Synthesis of graphene nanosheetsviaoxalic acid-induced chemical reduction of exfoliated graphite oxide
Peng
Song
a,
Xiaoyan
Zhang
a,
Mingxuan
Sun
a,
Xiaoli
Cui
*a and
Yuehe
Lin
*b
aDepartment of Materials Science, Fudan University, Shanghai, 200433, P R China. E-mail: xiaolicui@fudan.edu.cn; Fax: +86-21-65642682; Tel: +86-21-65642397
bPacific Northwest National Laboratory, Richland, Washington 99352, USA. E-mail: yuehe.lin@pnl.gov
First published on 12th December 2011
Abstract
Preparing high-quality graphene through reduction of graphene oxide (GO) by oxalic acid is demonstrated in this paper. Transmission electron microscopy, scanning electron microscopy, Fourier transform infrared spectrometry, X-ray diffraction and Raman spectrometry were taken to confirm the reduction of GO and the formation of graphene under these mild conditions. Thermogravimetric analysis and conductivity measurements further testify the excellent thermal stability and conductivity of the obtained graphene. A possible mechanism for the reduction process was also proposed. Furthermore, a Pt–graphene composite was fabricated on a glassy carbon electrode and excellent electrocatalytic activity towards methanol oxidation was observed. With advantages of low toxicity, simple purification process and high quality of the product, oxalic acid provides a feasible route to prepare graphene from GO under mild conditions, thus facilitating the use of graphene-based materials for large-scale applications.
1. Introduction
Graphene, a two-dimensional carbon material with one-atom thickness,1 has given rise to intense interest due to its unique properties, including large surface area, ultrahigh carrier mobility, excellent mechanical properties and thermal stability.2–4 It has shown great application value in various fields, such as field effect transistors,5 sensors,6fuel cells,7 solar cells,8,9 supercapacitors,10 transparent electrodes11 and composite materials.12–14A facile approach to prepare graphene with desirable properties is of great importance to the study and application of graphene. Unfortunately, it is still a great challenge to achieve this goal so far. Several methods have been developed for the preparation of graphene, including micromechanical cleavage,1 epitaxial growth,15 solution-based reduction of GO,16 and chemical vapor deposition.17,18 Among all the strategies pursued, reduction of GO is considered to be the most promising approach to produce graphene economically in large scale. Typically, graphene is obtained from oxidation of graphite powder by a modified Hummers method and subsequent reduction process. Much endeavor has been devoted to the reduction of GO and various approaches have been adopted. Electrochemical reduction,19–21 photocatalytic reduction,22,23 thermal reduction,24,25 chemical reduction with reducing agents like hydrazine hydrate,26,27NaBH4,28urea,29vitamin C,30aluminum powder,31alcohols,32sulfur-containing compounds,33sodium hydrosulfite34 and carbon monoxide35 have been demonstrated to be viable for the reduction process. Hydrazine hydrate is widely considered to be the most efficient reducing agent to date. However, the hydrazine hydrate reduction process must be conducted with great care for it is highly poisonous and explosive.36 Additionally, the as-prepared graphene is prone nitrogen-doping,33 which results in a decrease of the properties of graphene. At the same time, almost all chemical methods introduce impurities into graphene inevitably, thus affecting its properties and practical application.
The development of an efficient, low-toxicity, economic reducing agent is of great significance to the preparation and application of graphene in a large scale. Oxalic acid is a reducing organic acid widely used in redox reactions with low toxicity. Oxalic acid sublimates rapidly above 125 °C, which means it can be easily removed after the reaction. No impurity elements would be introduced into the product when using oxalic acid to reduce GO for the preparation of graphene, for its simple constitution of elements of C, H and O. So far, no investigation into the preparation of graphene employing oxalic acid as the reducing agent has been reported. Herein, we propose a facile approach to prepare high quality graphenevia employing oxalic acid as the reducing agent under mild conditions. Compared with other chemical reduction methods reported before, this method is a combination of advantages including low toxicity, mild synthesis conditions, simple purification process and low cost.
2. Experimental
2.1 Preparation and chemical reduction of GO
A graphene oxide colloidal suspension was first prepared from purified graphite powder by a modified Hummers method.27,37 Then GO was reduced by oxalic acid in the route described below: 6.0 mL of GO suspension (2.5 mg mL−1) was diluted with deionized water to 50 mL, and then 3.78 g of oxalic acid was added to the aqueous solution. The mixture was heated to 75 °C under the assistance of magnetic stirring for 18 h. Residual oxalic acid was removed by heating at 150 °C for 1 h, at which temperature it sublimates quickly. The product was rinsed with deionized water several times and thus reduced graphene oxide (RGO) was obtained.2.2 Characterization of samples
Transmission electron microscopy (TEM, JEOL, JEM-2011) and scanning electron microscopy (SEM, S-4800, Hitachi) were utilized to investigate the microstructure of RGO. Fourier transform infrared spectra (FT-IR, Nicolet Nexus 470, Nicolet) were recorded to analyze functional groups and their changes occurring during the reduction process. X-Ray diffraction (XRD, Bruker D8 advanced diffractometer) was applied to characterize the crystallographic structure of samples with a scanning rate of 4° min−1. Raman spectra (Dilor Labram-1B microspectrometer with 632.8 nm laser excitation) were recorded to further study the crystallographic structure of RGO. Thermogravimetric analysis (TGA, TGA7 thermogravimetric analyzer, Perkin Elmer instruments) was used to study thermal stability of RGO under N2 atmospheres with a heating rate of 10 °C min−1. The electrical conductivity of RGO paper was measured with a four-point probe method.2.3 Electrochemical measurements
Graphene modified Pt composite electrode (Pt–RGO) was prepared as follows: the as-prepared RGO was dispersed in ethanol and ultrasonicated to form a suspension with a concentration of 0.5 mg mL−1. The glassy carbon (GC) electrode was polished with alumina slurry to a mirror finish and ultrasonically cleaned with deionized water. 20 μL of the suspension was directly cast on the surface of GC electrode, followed by solvent evaporation at room temperature. Pt particles were then deposited on the RGO modified GC electrode with the chronpotentiometry method in the 1.25 mM H2PtCl6 + 0.5 M H2SO4 aqueous solution, with a cathodic current of 0.02 mA and deposition time of 800 s. The electrode was rinsed with deionized water and thus the Pt–RGO composite electrode was obtained.The electrochemical measurements were carried out in a standard three-electrode system, with the Pt–RGO composite, Pt foil and Ag/AgCl as the working electrode, counter electrode and reference electrode, respectively. Cyclic voltammograms (CV) were recorded in 0.5 M CH3OH + 0.5 M H2SO4 aqueous solution at a scanning rate of 50 mV s−1, with the potential ranging between −0.1 V and 1.0 V. All measurements were carried out under ambient conditions and all potentials were reported versus the Ag/AgCl reference electrode.
3. Results and discussion
Presence of a mass of oxygenated groups in GO sheets makes it strongly hydrophilic, which allows GO to readily swell and disperse in water. Color change of the aqueous solution during the reduction process can provide the most intuitive evidence of the formation of graphene from GO. As shown in Fig. 1, the yellow brown GO suspension finally turned black after reaction with oxalic acid for 18 h at 75 °C, suggesting that an efficient reduction of GO has taken place. Along with the change of color, the as-prepared graphene sheets aggregate into thicker flakes, which also indicate that hydrophilic functional groups (hydroxyl, carboxyl, et al.) were removed to a great extent during the reduction process. | ||
| Fig. 1 Photograph of GO suspension (a) and RGO suspension (b) dispersed in ethanol. | ||
A typical wrinkled morphology was observed in the TEM image of RGO, as shown in Fig. 2(a), and it resulted from the multiplicity of chemical carbon bonding in a single carbon layer or a few carbon layers.38 The well-defined diffraction spots in the corresponding SAED pattern indicated the multilayer structure of the as-prepared RGO and its well-formed crystal lattice. A distinct layered structure of RGO was observed in its side-view SEM image and some ultrathin graphene sheets can be recognized in the top-view SEM image. The folded edge of RGO as shown in Fig. 2(c) endows itself with extremely high toughness, which is in great agreement with the outstanding mechanical property of graphene.
 | ||
| Fig. 2 TEM image of RGO (a) (inset show the high resolution TEM image) and the relative selected area electron diffraction pattern (SAED) (b); SEM image of RGO: side view (c) and top view (d). | ||
Since the oxygenated functional groups are infrared active, FT-IR spectra can give a qualitative measure of the deoxygenating reaction. Fig. 3 shows the typical FT-IR spectra of GO and RGO. The bulk of oxygen-containing functional groups decreased dramatically after reduction. For GO, the strong and broad peak around 3410 cm−1 was attributed to hydroxyl groups on the plane, and the peak at 1729 cm−1 was assigned to carboxyl groups situated at the edges of GO sheets. The peak located at 1623 cm−1 was associated with aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds. Additionally, the peaks at 1222 cm−1 and 1054 cm−1 were ascribed to the vibration of C–O–C and C–O, respectively.39 As for the resultant RGO, the peak at 1729 cm−1, 1222 cm−1, 1054 cm−1 disappeared completely and the broad peak around 3410 cm−1 weakened obviously, suggesting that most oxygen-containing functional groups were removed from GO. At the same time, a new peak emerged at 1565 cm−1, as a result of the restoration of the highly conjugated structure like that of graphite after reduction. The greater loss of oxygen-containing functional groups compared with benzyl alcohol32 and trioctylphosphine40 demonstrates oxalic acid is a more effective reducing agent for the preparation of graphene.
C bonds. Additionally, the peaks at 1222 cm−1 and 1054 cm−1 were ascribed to the vibration of C–O–C and C–O, respectively.39 As for the resultant RGO, the peak at 1729 cm−1, 1222 cm−1, 1054 cm−1 disappeared completely and the broad peak around 3410 cm−1 weakened obviously, suggesting that most oxygen-containing functional groups were removed from GO. At the same time, a new peak emerged at 1565 cm−1, as a result of the restoration of the highly conjugated structure like that of graphite after reduction. The greater loss of oxygen-containing functional groups compared with benzyl alcohol32 and trioctylphosphine40 demonstrates oxalic acid is a more effective reducing agent for the preparation of graphene.
 | ||
| Fig. 3 FT-IR spectra of GO (a) and RGO (b). | ||
XRD patterns of GO and RGO are shown in Fig. 4. The XRD pattern of GO represents a sharp peak at 11.9°, corresponding to an interlayer spacing of 7.43 Å. The enlarged interlayer distance compared with pristine graphite (3.37 Å) is ascribed to the introduction of oxygen-containing functional groups on the surface of graphite after oxidation. Accordingly, the XRD pattern of RGO has a broad peak centered at 21.9°, indicating that its interlayer spacing has decreased to 4.05 Å due to the partial removal of oxygen-containing functional groups. In addition, the obviously broadened peak and decreased peak intensity of RGO can be the result of the turbostratic arrangement of RGO stacked sheets in small size25 and formation of new crystal structure different from that of graphite.34
 | ||
| Fig. 4 XRD patterns of GO (a) and RGO (b). | ||
Raman spectrometry is an efficient tool to characterize the reduction degree of GO for its strong sensitivity to electronic structure. As shown in Fig. 5, both spectra of GO and RGO show the existence of D band and G band, which are ascribed to a breathing mode of κ-point photons of A1g symmetry and the first order scattering of the E2g phonon of sp2 C atoms, respectively.41,42 The G band of GO located at 1587 cm−1, while for RGO, it moved to 1594 cm−1, which is closer to that of pristine graphite, indicating that the original GO was reduced. At the same time, the D band of RGO shifted to 1327 cm−1 compared with that of GO located at 1334 cm−1, meaning the defect of the sample and the size of the in-plane sp2 domain.39 The intensity ratio of the D band and G band (ID/IG) improved from 1.2 (GO) to 1.3 (RGO) after reduction. The increase of the ratio was due to the decrease of the sp2 in-plane domain induced by the introduction of defects and disorder of the sp2 domains.27,43 One thing that deserves to be mentioned is that the increase of the ratio in our case is not as prominent as similar experiments reported before,31,33 suggesting that defects caused by the reduction process were somehow healed at the same time and oxalic acid is a better reducing agent. In addition, a broadened 2D band around 2700 cm−1 appeared in the spectra of RGO only, which is seen as a distinct band of graphene induced by a two-phonon resonant scattering process.44
 | ||
| Fig. 5 Raman spectra of GO (a) and RGO (b). | ||
The thermal stability of RGO was investigated by TGA and the TG curves of GO and RGO are shown in Fig. 6. For GO, three significant stages for mass loss were observed, about 15% mass loss around 100 °C was ascribed to the removal of absorbed water, 30% mass loss around 190 °C and 15% mass loss around 500 °C were attributed to pyrolysis of the labile oxygen-containing functional groups. Meanwhile, significantly decreased mass loss has been observed for RGO with a temperature increase. 10% water removal-induced mass loss was observed, and the 15% mass loss around 190 °C is probably due to pyrolysis of residual functional groups. Oxygen-containing functional groups were largely decomposed during the reduction process, thus improving the thermal stability of GO, which is consistent with the excellent thermal stability of graphene.
 | ||
| Fig. 6 TGA curves of GO (a) and RGO (b). | ||
The improvement of electrical conductivity is considered to be a potent indicator of the extent to which π–π conjugated structure is restored after reduction. The square resistance (345 Ω/m2) and thickness (2.94 μm) of the RGO paper was determined with a four-point probe method and side-view SEM image, respectively. RGO paper with an electrical conductivity about 1000 S m−1 was obtained after oxalic acid-induced reduction, and it is approximately six orders of magnitude higher than that of the GO precursor. The relatively higher conductivity of the as-prepared RGO compared with those prepared by NaBH4,28 hydrohalic acids,16 L-ascorbic45 further proves oxalic acid a competitive reducing agent in the preparation of graphenevia chemical reduction.
A possible mechanism for the reduction of GO by oxalic acid involves two steps, as discussed below. In the first step, oxalic acid releases electrons and gets oxidized. The released electrons then interact with hydroxyl and carboxyls on the surface of GO sheets, thus converting GO to RGO, as shown in the second step.
| C2O42− → CO2 + 2e− | (Step 1) |
| GO + e− → RGO | (Step 2) |
In order to characterize the features of as-prepared RGO, the Pt–RGO composite electrode was fabricated and its electrocatalytic activity was characterized with cyclic voltammetry in 0.5 M CH3OH + 0.5 M H2SO4 aqueous solution. Fig. 7(A) shows the typical cyclic voltammogram (CV) as a function of the number of cycles. The onset potential of methanol electrooxidation occurs at 0.065 V. The Pt–RGO electrode exhibits a peak current at 0.617 V in the forward scan, as a result of methanol oxidation. In the backward scan, the composite electrode shows a second oxidation peak current at 0.452 V, which is attributed to the oxidation of incompletely oxidized carbonaceous species. Compared with previous reports,46,47 the Pt–RGO composite electrode here shows a negative onset oxidation potential and higher peak current, demonstrating a better electrocatalytic activity towards methanol oxidation of the prepared electrode.
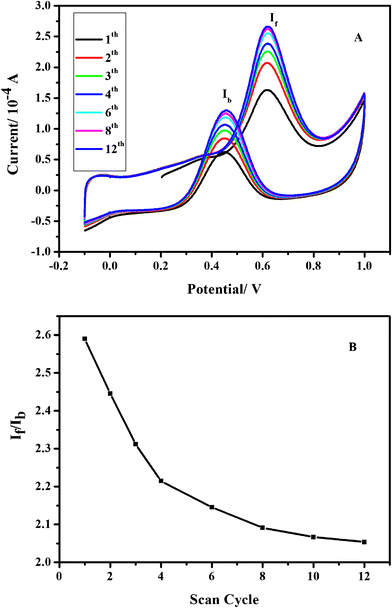 | ||
| Fig. 7 A series of cyclic voltammograms for the oxidation of 0.5 M methanol on Pt–RGO electrode in 0.5 M H2SO4 at a scan rate of 50 mV s−1 (A). The ratio of If and Ib as a function of the number of scan cycle for the composite electrode (B). | ||
The ratio of forward anodic peak current (If) and backward anodic peak current (Ib) is also an indicator of the efficiency of catalysts. As shown in Fig. 7(B), the ratio decreases from 2.59 for the first cycle to 2.05 for the 12th cycle. A higher value of the ratio compared with Pt–MWCNTs,47Pt1–Pd3Co1–MWCNTs48 indicates a more complete oxidation of methanol on the Pt–RGO electrode.
Conclusions
Using oxalic acid as reducing agent, we have successfully prepared graphene from GO at 75 °C. This approach provides a new way to produce graphene with relatively high quality. Platinum deposited on as-prepared graphene has been shown to possess a higher electrocatalytic activity for the methanol oxidation reaction. The results presented in this paper demonstrate the use of oxalic acid as an efficient way to prepare graphene. Considering its advantages over other reducing agents, oxalic acid shows great potential application in preparing graphene with good quality in large scale.Acknowledgements
This work is supported by the National Basic Research Program of China (No.2011CB933300, 2012CB934300), the Shanghai Science Technology Commission (No. 1052 nm01800) and Fudan’s Undergraduate Research Opportunities Program (No. 10073). Dr. Y. Lin would like to acknowledge the support from a LDRD program at Pacific Northwest National Laboratory (PNNL). PNNL is operated by Battelle for DOE under Contract DE-AC05-76RL01830. The authors would like to thank the editorial office (Royal Society of Chemistry) for the recommendation on our manuscript. We also appreciate the referee’s very valuable comments, which have greatly improved the quality of the manuscript.References
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666 CrossRef CAS.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183 CrossRef CAS.
- S. Stankovich, D. A. Dikin, G. H. B. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282 CrossRef CAS.
- F. Schedin, A. K. Geim, S. V. Morozov, E. W. Hill, P. Blake, M. I. Katsnelson and K. S. Novoselov, Nat. Mater., 2007, 6, 652 CrossRef CAS.
- Y. Lu, B. Goldsmith, D. R. Strachan, J. H. Lim, Z. T. Luo and A. T. C. Johnson, Small, 2010, 6, 2748 Search PubMed.
- J. T. Robinson, F. K. Perkins, E. S. Snow, Z. Q. Wei and P. E. Sheehan, Nano Lett., 2008, 8, 3137 CrossRef CAS.
- B. Seger and P. V. Kamat, J. Phys. Chem. C, 2009, 113, 7990 CrossRef CAS.
- X. Wang, L. J. Zhi and K. Mullen, Nano Lett., 2008, 8, 323 CrossRef CAS.
- X. M. Li, H. W. Zhu, K. L. Wang, A. Y. Cao, J. Q. Wei, C. Y. Li, Y. Jia, Z. Li, X. Li and D. H. Wu, Adv. Mater., 2010, 22, 2743 CrossRef CAS.
- S. Y. Yang, K. H. Chang, H. W. Tien, Y. F. Lee, S. M. Li, Y. S. Wang, J. Y. Wang, C. C. M. Ma and C. C. Hu, J. Mater. Chem., 2011, 21, 2374 RSC.
- K. S. Kim, Y. Zhao, H. Jang, S. Y. Lee, J. M. Kim, K. S. Kim, J. H. Ahn, P. Kim, J. Y. Choi and B. H. Hong, Nature, 2009, 457, 706 CrossRef CAS.
- H. L. Fan, L. L. Wang, K. K. Zhao, N. Li, Z. J. Shi, Z. G. Ge and Z. X. Jin, Biomacromolecules, 2010, 11, 2345 CrossRef CAS.
- D. D. Kulkarni, I. Choi, S. S. Singamaneni and V. V. Tsukruk, ACS Nano, 2010, 4, 4667 CrossRef CAS.
- M. A. Rafiee, J. Rafiee, Z. Wang, H. H. Song, Z. Z. Yu and N. Koratkar, ACS Nano, 2009, 3, 3884 CrossRef CAS.
- P. W. Sutter, J. I. Flege and E. A. Sutter, Nat. Mater., 2008, 7, 406 CrossRef CAS.
- S. F. Pei, J. P. Zhao, J. H. Du, W. C. Ren and H. M. Cheng, Carbon, 2010, 48, 4466 CrossRef CAS.
- S. Lee, K. Lee and Z. H. Zhong, Nano Lett., 2010, 10, 4702 CrossRef CAS.
- Y. Zhang, L. Gomez, F. N. Ishikawa, A. Madaria, K. Ryu, C. Wang, A. Badmaev and C. W. Zhou, J. Phys. Chem. Lett., 2010, 1, 3101 Search PubMed.
- Y. Y. Shao, J. Wang, M. Engelhard, C. M. Wang and Y. H. Lin, J. Mater. Chem., 2010, 20, 743 RSC.
- S. J. An, Y. W. Zhu, S. H. Lee, M. D. Stoller, T. Emilsson, S. Park, A. Velamakanni, J. An and R. S. Ruoff, J. Phys. Chem. Lett., 2010, 1, 1259 Search PubMed.
- H. L. Guo, X. F. Wang, Q. Y. Qian, F. B. Wang and X. H. Xia, ACS Nano, 2009, 3, 2653 CrossRef CAS.
- L. J. Cote, R. C. Silva and J. X. Huang, J. Am. Chem. Soc., 2009, 131, 11027 CrossRef CAS.
- G. Williams, B. Seger and P. V. Kamat, ACS Nano, 2008, 2, 1487 CrossRef CAS.
- Y. W. Zhu, M. D. Stoller, W. W. Cai, A. Velamakanni, R. D. Piner, D. Chen and R. S. Ruoff, ACS Nano, 2010, 4, 1227 CrossRef CAS.
- S. Dubin, S. Gilje, K. Wang, V. C. Tung, K. Cha, A. S. Hall, J. Farrar, R. Varshneya, Y. Yang and R. B. Kaner, ACS Nano, 2010, 4, 3845 CrossRef CAS.
- X. F. Gao, J. Jang and S. Nagase, J. Phys. Chem. C, 2010, 114, 832 CrossRef CAS.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558 CrossRef CAS.
- H. J. Shin, K. K. Kim, A. Benayad, S. M. Yoon, H. K. Park, I. S. Jung, M. H. Jin, H. K. Jeong, J. M. Kim, J. Y. Choi and Y. H. Lee, Adv. Funct. Mater., 2009, 19, 1987 CrossRef CAS.
- S. Wakeland, R. Martinez, J. K. Grey and C. C. Luhrs, Carbon, 2010, 48, 3463 CrossRef CAS.
- M. J. F. Merino, L. Guardia, J. I. Paredes, S. V. Rodil, P. S. Fernandez, A. M. Alonso and J. M. D. Tascon, J. Phys. Chem. C, 2010, 114, 6426 CrossRef CAS.
- Z. J. Fan, K. Wang, T. Wei, J. Yan, L. P. Song and B. Shao, Carbon, 2010, 48, 1686 CrossRef CAS.
- D. R. Dreyer, S. Murali, Y. W. Zhu, R. S. Ruoff and C. W. Bielawski, J. Mater. Chem., 2011, 21, 3443 RSC.
- W. F. Chen, L. F. Yan and P. R. Bangal, J. Phys. Chem. C, 2010, 114, 19885 CrossRef CAS.
- T. N. Zhou, F. Chen, K. Liu, H. Deng, Q. Zhang, J. W. Feng and Q. Fu, Nanotechnology, 2011, 22, 045704 CrossRef.
- C. D. Kim, B. K. Min and W. S. Jung, Carbon, 2009, 47, 1610 CrossRef CAS.
- A. Furst, R. C. Berlo and S. Hooton, Chem. Rev., 1965, 65, 51 CrossRef CAS.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- J. C. Meyer, A. K. Geim, M. I. Katsnelson, K. S. Novoselov, T. J. Booth and S. Roth, Nature, 2007, 446, 60 CrossRef CAS.
- S. Park, J. An, R. D. Piner, I. Jung, D. X. Yang, A. Velamakanni, S. T. Nguyen and R. S. Ruoff, Chem. Mater., 2008, 20, 6592 CrossRef CAS.
- J. C. Liu, H. Jeong, J. Z. Liu, K. Lee and J. Y. Park, Carbon, 2010, 48, 2282–2289 CrossRef CAS.
- K. N. Kudin, B. Ozbas, H. C. Schniepp, R. K. Prud'homme, I. A. Aksay and R. Car, Nano Lett., 2008, 8, 36 CrossRef CAS.
- F. Tuinstra and J. L. Koenig, J. Chem. Phys., 1970, 53, 1126 CrossRef CAS.
- G. X. Wang, J. Yang, J. Park, X. L. Gou, B. Wang, H. Liu and J. Yao, J. Phys. Chem. C, 2008, 112, 8192 CrossRef CAS.
- D. A. Sokolov, K. R Shepperd and T. M. Orlando, J. Phys. Chem. Lett., 2010, 1, 2633 Search PubMed.
- J. L. Zhang, H. J. Yang, G. X. Shen, P. Cheng, J. Y. Zhang and S. W. Guo, Chem. Commun., 2010, 46, 1112 RSC.
- Y. W. Ou, X. L. Cui, X. Y. Zhang and Z. Y. Jiang, J. Power Sources, 2010, 195, 1365 Search PubMed.
- Y. Zhao, L. Z. Fan, H. Z. Zhong, Y. F. Li and S. H. Yang, Adv. Funct. Mater., 2007, 17, 1537 CrossRef CAS.
- Y. M. Guo, C. G. Hu, L. Yang, Z. Y. Bai, K. Wang and S. J. Chao, Electrochem. Commun., 2011, 13, 886 Search PubMed.
| This journal is © The Royal Society of Chemistry 2012 |