 5.1%. Found: C 64.1, H 5.0%.
Crystals of compound 8 were grown by slow diffusion of ethanol into a dichloromethane solution of the complex. Data were collected using an Oxford Diffraction Xcalibur 3 diffractometer, and the structures were refined based on F2 using the SHELXTL and SHELX-97 program systems.32Crystal data for 8: C56H53O4P3RuS2·CH2Cl2, M = 1133.01, triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2), a = 10.4330(3), b = 14.2442(3), c = 18.2189(5) Å, α = 80.194(2), β = 84.401(3), γ = 87.677(2)°, V = 2654.43(12) Å3, Z = 2, Dc = 1.418 g cm−3, μ(Cu-Kα) = 5.269 mm−1, T = 173 K, pale yellow tablets, Oxford Diffraction Xcalibur PX Ultra diffractometer; 10267 independent measured reflections (Rint = 0.0332), F2 refinement, R1(obs) = 0.0356, wR2(all) = 0.0925, 9052 independent observed absorption-corrected reflections [|Fo| > 4σ(|Fo|), 2θmax = 145°], 652 parameters. CCDC 834118.
(no. 2), a = 10.4330(3), b = 14.2442(3), c = 18.2189(5) Å, α = 80.194(2), β = 84.401(3), γ = 87.677(2)°, V = 2654.43(12) Å3, Z = 2, Dc = 1.418 g cm−3, μ(Cu-Kα) = 5.269 mm−1, T = 173 K, pale yellow tablets, Oxford Diffraction Xcalibur PX Ultra diffractometer; 10267 independent measured reflections (Rint = 0.0332), F2 refinement, R1(obs) = 0.0356, wR2(all) = 0.0925, 9052 independent observed absorption-corrected reflections [|Fo| > 4σ(|Fo|), 2θmax = 145°], 652 parameters. CCDC 834118.
[Ru{CHCH(HO)C6H10}{κ2-S2P(OEt)2}(CO)(PPh3)2] (9)
[RuH{κ2-S2P(OEt)2}(CO)(PPh3)2] (100 mg, 0.121 mmol) and HCC(HO)C6H10 (32 mg, 0.258 mmol) were dissolved in dichloromethane (20 mL) and stirred for 15 min. Rotary evaporation of all solvent, and trituration in petroleum ether (10 mL) led to precipitation of the yellow product. This was washed with petroleum ether (10 mL) and dried. Yield: 71 mg (61%). IR: 3571 (νO–H), 1925 (νCO), 1571, 1313, 1020, 944, 659 cm−1. NMR (CD2Cl2) 1H: 0.78–1.34 (m, 10H, Cy), 0.87 (t, 6H, OCCH3, JHH = 7.0 Hz), 1.61 (s, 1H, OH), 2.90–3.12 (m×2, 4H, OCH2), 4.79 (d, 1H, Hβ, JHH = 16.4 Hz), 6.58 (d, 1H, Hα, JHH = 16.4 Hz), 7.38–7.58 (m, 30H, PC6H5) ppm. 31P NMR: 31.6 (s, PPh3), 95.3 (s, S2P) ppm. MS (FAB +ve) m/z (abundance): 702(12) [M − PPh3]+, 685(15) [M − OH − PPh3]+, 656(15) [M − CO − OH − PPh3]+. Calcd. for C49H53O4P3RuS2: C 61.1, H 5.5%. Found: C 61.2, H 5.6%.
[Ru(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e002.gif) CC6H4Me-4){κ2-S2P(OEt)2}(CO)(PPh3)2] (10)
CC6H4Me-4){κ2-S2P(OEt)2}(CO)(PPh3)2] (10)
a) [RuH{κ2-S2P(OEt)2}(CO)(PPh3)2] (40 mg, 0.049 mmol) and HCCC6H4Me-4 (11.3 mg, 0.097 mmol) were dissolved in dichloromethane (10 mL). The reaction mixture was stirred for 3 h. Rotary evaporation of solvent led to precipitation of the dark yellow product. This was recrystallised in dichloromethane (10 mL) and methanol (10 mL) and then washed with methanol (10 mL) and petroleum ether (10 mL) before drying. Yield: 5 mg (11%). b) [Ru(CHCHC6H4Me-4){κ2-S2P(OEt)2}(CO)(PPh3)2] (50 mg, 0.052 mmol) and HCCC6H4Me-4 (12 mg, 0.103 mmol) were dissolved in toluene (10 mL) and the reaction mixture heated at reflux for 10 min. All solvent was removed and the residue dissolved in dichloromethane (10 mL) and ethanol (5 mL). Rotary evaporation to a solvent volume of approximately 3 mL led to precipitation of the dark yellow product. This was washed with ethanol (10 mL) and petroleum ether (10 mL) and dried. Yield: 32 mg (65%). IR: 2105 (νCC), 1936 (νCO), 1502, 1018, 947, 816, 786, 765, 675 cm−1. NMR (CD2Cl2) 1H: 0.93 (t, 6H, OCCH3, JHH = 7.1 Hz), 2.23 (s, 3H, tolyl–CH3), 3.01–3.18 (m, 4H, OCH2), 6.44, 6.83 (AB, 4H, C6H4, JAB = 7.8 Hz), 7.36–7.87 (m, 30H, C6H5) ppm. 13C: 204.4 (t, CO, JPC = 15.6 Hz), 135.7 (tv, o/m–PC6H5, JPC = 5.0 Hz), 134.2 (s, tolyl–C4), 133.6 (tv, ipso–PC6H5, JPC = 4.4 Hz), 130.4 (s, tolyl–C2,6), 129.8 (s, p-PC6H5), 128.5 (s, tolyl–C2,6), 127.7 (tv, o/m–PC6H5, JPC = 4.8 Hz), 126.4 (s, tolyl–C1), 115.8 (s, Cβ), 108.1 (t, Cα, JPC = 21.0 Hz), 62.0 (d, OCH2, JPC = 7.5 Hz), 21.2 (d, tolyl–CH3), 15.7 (d, OCH2, JPC = 8.4 Hz) ppm. 31P NMR: 30.5 (s, PPh3), 94.7 (s, S2P) ppm. MS (FAB +ve) m/z (abundance): 954(2) [M]+, 926(12) [M − CO]+, 692 (58) [M − PPh3]+. Calcd. for C51H47O3P3RuS2: C 63.0, H 5.0%. Found: C 63.1, H 4.9%.
[Ru(CCBut){κ2-S2P(OEt)2}(CO)(PPh3)2] (11)
[Ru(CHCHC6H4Me-4){κ2-S2P(OEt)2}(CO)(PPh3)2] (50 mg, 0.052 mmol) and HCCBut (13 mg, 0.158 mmol) were dissolved in tetrahydrofuran (20 mL) and heated at reflux for 2 h. After cooling to room temperature, all solvent was removed from mixture by rotary evaporation, and the solid dissolved in dichloromethane (10 mL). Ethanol (10 mL) was added and subsequent rotary evaporation led to precipitation of the yellow product. This was washed with ethanol (10 mL) and petroleum ether (10 mL) and dried. Yield: 32 mg (67%). IR: 2113 (νCC), 1943 (νCO), 1249, 1013, 949, 791, 660 cm−1. NMR (CD2Cl2) 1H: 0.76 (s, 9H, But), 0.93 (t, 6H, OCCH3, JHH = 7.1 Hz), 3.06–3.15 (m, 4H, OCH2), 7.36–7.90 (m, 30H, C6H5) ppm. 31P NMR: 30.4 (s, PPh3), 94.9 (s, S2P) ppm. MS (ES +ve) m/z (abundance): 839(6) [M − acetylide]+. Calcd. for C47H49O3P3RuS2: C 61.4, H 5.4%. Found: C 61.1, H 5.3%.
[RuCl{κ2-S2P(OEt)2}(CO)(PPh3)2] (12)
a) The reaction between [Ru(CHCHC6H4Me-4){κ2-S2P(OEt)2}(CO)(PPh3)2] (50 mg, 0.052 mmol) and HCCBut (20 mg, 0.244 mmol) in 1,2-dichloroethane (20 mL) formed a white side product in low yield. Spectroscopic analysis helped clarify its formulation as [RuCl{κ2-S2P(OEt)2}(CO)(PPh3)2]. b) [RuH{κ2-S2P(OEt)2}(CO)(PPh3)2] (50 mg, 0.061 mmol) and N–chlorosuccinimide (16 mg, 0.120 mmol) were dissolved in dichloromethane (10 mL) and methanol (5 mL). The reaction mixture was stirred for 40 min. Removal of the solvent and trituration in diethylether (5 mL) led to isolation of a colourless product. Yield: 46 mg (87%). IR: 1965 (νCO), 1089, 1011, 951, 774, 745, 649 cm−1. NMR (CD2Cl2) 1H: 1.31 (t, 6H, OCCH3, JHH = 7.2 Hz), 4.11, 4.29 (m×2, 4H, OCH2), 7.24–7.54 (m, 30H, C6H5) ppm. 31P NMR: 36.8 (s, PPh3), 103.6 (s, S2P) ppm. MS (FAB +ve) m/z (abundance): 874(2) [M]+, 839 (63) [M − Cl]+. Calcd. for C41H40ClO3P3RuS2: C 56.3, H 4.6%. Found: C 56.0, H 4.3%.
[Ru(η3-PhCC–CCHPh){κ2-S2P(OEt)2}(CO)(PPh3)] (13)
[Ru{C(CCPh)CHPh}Cl(CO)(PPh3)2] (100 mg, 0.112 mmol) and (NH4)[S2P(OEt)2] (25 mg, 0.123 mmol) were dissolved in dichloromethane (20 mL) and ethanol (10 mL). The reaction mixture was stirred for 1 h, showing a colour change from orange to yellow. Rotary evaporation to a solvent volume of approximately 10 mL led to precipitation of the bright yellow product. This was washed with ethanol (10 mL) and petroleum ether (10 mL) and dried. Yield: 26 mg (30%). IR: 1921 (νCO), 1191, 1019, 950, 862, 664 cm−1. NMR (CD2Cl2) 1H: 1.31 (t, 6H, OCCH3, JHH = 7.1 Hz), 4.04–4.12 (m, 4H, OCH2), 6.39 (s, 1H, Hβ), 7.13–7.72 (m, 25H, C6H5) ppm. 31P NMR: 52.5 (s, PPh3), 100.5 (s, S2P) ppm. MS (ES +ve) m/z (abundance): 780(12) [M]+. Calcd. for C39H36O3P2RuS2: C 60.1, H 4.7%. Found: C 59.8, H 4.4%.
[Ru(CPhCHPh){μ,κ1,κ2-S2P(OEt)2}(CO)(PPh3)]2 (14)
[Ru(CPhCHPh)Cl(CO)(PPh3)2] (100 mg, 0.115 mmol) and (NH4)[S2P(OEt)2] (26 mg, 0.127 mmol) were dissolved in dichloromethane (10 mL) and methanol (15 mL). The reaction mixture was stirred for 1 h. Rotary evaporation to a solvent volume of approximately 10 mL led to precipitation of the pale brown product. This was washed with methanol (10 mL) and petroleum ether (10 mL) and dried. Yield: 46 mg (53%). IR: 1965 (νCO), 1163, 1089, 1011, 950, 796, 745, 651 cm−1. NMR (CD2Cl2) 1H: 1.33 (t, 12H, OCCH3, JHH = 7.1 Hz), 4.11, 4.29 (m×2, 8H, OCH2), 7.24–7.55 (m, C6H5 + Hβ, 50H + 2H) ppm. 31P NMR: 36.9 (s, PPh3), 103.6 (s, SPO) ppm. MS (FAB +ve) m/z (abundance): not diagnostic. Calcd. for C74H72O6P4Ru2S4: C 58.8, H 4.8%. Found: C 58.6, H 4.7%.
[Ru(CPhCHPh){μ,κ1,κ2-S2P(OEt)2}(CS)(PPh3)]2 (15)
[Ru(CPhCHPh)Cl(CS)(PPh3)2] (55 mg, 0.062 mmol) and (NH4)[S2P(OEt)2] (14 mg, 0.068 mmol) were dissolved in dichloromethane (10 mL) and methanol (15 mL). The reaction mixture was stirred for 20 min. Rotary evaporation to a solvent volume of approximately 10 mL led to precipitation of the orange product. This was washed with methanol (10 mL) and petroleum ether (10 mL) and dried. Yield: 37 mg (77%). IR: 1594, 1386, 1280 (νCS), 1187, 1014, 967, 945, 840, 779, 706, 645 cm−1. NMR (CD2Cl2) 1H: 1.46 (t, OCCH3, 12H, JHH = 6.8 Hz), 4.26–4.33 (m×2, 8H, OCH2), 6.81 (s, 2H, Hβ), 6.87–7.47 (m, 50H, C6H5) ppm. 31P NMR: 45.6 (s, PPh3), 99.4 (s, S2P) ppm. MS (ES +ve) m/z (abundance): 772(14) [Ru(CPhCHPh){S2P(OEt)2}(CS)(PPh3)]+. Calcd. for C74H72O4P4Ru2S6: C 57.6, H 4.7%. Found: C 57.8, H 4.6%.
[Ru(η2-SCCPhCHPh){κ2-S2P(OEt)2}(CO)(PPh3)] (16)
Carbon monoxide gas was bubbled through a yellow solution of 15 (40 mg, 0.026 mmol) in dichloromethane, resulting in a red colour change. All solvent was removed and the residue triturated in petroleum ether (10 mL) to yield a red solid. This was washed with petroleum ether (10 mL) and dried. Yield: 39 mg (94%). IR: 1910 (νCO), 1584, 1567, 1385, 1256 (νC–S), 1205, 1144, 1014, 956, 816, 791, 639 cm−1. NMR (CD2Cl2) 1H: 1.40 (t, 6H, OCCH3, JHH = 7.0 Hz), 4.13–4.31 (m, 4H, OCH2), 6.77–7.67 (m, 25H, C6H5), 7.94 (s, 1H, CPhCHPh) ppm. 31P NMR: 49.7 (s, PPh3) 102.7 (s, S2P) ppm. MS (FAB +ve) m/z (abundance): 801(28) [M]+, 772 (100) [M − CO]+. Calcd. for C38H36O3P2RuS3: C 57.1, H 4.5%. Found: C 57.2, H 4.4%.
[Ru(CHCHC6H4Me-4){κ2-SOP(OEt)2}(CO)(PPh3)2] (17)
[Ru(CHCHC6H4Me-4)Cl(CO)(BTD)(PPh3)2] (100 mg, 0.106 mmol) and K[SOP(OEt)2] (24 mg, 0.115 mmol) were dissolved in dichloromethane (20 mL) and ethanol (10 mL). The reaction mixture was stirred for 1 h. Rotary evaporation to a solvent volume of approximately 10 mL led to precipitation of the yellow product. This was washed with ethanol (10 mL) and petroleum ether (10 mL) and dried. Yield: 86 mg (86%). IR: 1927 (νCO), 1029, 968, 949, 786, 649 cm−1. NMR (CD2Cl2) 1H: 0.75 (t, 6H, OCCH3, JHH = 7.1 Hz), 2.16 (s, 3H, tolyl–CH3), 3.11–3.21 (m, 4H, OCH2), 5.88 (d, 1H, Hβ, JHH = 16.2 Hz), 6.31, 6.78 (AB, 4H, C6H4, JAB = 7.9 Hz), 7.27–7.70 (m, 30H, C6H5) ppm. 31P NMR: 29.7 (s, PPh3), 48.8 (s, S2P) ppm. MS (ES +ve) m/z (abundance): 939(1) [M]+, 678(10) [M − PPh3]+. Calcd. for C50H49O4P3RuS: C 63.9, H 5.2%. Found: C 64.2, H 5.2%.
Acknowledgements
We thank Johnson Matthey Ltd for a generous loan of ruthenium trichloride. We are grateful to Prof Andrea Sella (University College London) for invaluable assistance with molecular mass determinations.References
- (a) G. Hogarth, Prog. Inorg. Chem., 2005, 53, 71–561 CrossRef CAS; (b) J. Cookson and P. D. Beer, Dalton Trans., 2007, 1459–1472 RSC; (c) R. P. Burns, F. P. McCullough and C. A. McAuliffe, Adv. Inorg. Chem. Radiochem., 1980, 23, 211–280 CAS; (d) D. Coucouvanis, Prog. Inorg. Chem., 1970, 11, 233–371 CrossRef CAS; (e) D. Coucouvanis, Prog. Inorg. Chem., 1979, 26, 301–469 CrossRef CAS; (f) E. R. T. Tiekink and I. Haiduc, Prog. Inorg. Chem., 2005, 54, 127–319 CrossRef CAS; (g) H. Loumrhari, J. Ros and M. R. Torres, Polyhedron, 1991, 10, 421–427 CrossRef CAS; (h) Y. H. Lin, N. H. Leung, K. B. Holt, A. L. Thompson and J. D. E. T. Wilton-Ely, Dalton Trans., 2009, 7891–7901 RSC.
- (a) J. D. E. T. Wilton-Ely, D. Solanki and G. Hogarth, Eur. J. Inorg. Chem., 2005, 4027–4030 CrossRef CAS; (b) E. R. Knight, D. Solanki, G. Hogarth, K. B. Holt, A. L. Thompson and J. D. E. T. Wilton-Ely, Inorg. Chem., 2008, 47, 9642–9653 CrossRef; (c) E. R. Knight, A. R. Cowley, G. Hogarth and J. D. E. T. Wilton-Ely, Dalton Trans., 2009, 607–609 RSC; (d) E. R. Knight, N. H. Leung, Y. H. Lin, A. R. Cowley, D. J. Watkin, A. L. Thompson, G. Hogarth and J. D. E. T. Wilton-Ely, Dalton Trans., 2009, 3688–3697 RSC; (e) E. R. Knight, N. H. Leung, A. L. Thompson, G. Hogarth and J. D. E. T. Wilton-Ely, Inorg. Chem., 2009, 48, 3866–3874 CrossRef CAS; (f) M. J. Macgregor, G. Hogarth, A. L. Thompson and J. D. E. T. Wilton-Ely, Organometallics, 2009, 28, 197–208 CrossRef CAS; (g) S. Naeem, E. Ogilvie, A. J. P. White, G. Hogarth and J. D. E. T. Wilton-Ely, Dalton Trans., 2010, 39, 4080–4089 RSC; (h) S. Naeem, A. J. P. White, G. Hogarth and J. D. E. T. Wilton-Ely, Organometallics, 2010, 29, 2547–2556 CrossRef CAS; (i) K. Oliver, G. Hogarth, A. J. P. White and J. D. E. T. Wilton-Ely, Dalton Trans., 2011, 40, 5852–5864 RSC.
- (a) S. Naeem, L. Delaude, A. J. P. White and J. D. E. T. Wilton-Ely, Inorg. Chem., 2010, 49, 1784–1793 CrossRef CAS; (b) S. Naeem, A. L. Thompson, L. Delaude and J. D. E. T. Wilton-Ely, Chem.–Eur. J., 2010, 16, 10971–10974 CrossRef CAS; (c) S. Naeem, A. L. Thompson, L. Delaude and J. D. E. T. Wilton-Ely, Dalton Trans., 2011, 40, 3737–3747 RSC; (d) E. Y. Chia, S. Naeem, L. Delaude, A. J. P. White and J. D. E. T. Wilton-Ely, Dalton Trans., 2011, 40, 6645–6658 RSC.
- A. M. Barnes, K. D. Bartle and V. R. A. Thibon, Tribol. Int., 2001, 34, 389–395 CrossRef CAS.
- A. I. Busev and M. I. Ivanyutin, Trudy Komissii po Analiticheskoi Khimii, Akademiya Nauk SSSR, 1960, 11, 172–91 CAS.
- (a) H. Bode and W. Arnswald, Fresenius' Z. Anal. Chem., 1962, 185, 179–201 CrossRef CAS; (b) K. Hayashi, Y. Sasaki, S. Tagashira, Y. Murakami and T. Suehiro, Anal. Sci., 1987, 3, 55–7 CrossRef CAS.
- R. Fitoussi, S. Lours and C. Musikas, Eur. Pat. Appl., 1980, EP 14629 A1, 19800820 Search PubMed.
- F. Faraone and V. Marsala, Inorg. Chim. Acta, 1976, 17, 217–20 CrossRef CAS.
- (a) V. K. Jain and V. S. Jakkal, J. Organomet. Chem., 1996, 515, 81–87 CrossRef CAS; (b) W.-H. Leung, K.-K. Lau, Q.-F. Zhang, W.-T. Wong and B. Tang, Organometallics, 2000, 19, 2084–2089 CrossRef CAS; (c) S. Wu, Z. Liu, Y. Zhang and P. Li, Gazz. Chim. Ital., 1993, 123, 647–50 CAS; (d) V. K. Jain, Transition Met. Chem., 1993, 18, 312–14 CrossRef CAS; (e) D. R. Robertson and T. A. Stephenson, J. Chem. Soc., Dalton Trans., 1978, 486–95 RSC; (f) D. R. Robertson and T. A. Stephenson, J. Organomet. Chem., 1976, 107, C46–C48 CrossRef CAS; (g) J. Perez, L. Riera, V. Riera, S. Garcia-Granda, E. Garcia-Rodriguez and D. Miguel, Chem. Commun., 2002, 384–385 RSC.
- A. E. D. McQueen, A. J. Blake, T. A. Stephenson, M. Schröder and L. J. Yellowlees, J. Chem. Soc., Chem. Commun., 1988, 1533–5 RSC.
- A. J. Deeming, C. Forth and G. Hogarth, Transition Met. Chem., 2006, 31, 42–45 CrossRef CAS.
- (a) H. C. Tong, C. Y. C. Hsu, Y. R. Liang, Y. H. Lo and C. H. Lin, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, E64, m1372–m1373 CAS; (b) K.-B. Shiu, S.-J. Yu, Y. Wang and G.-H. Lee, J. Organomet. Chem., 2002, 650, 37–42 CrossRef CAS; (c) A. J. Deeming, C. Forth and G. Hogarth, J. Organomet. Chem., 2006, 691, 79–85 CrossRef CAS.
- (a) V. K. Jain, Indian J. Chem., 1993, 32A, 1034–6 CAS; (b) X. Liu, Q.-F. Zhang and W.-H. Leung, J. Coord. Chem., 2005, 58, 1299–1305 CrossRef CAS; (c) A. A. S. El Kahldy and J. R. Durig, Phosphorus, Sulfur Silicon Relat. Elem., 2000, 165, 197–204 CrossRef CAS.
- (a) A. A. S. El Khaldy and J. R. Durig, Phosphorus, Sulfur Silicon Relat. Elem., 2001, 175, 227–236 CrossRef CAS; (b) P. U. Jain, P. Munshi, M. G. Walawalkar, S. P. Rath, K. K. Rajak and G. K. Lahiri, Polyhedron, 2000, 19, 801–808 CrossRef CAS; (c) S. S. Kulkarni, B. K. Santra, P. Munshi and G. K. Lahiri, Polyhedron, 1998, 17, 4365–4377 CrossRef CAS.
- (a) A. A. S. El-Khaldy and J. R. Durig, Phosphorus, Sulfur Silicon Relat. Elem., 1998, 134, 365–371 Search PubMed; (b) S. W. Ng, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2002, E58, m18–m19 CAS; (c) C. K. Joergensen, Acta Chem. Scand., 1962, 16, 1048–9 CrossRef CAS.
- E. P. L. Tay, S. L. Kuan, W. K. Leong and L. Y. Goh, Inorg. Chem., 2007, 46, 1440–1450 CrossRef CAS.
- (a) G. J. Bridger, S. P. Fricker, M. J. Abrams, I. Mayers, U.S. Pat. Appl. Publ. ( 2002), US 20020193363 A1 20021219 Search PubMed; (b) S. Fricker, M. J. Abrams, G. Bridger, R. Skerlj, I. Baird and B. R. Cameron, PCT Int. Appl., 2000, WO 2000056743 A1, 20000928 Search PubMed.
- M. R. Torres, A. Vegas, A. Santos and J. Ros, J. Organomet. Chem., 1986, 309, 169–177 CrossRef CAS.
- H. Werner, M. A. Esteruelas and H. Otto, Organometallics, 1986, 5, 2295–2299 CrossRef CAS.
- (a) M. R. Torres, A. Vegas, A. Santos and J. Ros, J. Organomet. Chem., 1987, 326, 413–421 CrossRef CAS; (b) M. R. Torres, A. Santos, J. Ros and X. Solans, Organometallics, 1987, 6, 1091–1095 CrossRef; (c) A. M. Castaño, A. M. Echavarren, J. López and A. Santos, J. Organomet. Chem., 1989, 379, 171–175 CrossRef; (d) J. López, A. Romero, A. Santos, A. Vegas, A. M. Echavarren and P. Noheda, J. Organomet. Chem., 1989, 373, 249–258 CrossRef; (e) J. Montoya, A. Santos, A. M. Echavarren and J. Ros, J. Organomet. Chem., 1990, 390, C57–C60 CrossRef CAS; (f) H. Loumrhari, J. Ros, M. R. Torres, A. Santos and A. M. Echavarren, J. Organomet. Chem., 1991, 411, 255–261 CrossRef CAS; (g) A. M. Echavarren, J. López, A. Santos and J. Montoya, J. Organomet. Chem., 1991, 414, 393–400 CrossRef CAS; (h) J. Montoya, A. Santos, J. López, A. M. Echavarren, J. Ros and A. Romero, J. Organomet. Chem., 1992, 426, 383–398 CrossRef CAS; (i) A. Santos, J. Lopez, L. Matas, J. Ros, A. Galan and A. M. Echavarren, Organometallics, 1993, 12, 4215–4218 CrossRef CAS; (j) B. Gómez-Lor, A. Santos, M. Ruiz and A. M. Echavarren, Eur. J. Inorg. Chem., 2001, 2305–2310 CrossRef.
- (a) J. C. Cannadine, A. F. Hill, A. J. P. White, D. J. Williams and J. D. E. T. Wilton-Ely, Organometallics, 1996, 15, 5409–5415 CrossRef CAS; (b) A. F. Hill, C. T. Ho and J. D. E. T. Wilton-Ely, Chem. Commun., 1997, 2207–2208 RSC; (c) R. B. Bedford, A. F. Hill, C. Jones, A. J. P. White and J. D. E. T. Wilton-Ely, J. Chem. Soc., Dalton Trans., 1997, 139–140 RSC; (d) A. F. Hill, A. J. P. White, D. J. Williams and J. D. E. T. Wilton-Ely, Organometallics, 1998, 17, 4249–4258 CrossRef CAS; (e) R. B. Bedford, A. F. Hill, C. Jones, A. J. P. White, D. J. Williams and J. D. E. T. Wilton-Ely, Organometallics, 1998, 17, 4744–4753 CrossRef CAS; (f) J. D. E. T. Wilton-Ely, P. J. Pogorzelec, S. J. Honarkhah and D. A. Tocher, Organometallics, 2005, 24, 2862–2874 CrossRef CAS; (g) J. D. E. T. Wilton-Ely, S. J. Honarkhah, M. Wang and D. A. Tocher, Dalton Trans., 2005, 1930–1939 RSC; (h) J. D. E. T. Wilton-Ely, M. Wang, S. J. Honarkhah and D. A. Tocher, Inorg. Chim. Acta, 2005, 358, 3218–3226 CrossRef CAS; (i) J. D. E. T. Wilton-Ely, M. Wang, D. Benoit and D. A. Tocher, Eur. J. Inorg. Chem., 2006, 3068–3078 CrossRef CAS; (j) A. R. Cowley, A. L. Hector, A. F. Hill, A. J. P. White, D. J. Williams and J. D. E. T. Wilton-Ely, Organometallics, 2007, 26, 6114–6125 CrossRef CAS; (k) J. C. Green, A. L. Hector, A. F. Hill, S. Lin and J. D. E. T. Wilton-Ely, Organometallics, 2008, 27, 5548–5558 CrossRef CAS; (l) A. F. Hill and J. D. E. T. Wilton-Ely, J. Chem. Soc., Dalton Trans., 1999, 3501–3510 Search PubMed; (m) K. J. Harlow, A. F. Hill and T. Welton, J. Chem. Soc., Dalton Trans., 1999, 1911–1912 RSC.
- (a) G. Jia, W. F. Wu, R. C. Y. Yeung and H. P. Xia, J. Organomet. Chem., 1997, 539, 53–59 CrossRef CAS; (b) S. H. Liu, Y. Chen, K. L. Wan, T. B. Wen, Z. Zhou, M. F. Lo, I. D. Williams and G. Jia, Organometallics, 2002, 21, 4984–4992 CrossRef CAS; (c) S. H. Liu, H. Xia, T. B. Wen, Z. Zhou and G. Jia, Organometallics, 2003, 22, 737–743 CrossRef CAS; (d) S. H. Liu, H. Xia, K. L. Wan, R. C. Y. Yeung, Q. Y. Hu and G. Jia, J. Organomet. Chem., 2003, 683, 331–336 CrossRef CAS; (e) H. Xia, T. B. Wen, Q. Y. Hu, X. Wang. X. Chen, L. Y. Shek, I. D. Williams, K. S. Wong, G. K. L. Wong and G. Jia, Organometallics, 2005, 24, 562–569 CrossRef CAS; (f) S. H. Liu, Q. Y. Hu, P. Xue, T. B. Wen, I. D. Williams and G. Jia, Organometallics, 2005, 24, 769–772 CrossRef CAS.
- (a) H. Werner, S. Jung, P. Gonzalez-Herrero, K. Ilg and J. Wolf, Eur. J. Inorg. Chem., 2001, 1957–1961 CrossRef CAS; (b) S. Jung, K. Ilg, J. Wolf and H. Werner, Organometallics, 2001, 20, 2121–2123 CrossRef CAS; (c) R. Castarlenas, M. A. Esteruelas and E. Oñate, Organometallics, 2001, 20, 2294–2302 CrossRef CAS; (d) R. Castarlenas, M. A. Esteruelas and E. Oñate, Organometallics, 2001, 20, 3283–3292 CrossRef CAS; (e) H. Werner, W. Stüer, S. Jung, B. Weberndörfer and J. Wolf, Eur. J. Inorg. Chem., 2002, 1076–1080 CrossRef CAS; (f) S. Jung, C. D. Brandt, J. Wolf and H. Werner, Dalton Trans., 2004, 375–383 RSC; (g) S. Jung, K. Ilg, C. D. Brandt, J. Wolf and H. Werner, Eur. J. Inorg. Chem., 2004, 469–480 CrossRef CAS; (h) B. Eguillar, M. A. Esteruelas, M. Oliván and E. Oñate, Organometallics, 2005, 24, 1428–1438 CrossRef; (i) T. Bolano, R. Castarlenas, M. A. Esteruelas and E. Oñate, J. Am. Chem. Soc., 2006, 128, 3965–3973 CrossRef CAS; (j) M. A. Esteruelas, A. M. López and E. Oñate, Organometallics, 2007, 26, 3260–3263 CrossRef CAS.
- (a) H. Loumrhari, J. Ros, R. Yanez and M. R. Torres, J. Organomet. Chem., 1991, 408, 233–239 CrossRef CAS; (b) A. Pedersen, M. Tilset, K. Folting and K. G. Caulton, Organometallics, 1995, 14, 875–888 CrossRef CAS; (c) J. N. Coalter, W. E. Streib and K. G. Caulton, Inorg. Chem., 2000, 39, 3749–3756 CrossRef CAS; (d) A. V. Marchenko, H. Gérard, O. Eisenstein and K. G. Caulton, New J. Chem., 2001, 25, 1244–1255 RSC; (e) A. V. Marchenko, H. Gérard, O. Eisenstein and K. G. Caulton, New J. Chem., 2001, 25, 1382–1388 RSC; (f) D. J. Huang, K. B. Renkema and K. G. Caulton, Polyhedron, 2006, 25, 459–468 CrossRef CAS; (g) J. Maurer, B. Sarkar, B. Schwederski, W. Kaim, R. F. Winter and S. Záliš, Organometallics, 2006, 25, 3701–3712 CrossRef CAS; (h) J. Maurer, B. Sarkar, W. Kaim, R. F. Winter and S. Záliš, Chem.–Eur. J., 2007, 13, 10257–10272 CrossRef CAS; (i) J. Maurer, M. Linseis, B. Sarkar, B. Schwederski, M. Niemeyer, W. Kaim, S. Záliš, C. Anson, M. Zabel and R. F. Winter, J. Am. Chem. Soc., 2008, 130, 259–268 CrossRef CAS; (j) M. Pichlmaier, R. F. Winter, M. Zabel and S. Záliš, J. Am. Chem. Soc., 2009, 131, 4892–4903 CrossRef CAS; (k) K. Kowalski, M. Linseis, R. F. Winter, M. Zabel, S. Záliš, H. Kelm, H.-J. Kruger, B. Sarkar and W. Kaim, Organometallics, 2009, 28, 4196–4209 CrossRef CAS; (l) R. B. Bedford and C. S. J. Cazin, J. Organomet. Chem., 2000, 598, 20–23 CrossRef CAS.
- (a) M. C. J. Harris and A. F. Hill, Organometallics, 1991, 10, 3903–3906 CrossRef CAS; (b) A. F. Hill and J. D. E. T. Wilton-Ely, Organometallics, 1997, 16, 4517–4518 CrossRef CAS.
- (a) For an overview of vinyl chemistry of ruthenium(II), see: A. F. Hill, in Comprehensive Organometallic Chemistry II; E. W. Abel, F. G. A Stone and G. Wilkinson, ed.; Pergamon Press: Oxford, U.K., 1995; Vol. 7 Search PubMed; (b) M. K. Whittlesey, in Comprehensive Organometallic Chemistry III; R. H. Crabtree, D. M. P. Mingos, M. I. Bruce, ed.; Elsevier: Oxford, U.K., 2006, 6 Search PubMed.
- (a) Y. Wakatsuki, H. Yamazaki, N. Kumegawa, T. Satoh and J. Y. Satoh, J. Am. Chem. Soc., 1991, 113, 9604–9610 CrossRef CAS; (b) G. Jia, A. L. Rheingold and D. W. Meek, Organometallics, 1989, 8, 1378–1380 CrossRef CAS; (c) G. Jia and D. W. Meek, Organometallics, 1991, 10, 1444–1450 CrossRef CAS; (d) N. W. Alcock, A. F. Hill, R. P. Melling and A. R. Thompsett, Organometallics, 1993, 12, 641–648 CrossRef CAS.
- (a) R. Signer, Annalen, 1930, 478, 246 CAS; (b) E. P. Clark, Ind. Eng. Chem., Anal. Ed., 1941, 13, 820 CrossRef CAS.
- F. H. Allen, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, in International Tables for Crystallography; E. Prince, Ed.; Springer: Berlin, 2006, Vol. C, 790–811 Search PubMed.
- N. W. Alcock, A. F. Hill and M. S. Roe, J. Chem. Soc., Dalton Trans., 1990, 1737–1740 RSC.
- (a) A. F. Hill and R. P. Melling, J. Organomet. Chem., 1990, 396, C22–C24 CrossRef CAS; (b) A. F. Hill, M. C. J. Harris and R. P. Melling, Polyhedron, 1992, 11, 781–787 CrossRef CAS.
- SHELX-97, G. Sheldrick, Institut Anorg. Chemie, Tammannstr. 4, D37077 Göttingen, Germany, 1998.
Footnote |
| † Electronic supplementary information (ESI) available: CIF file with crystallographic data for the structure of 8 and further information on the molecular weight determinations. CCDC reference numbers 834118. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1ra00973g |
| This journal is © The Royal Society of Chemistry 2012 |