An evaluation of desulfurization technologies for sulfur removal from liquid fuels
Vimal
Chandra Srivastava
*
Department of Chemical Engineering, Indian Institute of Technology Roorkee, Roorkee -247667, Uttarakhand, India. E-mail: vimalcsr@yahoo.co.in; vimalfch@iitr.ernet.in; Fax: +91-1332-276535, 273560; Tel: +91-1332-285889
First published on 5th December 2011
Abstract
Sulfur compounds represent one of the most common impurities present in the crude oil. Sulfur in liquid fuel oil leads directly to the emission of SO2 and sulfate particulate matter (SPM) that endangers public health and community property; and reduces the life of the engine due to corrosion. Furthermore, the sulfur compounds in the exhaust gases of diesel engines can significantly impair the emission control technology designed to meet NOx and SPM emission standards. The research efforts for developing conventional hydrodesulfurization and alternative desulfurization methods such as selective adsorption, biodesulfurization, oxidation/extraction (oxidative desulfurization), etc. for removing these refractory sulfur compounds from petroleum products are on the rise. Research laboratories and refineries are spending huge amounts of money in finding a viable and feasible solution to reduce sulfur to a concentration of less than 10 mg l−1. This paper reviews the current status in detail of various desulphurization techniques being studied worldwide. It presents an overview of novel emerging technologies for ultra-deep desulfurization so as to produce ultra-low-sulfur fuels.
1. Introduction
Energy production is one of the most pressing issues of modern times. Economic activity and energy usage are intimately linked. The production of useful goods and services requires energy. Fossil fuels are the most widely used sources of energy in the world. Although the percentage of energy obtained from fossil fuels declined over recent years, the share of world energy from fossil fuels is still over 82%, half of which comes from petroleum.1 Crude oil, a complex mixture of organic liquids, is the largest source of energy. Major portions of the crude oil are used as transportation fuels such as gasoline, diesel and jet fuel. It occurs naturally in the ground and was formed millions of years ago. The sulfur content and the American Petroleum Institute (API) gravity are two properties which have a great influence on the value of the crude oil. The sulfur content is expressed as a percentage of sulfur by weight and varies from less than 0.1% to greater than 5% depending on the type and source of crude oils.2 Sulfur compounds exist in various forms and can be classified into four main groups: mercaptans, sulfides, disulfides and thiophenes (THs).Sulfur compounds are undesirable in refining process as they tend to deactivate some catalysts used in crude oil processing and cause corrosion problems in pipeline, pumping, and refining equipments. Table 1 shows the level of sulfur in global supplies of crude oil. Naturally occurring sulfur compounds left in fuels lead to the emission of sulfur oxide gases. These gases react with water in the atmosphere to form sulfates and acid rain which damages buildings, destroys automotive paint finishes, acidifies soil, and ultimately leads to loss of forests and various other ecosystems.3 Sulfur emissions also cause respiratory illnesses, aggravate heart disease, trigger asthma, and contribute to formation of atmospheric particulates.4 Automobiles are also adversely affected by presence of sulfur compounds in liquid fuels. Sulfur levels in automotive fuels have a profound effect on the efficacy of catalytic converters.
| Region | Crude Oil gravity (API) | Sulfur weight (%, 1990) | Production (tpd) | Crude oil gravity (API) | Sulfur weight (%, 2010) | Production (tpd) |
|---|---|---|---|---|---|---|
| Alaska | 26.970 | 1.11 | 1954 | 28.340 | 0.99 | 1645 |
| Canada | 31.400 | 1.52 | 2000 | 32.000 | 1.62 | 2500 |
| California | 17.430 | 1.59 | 970 | 18.730 | 2.60 | 951 |
| Rest of USA | 35.110 | 0.86 | 4510 | 36.930 | 0.88 | 2470 |
| Africa | 31.280 | 0.17 | 7000 | 32.640 | 0.18 | 6100 |
| Europe | 33.200 | 1.09 | 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 330 330 |
33.700 | 1.10 | 15530 |
| Latin America | 25.060 | 1.62 | 7770 | 27.100 | 1.82 | 9850 |
| Middle East | 33.730 | 1.69 | 29100 |
34.350 | 1.71 | 35760 |
| Far East | 33.800 | 1.09 | 16330 |
37.300 | 1.10 | 15530 |
| World Average | 31.300 | 1.13 | 70800 |
32.810 | 1.27 | 83450 |
The current industrial method for removal of sulfur from fuels is hydrodesulfurization (HDS), which is a high temperature, high pressure catalytic process. This makes HDS a very costly option for deep desulfurization. Moreover, HDS is not effective for removing heterocyclic sulfur compounds such as dibenzothiophene (DBT) and its derivatives, especially 4,6-dimethyldibenzothiophene (4,6-DMDBT). Deep desulfurization of gasoline (from 500 to <10 ppm sulfur) is restricted largely by DMDBT, which is the least reactive sulfur compounds. Oxidative desulfurization (ODS), oxidation–extraction desulfurization (OEDS), adsorptive desulfurization and bio-desulfurization (BDS) are the other desulfurization techniques that have the potential to produce ultra clean fuels. In ODS, the sulfur containing compounds is oxidized to sulfone by chemical reaction using an oxidant viz. H2O2, H2SO4, etc. The sulfone compound is then easily extracted from the fuel due to its higher polarity. In the adsorption process, the adsorbents used in the process selectively grab the sulfur. The active adsorbent is placed on a porous, non-reactive substrate that allows the greatest surface area for adsorption. Adsorption occurs when the sulfur molecules attach to the adsorbent on the substrate and remain there separate from the fuel. BDS has drawn wide attention recently because of its green processing of fossil fuel. However, the slowness of the removal process is a major hindrance in the use of BDS process.
Today, the strongest motivation for the reduction of sulfur in fuels is due to environmental regulations which are imposing stringent limits for sulfur levels in transportation fuels. New techniques are required to remove the sulfur from lower quality feed stocks to ensure that energy is available at a reasonable cost. This paper reviews the current status and details of various desulphurization techniques being studied worldwide to remove sulfur compounds from liquid fuels and aims to identify the research gaps in these techniques.
2. Effect of sulfur on the environment
The pollution emitted by vehicular engines greatly affects the air quality. The sulfur compounds are converted to sulfur oxides by combustion, and these ultimately lead to acid rain.5–7 Sulfur dioxide and other combustion related pollutants from sulfur compound containing fuels lead to environmental concerns such as smog, global warming and water pollution.8 Even with more stringent heavy-duty highway engine standards, these engines will continue to emit large amounts of nitrogen oxides (NOx) and particulate matter (PM) both of which contribute to serious public health problems. The PM present in diesel exhaust is likely to cause high levels of lung cancer in humans. Other health effects include aggravation of respiratory and cardiovascular diseases, aggravation of existing asthma, chronic bronchitis and decreased lung function. Therefore, desulfurization of fuels is extremely important in the petroleum industry.The removal of sulfur from petroleum is also necessary from industrial point of view. The automobile manufacturers demand removal of sulfur containing compounds out of petroleum in order to reduce overall emissions from vehicles. This is because sulfur compounds poison the catalytic converters that reduce particulates and NOx emissions.9 Sulfur affects these emission control devices by strongly adsorbing to the precious metal catalysts, preventing the adsorption and reaction of hydrocarbons, NOx, and carbon monoxide.10
In addition, sulfur compounds in petroleum also cause corrosion to parts of internal combustion engines and refineries because of the formation of the oxyacids of sulfur from combustion products.11 Also, sulfur compounds are undesirable in refining processes because they tend to deactivate some catalysts used downstream and upgrading of hydrocarbons.12 Moreover, sulfur compounds contribute to the formation of gummy deposits in liquid petroleum products.
3. Standards
The United States Environmental Protection Agency (US EPA) mandated reduction of the sulfur content of diesel fuel and gasoline. In 2006, EPA mandated maximum sulfur content of 15 ppm in diesel fuel.10Table 2 shows the journey of emission standards the worldwide.13 According to Euro V norms the fuel sulfur content will have to be reduced to as low as 10 ppm in near future. Sulfur regulations that took effect in Canada and the USA in June 1, 2006 reduced the sulfur content in on-road diesel fuel and gasoline from 500 mg kg−1 and 350 mg kg−1 to 15 mg kg−1 and 30 mg kg−1, respectively.14,15 Currently, the maximum allowable sulfur content in diesel in USA and Europe is 10 mg kg−1.16,17| Country | 96 | 97 | 98 | 99 | 00 | 01 | 02 | 03 | 04 | 05 | 06 | 07 | 08 | 09 | 10 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| California (US) | CARB (90 ppm) | CARB (80 ppm) | CARB (15 ppm) | ||||||||||||
| USA | EPA (500 ppm) | 300 ppm | 80 ppm | 15 (ppm) | |||||||||||
| EU | Euro II (500 ppm) | E III (150 ppm) | E IV 50 ppm | E V 10 ppm | |||||||||||
| China (H) | E I | Euro II | Euro III | Euro IV | |||||||||||
| Thailand | Euro I | E II | Euro III | E IV | |||||||||||
| Singapore | Euro I | E II | Euro III | E IV | |||||||||||
| Malaysia | Euro I | Euro II | E III | ||||||||||||
| India (metros) | E I | Euro II | Euro III | E IV | |||||||||||
| India | Euro I | Euro II | E III | ||||||||||||
| Nepal | Euro I | ||||||||||||||
| Philippines | Euro I | ||||||||||||||
| Indonesia | Euro II | ||||||||||||||
The latest regulation in India, which was regulated in 2010 for major cities of India, reduced the gasoline sulfur content from 150 ppm to 50 ppm and the diesel sulfur content from 350 ppm to 50 ppm. Refineries in India are facing major challenges to meet the fuel sulfur specification along with the required reduction of aromatics contents.18 In coming sections, details of various sulfur removal technologies like HDS, ODS, OEDS, adsorptive removal and BDS are discussed.
4. Hydrodesulfurization
4.1. Introduction
HDS is used traditionally in refineries to reduce the sulfur content in fuels. Interest in HDS was initially stimulated by the availability of hydrogen from catalytic reformers.19 Typically, the HDS process involves catalytic treatment with hydrogen to convert the various sulfur compounds to H2S and sulfur-free organic compounds at high temperature and partial pressure of hydrogen.20–21 Conventional catalytic HDS method for reducing sulfur content requires severe conditions of operation. In refineries, the H2S resulting from the HDS reaction is eventually converted to elemental sulfur by a modified version of the Claus process.224.2. Reaction conditions for HDS reaction
The HDS reaction in refineries is carried out in trickle-bed reactors. These reactors are commonly operated at temperatures in the range 300–450 °C, and at H2 pressures of 3.0–5.0 MPa, usually with CoMo/Al2O3 or NiMo/A12O3 catalysts.7,23 Under these harsh conditions, olefins get hydrogenated, leading to a loss of octane rating and excess hydrogen consumption. Under mild HDS conditions, H2S can react with olefins in the reactor to create recombinant mercaptans which are linear or branched thiols of typically 5–12 carbons. The formation of recombinant mercaptans causes sulfur to be retained in the product, limiting the effectiveness of the HDS process. Further research in HDS catalysis and process designs are being carried out so as to increase the sulfur removal and still maintain the fuel quality at some minimum specification.In the fuel cell systems, HDS reactions are preferable to be carried out at atmospheric pressure. Since hydrogen is valuable reactant in the fuel cell, it is desirable to run the HDS reaction with a low H2/fuel ratio. HDS has been adapted for phosphoric acid fuel cell (PAFC) systems, which operate with natural gas.24 HDS has also been investigated for use in conjunction with a direct carbonate fuel cell.25 The mechanism of HDS is discussed in subsequent section. At atmospheric pressure, the HDS reaction proceeds only via hydrogenolysis route and not via the hydrogenation (HYD) route26 which is considered to be the effective route in desulfurizing the refractory sulfur compounds such as 4,6-DMDBT.
At atmospheric pressure and at temperatures >300 °C, the hydrogenation of aromatic rings is thermodynamically limited. This is advantageous in the fact that the catalysts active sites don't get occupied for the hydrogenation of aromatic compounds. However, inhibition in the presence of aromatics has been noticed even when they are not hydrogenated. Studies to determine the optimum operational pressure for the HDS unit for fuel cell application are, therefore, scope of interest for further research.
4.3. Mechanism of HDS
It is generally accepted that the HDS proceeds through the reaction network proposed by Houalla et al.27–28 In general, the reaction mechanism of DBT and 4,6-DMDBT through HDS process is suggested to proceed via two main pathways. One is a DDS or hydrogenolysis pathway where sulfur is removed without abetting the aromatic rings. The other is via a hydrogenation desulfurization (HYD) pathway, in which aromatic rings of DBT compounds are preferentially hydrogenated to 4H-or 6H-DBT intermediates and are subsequently desulfurized.20The reaction mechanism for the HDS of DBT at 300 °C and 102 atm is illustrated in Fig. 1.28 The reaction of hydrogen with DBT gives biphenyl (BiPh) as predominant organic product. In the HYD pathway mechanism, the primary reaction products formed directly from DBT are tetrahydrodibenzothiophene (THDBT) and/or hexahydrodibenzothiophene (HHDBT). Both THDBT and HHDBT are very reactive intermediates and are difficult to isolate for detection. These compounds get further desulfurized to form cyclohexylbenzene (CHB) as the secondary product. This pathway is referred to as HYD pathway since the sulfur compound is hydrogenated prior to desulfurization. Direct C–S bond hydrogenolysis of DBT gives biphenyl via DDS pathway. Sequential hydrogenation of biphenyl produces CHB. Bicyclohexyl (BiCh) is the tertiary product formed in traces via the slow hydrogenation of CHB formed by any of the two pathways.
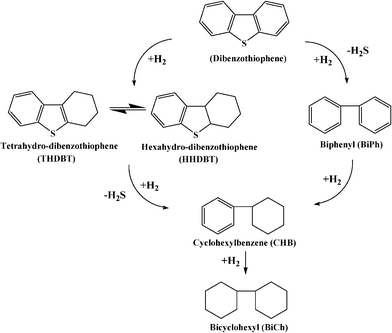 | ||
| Fig. 1 Pathways for the HDS of DBT at 300 °C and 102 atm in the presence of CoMo/Al2O3.28 | ||
The DDS reaction is faster than the HYD. However, the HYD pathway becomes relatively fast with an increase in H2S and/or H2 concentration in the reaction mixture; with an increase in methyl groups in the 4 or 4 and 6 positions; and with more active catalyst for hydrogenation e. g. NiMo/Al2O3vs. CoMo/Al2O3.27–29 Houalla et al.27–28 found that the activity of NiMo/Al2O3 catalyst (per unit surface area) was about twice as much as that of the CoMo/Al2O3. The yield of CHB, at a given conversion, was about three times higher with NiMo/Al2O3 catalysts than with CoMo/Al2O3. This indicates that HYD is a better route for increasing the desulfurization extent of the refractory compounds. The desulfurization rate of hindered compounds is greatly increased through the HYD route. Without one or both of the rings, the sulfur molecule becomes much more flexible, and the sulfur atom approaches the catalyst surface much easily and gets removed.
In general, when the unpaired electrons of the sulfur can resonate with the pi electrons of the organic structure, the energy of the carbon-sulfur bond (C–S) becomes practically identical with that of the carbon-carbon (C–C) bond.30 This leads to a reduction in the selectivity of the HDS process, and hydrogenation of carbon-carbon bonds happens. Saturated hydrocarbons lead to a lower-grade fuel, and require additional processing steps.
4.4. Effect of refractory organosulfur compounds on HDS
The effectiveness of the HDS process depends on the type of the sulfur compound. Nag et al.31 carried out HDS of various organosulfur compounds using sulfided CoO–MoO3/γ–Al2O3 as catalyst. They found that the following order of reactivity for HDS: TH > BTH > benzonaphthothiophene > tetrahydrobenzonaphthothiophene > DBT. The substitution of these compounds by ring alkylation further affected the reactivity. Kilanowski et al.32 carried out HDS of methyl-substituted DBTs (DBTs) at atmospheric pressure using sulfides CoMo/γ–A12O3. They showed that the reactivity of DBTs, with methyl substitution in different positions, decreases in the following order: 2,8-dimethylDBT (2, 8-DMDBT) > DBT (DBT) > 4-methyldibenthothiophene (4-MDBT) > 4,6-dimethylDBT (4,6-DMDBT). The reactivity trends for DBT, 4-MDBT, and 4,6-DMDBT were found to be similar under the industrial conditions with sulfided CoMo/Al2O3,27 CoMo/Al2O3–zeolite,33 and NiMo/Al2O3.34 The presence of aromatic compounds in the fuel further inhibited the activity towards HDS. Polar compounds such as nitrogen- and oxygen-containing compounds are also known to have strong inhibiting effects on HDS reactions.35–36 Egorova and Prins35 reported that even trace amount of naphthalene had an inhibitive effect on the HDS of DBT and 4,6-DMDBT at 340 °C and 5 MPa. It has been reported that H2S, produced by the HDS reaction itself, inhibits the HDS reaction in hydrocarbon fuels, especially in direct sulfur extraction reaction.37–39 However, inhibitive effect of H2S on the HDS reactions depends upon the sulfur content of the raw fuel feed. The inhibition to the HDS process is related to the competition between the inhibitors and the organosulfur compounds for adsorption on the catalyst active sites.Thus it may be said that reactivity of high boiling point organosulfur compounds such as BTHs and DBTs that are present in intermediate distillates like diesel, toward HDS reaction is substantially lower than that of the low boiling point compounds such as thiols, sulfides, and disulfides that are present in natural gas and light feedstocks.7,23,40 Aromatic, cyclic, and condensed multicyclic compounds are also considered to be more difficult to remove.41 The sterically hindered BTHs and DBTs are, therefore, the target compounds for HDS in most of the present studies. DBT, 4-methyl-DBT (4-MDBT) and 4,6-DMDBT etc., therefore, must be removed to reduce sulfur content in fuel.42
4.5. Catalysts for HDS
Table 3 summarizes recent researches conducted for optimization of HDS processes utilizing various catalysts and reactor configurations.43–57 HDS with Mo, Ni or W-based catalysts are widely used to reduce organosulfur compounds such as mercaptans, thioethers, and disulfides. The most common catalysts used in HDS are cobalt or nickel promoted molybdenum sulfide. Recent research in HDS aims to develop catalyst that can effectively remove the refractory organosulfur compounds. Subsequent section aims to discuss about various catalysts developed for HDS, their synthesis, characterization, activation and operating conditions.| Process | Sulfur Compound | Oil | Catalyst | System | S Conc. Co (ppm) | Optimum Conditions | % S Removal | Reference | |
|---|---|---|---|---|---|---|---|---|---|
| Temp. (°C) | Pressure (MPa) | ||||||||
| Deep HDS | Alkyl DBTs | Light Oil | Co-Mo/Al2O3 | Packed Bed | 4000 | 300 | — | 67% | 43 |
| Three stage HDS | Alkyl DBTs | Diesel | CoMo + NiMo | Packed Bed | 7060 | 250 | 2.9 | 97% | 44 |
| HDS | Alkyl DBTs | Heavy gas oil | Co-Mo-A1 | Trickle bed | 1800 | 360 | 5.5 | 20% | 45 |
| Deep HDS | Alkyl DBTs | Gas oil | Co-Mo/γ–A12O3 | Fixed bed | 13200 |
350 | 3 | 96% | 46 |
| HDS | Alkyl DBTs | Heavy gas oil | Co, Ni, Mo, W supported on Al2O3/SiO | Trickle Bed | 13300 |
320 | 5.4 | 80% | 47 |
| In situ H2 generation | BT | Water/Toluene emulsion | Mo | Batch autoclave | 350 | 340 | 20 | 99.5% | 48 |
| Staged HDS | Alkyl DBTs | Medium cycle oil | NiMoS/Al2O3 | Packed Bed | 4900 | 340 | 5 | 97% | 49 |
| Deep HDS | Alkyl DBTs | Light cycle oil | Co-Mo supported on MCM-41 | Packed Bed | 21900 |
350 | 4.5 | 57% | 50 |
| Lab Scale HDS | Alkyl DBTs | Middle distillates | NiMo/γ–Al2O3 | Trickle bed | 16740 |
350 | 4 | 90% | 51 |
| Deep HDS | Alkyl DBTs | Gas oil | CoMo/Al2O3 + NiMo/Al2O3 | Packed Bed | 14350 |
340 | 3 | 98% | 52 |
| HDS | Alkyl DBTs | Gas oil | NiMo sulfide on Al2O3–Si support | Packed Bed | 11780 |
340 | 4.9 | 94% | 53 |
| Deep HDS | Alkyl DBTs | Gas oil | CoMoS and NiMoS | Batch autoclave | 340 | — | 4.9 | 97% | 54 |
| Oil presaturated with H2 | Alkyl DBTs | Diesel | CoMo | Two phase reactor | 1200 | 400 | 7 | 99% | 55 |
| Lab Scale Deep HDS | Alkyl DBTs | Diesel | P and Ni–Al2O3 supported Mo oxycarbides | 520 | 340 | 4 | 50% | 56 | |
| HDS | Thiophene | n-Heptane | FeS–MoS supported on Al2O3 and carbon | — | 1000 | 280 | 0.1 | 30% | 57 |
The synergetic effects of the bimetallic catalysts like CoMo/Al2O3, NiMo/Al2O3, NiW/Al2O3, CoW/Al2O3, and PtPd/A12O3 has been extensively studied.35,62–64 Isoda et al.33 performed a study using a blend of CoMo/Al2O3 and Ru/Al2O3 catalysts, and compared its activity towards the HDS of 4,6-DMDBT in the presence of naphthalene to those of CoMo/Al2O3, NiMo/Al2O3, and Ru/A12O3. The blend catalyst showed the highest rate of HDS of 4,6-DMDBT through its selective hydrogenation without excessive hydrogenation of naphthalene. Lecrenay et al.65 found that the commercial NiMo/Al2O3 exhibited three times higher activity than that of the commercial CoMo/Al2O3 on the HDS reaction pathways of 4,6-DMDBT in decalin. This was ascribed to the higher hydrogenation activity of NiMo/A12O3 catalyst as compared to CoMo/A12O3. Steric hindrance is considered to be the reason behind the low reactivity of 4,6-DMDBT.66 Hydrogenation of the aromatic ring leads to easing of steric hindrance, thus, catalysts with higher hydrogenation capability show higher catalytic activity towards the HDS of 4,6-DMDBT. Li et al.67 investigated the HDS of tetrahydro-, hexahydro-, and dodecahydro-dimethyl DBT (DMDBT) over sulfided Mo and NiMo on γ–Al2O3 catalysts at 300 °C and 5 MPa. Tetrahydro-DMDBT reacted by hydrogenation to hexahydro-DMDBT, which in turn reacted to dodecahydro-DMDBT by hydrogenation and to 3,3-dimethylcyclohexylbenzene by desulfurization. All four diastereoisomers of hexahydro-DMDBT were observed, all of which interconverted rapidly during HDS. Rodriguez-Castellon et al.68 prepared HDS catalysts by incipient wetness impregnation of Ni–Mo(W) and Co–Mo(W) species over siliceous MCM-41 doped with zirconium. All the catalysts displayed a very good performance in the temperature range of 300–340 °C, with conversions between 49.0% and 92.6%. The Ni promoted catalysts displayed better performances than those of Co promoted catalysts in the HDS of DBT.
Yoosuk et al.69 carried out a comparative study of unsupported MoS2 and Me/MoS2 (Me![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Co, Ni) catalysts prepared by hydrothermal synthesis. Sakanishi et al.70 studied the HDS of 4,6-DMDBT using Ru and NiMo catalysts supported on carbon. The carbons examined in the study were carbon blacks, granular active carbons with moderate and large surface areas, and pitch-based activated carbon fibers (ACF) with large surface areas. The NiMo/C catalysts exhibited higher activity for the HDS of 4,6-DMDBT at relatively higher temperatures of 340–380 °C than a commercial NiMo/A12O3 catalyst regardless of the type of the carbon support. The main route was found to be the DDS. Nav et al.71 prepared Co/Mo catalysts supported on a Ti–loaded hexagonal mesoporous SBA-15 material. The Ti–loaded SBA-15 catalysts were more active than the Ti–free counterpart due to the enhancement of the DDS route in this reaction.
Co, Ni) catalysts prepared by hydrothermal synthesis. Sakanishi et al.70 studied the HDS of 4,6-DMDBT using Ru and NiMo catalysts supported on carbon. The carbons examined in the study were carbon blacks, granular active carbons with moderate and large surface areas, and pitch-based activated carbon fibers (ACF) with large surface areas. The NiMo/C catalysts exhibited higher activity for the HDS of 4,6-DMDBT at relatively higher temperatures of 340–380 °C than a commercial NiMo/A12O3 catalyst regardless of the type of the carbon support. The main route was found to be the DDS. Nav et al.71 prepared Co/Mo catalysts supported on a Ti–loaded hexagonal mesoporous SBA-15 material. The Ti–loaded SBA-15 catalysts were more active than the Ti–free counterpart due to the enhancement of the DDS route in this reaction.
Supported platinum has also been studied as a catalyst for HDS under standard industrial conditions. Qian et al.64 compared the activity of unsulfided Pt/A12O3 (3 wt%) with the conventional CoMo/Al2O3 catalyst. Both the catalysts were found to have similar activity towards HDS of DBT in decalin. Navarro et al.72 compared Pt-based catalysts with the commercial CoMo/A12O3 in HDS of commercial diesel fuel. It was found that both Pt/HY zeolite and R/ASA (amorphous silica-alumina) were more active than the CoMo/A12O3 catalyst. Robinson et al.73 used catalysts with high hydrogenation activity to desulfurize 4-ethyl, 6-methyl DBT (4-E, 6-MDBT). The HDS activity was found to be in the following order: Pt/ASA ≫ Pt/Al2O3 > NiW/Al2O3 ≫ CoMo/Al2O3 or NiMo/Al2O3. The superiority of Pt/ASA catalyst in HDS of 4-E, 6-MDBT was attributed to its superior hydrogenation activity. Both sulfides and unsulfided Pt/ASA were found to have similar catalytic activities. Reinhoudt et al.74 utilized unsulfided (reduced) Pt to hydrodesulfurize 4-E, 6-MDBT at 360 °C and 60 atm. The catalyst activity was found to reduce in the following order: Pt/ASA > CoMo/Al2O3 > Pt/Al2O3. Baldovino-Medrano et al.75 tested a Pt/γ–Al2O3 catalyst in simultaneous HDS of DBT and hydrodearomatization (HDA) of naphthalene reactions. Samples of it were subjected to different pretreatments: reduction, reduction–sulfidation, sulfidation with pure H2S and non-activation. The reduced catalyst presented the best performance, even comparable to that of Co(Ni)Mo catalysts.
In the past few decades there has been intense activity on oxides and mixed oxides as supports to Mo, CoMo, NiMo, W and NiW. These supports have been prepared using several methods, and effects of preparation on physico-chemical properties have been studied. The support and catalysts were characterized in few cases, however, most of the times only activity data is available. There is insufficient data to arrive at a consensus regarding the factors that are responsible for such increase in activity. Effect of catalyst support is not well researched and documented in the literature. This area is needed to be well understood for developing better catalyst for HDS.
Deactivation caused by loss of sulfur could be avoided by using catalysts that activate without sulfiding, while the deactivation caused by coking could be avoided if coke-tolerant or coke-preventing catalysts are used. The catalyst is activated before being used in the HDS process by sulfiding the metallic phase. The activation/sulfiding step is done by treating the catalyst with a mixture of HDS and H2 or a feed containing sulfur compounds and H2.19 Qian et al.64 and Reinhoudt et al.74 demonstrated that Pt based catalysts are sufficiently active without sulfiding the metal phase as compared to Mo, Ni, or Ru based catalysts.
It is generally recognized that the carbon-supported HDS catalysts have a potential advantage over alumina-based catalysts, particularly with respect to the low coking properties of the former which can result in prolonged catalyst life. Additional benefits can also be gained by recovering the active metals (cobalt, nickel, molybdenum or tungsten) from spent catalysts by burning off the carbon and the coke.
4.6. Commercial HDS and future challenges in refineries
HDS is a commercially proven refining process that passes a mixture of heated feadstock and hydrogen over catalysts to remove sulfur. Refineries desulfurize both distillate streams generated during direct distillation of crude oil and streams coming out from conversion units such as fluid catalytic cracking (FCC) and hydrocracker units. HDS can be performed either before FCC or after, depending on the refinery design. However, HDS must be performed before reforming, due to the poisoning effect of sulfur on Pt. In the HDS reactor, sulfur is reduced liberating H2S which is then removed from the flue gas by amine scrubbing. HDS is the primary desulfurization technology used today, although caustic washing to remove low molecular weight thiols is also performed. Most HDS operations also remove nitrogen compounds and some metal impurities.5Refineries meet the ultra low sulfur specifications on fuels that are produced from straight runs streams by controlling the hydrogenating conditions and selecting the appropriate catalysts. The difficulty however arises in the desulfurization of other steams that come from the conversion units, which mostly include the refracted sulfur compounds. Isoda et al.84 improved the currently practised HDS processes to remove sulfur from DBT derivatives by using a nickel-supported zeolite catalyst. This technology also has two steps. The reaction is carried out at 270 °C for 1 h and at a hydrogen pressure of 250 Pa in the first step, and then, the products are further desulfurized over a CoMo/A12O3 catalyst at 300 °C for 2 h at a hydrogen pressure of 250 Pa to remove 4,6-DMDBT. The disadvantage of the process is high temperature and pressure. Having more steps during the desulfurization also brings extra cost for the refineries.
There are several constraints in the HDS process. Meeting the sulfur requirement for gasoline is believed to be the greatest challenge for the refining business requiring substantial revamps to equipment or even construction of new units. This is due to the fact that most of the gasoline production in the market today is coming from cracked stocks that contain a larger concentration of compounds with aromatic rings and high olefin content, thus, making sulfur removal more difficult. The need to desulfurize the cracked stocks in addition to the straight-run streams is forcing the refiners to choose the most cost-effective technology.20–21,85–88 In India, refiners today decrease the sulfur content to about 50 ppm but as the sulfur limits are decreased, there will be a need to increase the number of HDS units, which will tremendously increase the demand for hydrogen. Higher temperatures and pressures will also be needed to remove the recalcitrant sulfur compounds which raise operating costs.89
HDS is limited in treating BTs and DBTs (especially DBTs having alkyl substituents on 4 and/or 6 positions). The production of light oil with very low levels of sulfur-containing compounds requires the application of severe operating conditions and the use of especially active catalysts.90–92 Moreover, the HDS process has reached a stage where increasing temperature and pressure are just not enough to remove last traces of sulfur without affecting the octane number.90
Another problem is that the increased use of the HDS process to extract sulfur from petroleum adds to the volume of H2S. This puts pressure on the Claus plant capacity, which produces hydrogen for the refineries.91
In the case of diesel, a two stage deep desulfurization process will most probably be sufficient to meet the 10 ppm sulfur target in future. The first stage can reduce the sulfur level to below 50 ppm with a second stage that could produce diesel product with 10 ppm sulfur or less. In some cases the first stage could be a conventional hydrogenating unit with moderate adjustments to the operating parameters. The second stage would require substantial modification of the desulfurization process, primarily through use of higher pressure, increasing hydrogen flow rate and purity, reducing space velocity, and choice of catalyst.88,92 Several academic and industrial research groups are working to improve the current HDS technology practised in refineries.
Although HDS processes have dominated desulfurization of petroleum in the past their cost and the requirements of the upcoming strict fuel specifications combine to motivate the development of innovative process technologies.
5. Oxidative desulfurization (ODS)
5.1. Introduction
ODS is a promising technology for the reduction of sulfur at low temperature (∼50 °C) and atmospheric pressure.93 In ODS, heavy sulfides are oxidized by adding one or two oxygen atoms to the sulfur using appropriate oxidants without breaking any carbon–sulfur bonds, yielding the sulfoxide and sulfone, respectively. These oxidized compounds are then extracted or adsorbed from the light oil due to their increased relative polarity. Thus, the ODS is basically a two stage process; oxidation, followed by liquid extraction.Oxidation of DBT to the corresponding sulfone can be represented as:40,42,94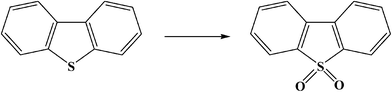
Oxidation of the DBT derivatives to the corresponding sulfones increases their polarity and molecular weight. This facilitates their separation by extraction,12 distillation,90 or adsorption.95 Any of these separation methods could be used for separation of sulfur from the organic phase.
Petroleum industries commonly employ solvent extraction techniques to remove sulfur and nitrogen compounds from light oil. The solvent is recovered and reused through a distillation process.88,96 However, the difference in polarity between sulfur and other aromatic hydrocarbons is very small. Therefore, employing only solvent extraction is accompanied by associated loss of useful hydrocarbons along with reduction of sulfur compounds removal.88,92,97–98 The selective oxidation approach of increasing the polarity of sulfur compounds followed by their removal by selective extraction, therefore, has received much great attention recently.
5.2. Oxidants in ODS
A significant research effort has been conducted on ODS over the years. Many oxidants have been investigated that include organic and inorganic peroxy acids, catalyzed hydro-peroxides, peroxy salts, NO2, tert-butyl-hydroperoxide, O3, etc.88,93,99–105ODS process were first employed using nitrogen dioxides as an oxidant, followed by extraction with methanol to remove both sulfur and nitrogen compounds from petroleum stocks. Few investigators have used an oxidizing gas containing nitrogen oxides for purifying hydrocarbon aqueous oils containing both sulfur and nitrogen compound.88,92,96 Attar and Corcoran106 oxidised diesel fuel with nitrogen oxides. Overall 70% conversion of sulfur compounds to sulfones was obtained at 140 °C. However, this technology yields many undesirable byproducts because of initiation of a very non-selective reaction by nitrogen oxides in the presence of oxygen. Also, there is always a safety problem due to the possibility of a rapid and explosive reaction.
Several peroxy organic acids (formic, acetic, propionic, performic, pertrifluoro acetic acids etc.) and Caro's acid (peroxysulfuric acid) have been employed at 95–125°F near atmospheric pressure for selective oxidation of organic-sulfur compounds.90,96,107 Liu et al.108 investigated the oxidation of model sulfur compound and diesel oil by K2FeO4 in water-phase, in organic acid and in the presence of phase-transfer catalysts. The results showed that the oxidation activity of BT and DBT was low in water-phase, even after adding phase-transfer catalyst to the system. This was because K2FeO4 reacted rapidly with water to form brown Fe(OH)3, thus, losing the oxidation ability. The oxidation activity of the BT and DBT increased markedly in acetic acid. Moreover, the addition of the solid catalyst to the acetic acid medium promoted oxidation of organic sulfur compounds. Conversions of the BT and DBT were 98.4% and 70.1%, respectively, under the condition of room temperature, atmospheric pressure, acetic acid/oil (v/v) = 1.0, K2FeO4/S (mol/mol) = 1.0 and catalyst/K2FeO4 (mol/mol) = 1.0. Under the same condition, 96.7% sulfur removal (457 ppm to 15.1 ppm) was obtained from diesel oil which was subjected to oxidation followed by furfural extraction. Nehlsen5 reported the oxidation and extraction of organo-sulfur compounds, including TH and alkyl sulfides with concentrated sulfuric acid. Sulfuric acid is not typically regarded as an oxidizing acid due to the stability of the sulfate ion. However, in the presence of sulfur atoms with lower oxidation states, such as those in sulfides, sulfate can be reduced. The reaction between H2S and concentrated sulfuric acid is fast and yields elemental sulfur, water, and SO2 as reaction products.
Shiraishi et al.109–111 accomplished desulfurization of sulfur compounds such as DBTs and BTs with alkylating agents, CH3I and AgBF4 in the presence of dichloromethane. This process was based on the formation and subsequent precipitation of S-alkylsulfonium salts. Main drawback of this process was methylation of other aromatic hydrocarbons. Shiraishi et al.112 reported desulfurization of light oils to less than 0.05 wt% based on the formation and subsequent adsorption of N-tosylsulfimides, produced by the reaction of the sulfur compounds in the light oils with chloramine T. The desulfurization of high-aromatic-content light oil was relatively more difficult due to the chlorination of the aromatic hydrocarbons by chloramine T which becomes competitive with chlorination of the sulfur compounds.
H2O2 is the most common oxidant because of its environmental friendliness. The oxidation of sulfur compounds with H2O2 has been studied over various catalytic systems such as HCOOH, CCl3COOH, polyoxomethalate CF3COOH, methyl-trioxorhenium(VII), and phosphotungstic acid, titano silicates, and solid base.92,94,99,112–118 Shiraishi et al.109 reported sulfur and nitrogen removal from light oils using hydrogen peroxide and acetic acid. Lanju et al.119 carried out ODS of simulated gasoline consisting of model sulfur compounds of TH and 3-methythiophene (3-MC4H4S) dissolved in n-heptane in hydrogen peroxide (H2O2) and formic acid oxidative system over metal oxide-loaded molecular sieve. The results showed that the sulfur removal rate of simulated gasoline was higher in H2O2/organic acid systems than in H2O2/inorganic acid systems. The cerium oxide-loaded molecular sieve was found very active catalyst for oxidation of simulated gasoline in this system. The sulfur removal rates of C4H4S and 3-MC4H4S were enhanced when phase transfer catalyst (PTC) was added. The sulfur removal rate of simulated gasoline reduced with the addition of cyclohexene and xylene into the solvent n-heptane. Al-Shahrani et al.120 utilized a catalytic system composed of Na2WO4, 30% H2O2 and CH3COOH for the deep removal of sulfur in diesel. Treatment of model solutions of octane containing DBT and 4,6-dimethyl DBT with the above ODS system showed 100% conversion of the THs to sulfones at 70 °C in less than 1 h. At modest temperatures and under atmospheric pressure, the catalytic system was effective for removing most of the last few hundred ppm of HDS-persistent organic sulfur containing compounds in diesel.
Transition metal based catalysts namely methyl trioxorhenium mixed molybdenum/tungsten oxides121 and tungsto phosphoric acid (TPA)122 have been used in conjunction with a hydrogen peroxide as oxidant. Herbstman and Patel123 also reported that the use of same catalyst followed by a high temperature thermal or KOH treatment at 250 °C accounted 43% desulfurization.
D'Alessandro et al.124 reported a catalytic system consisting of metal-sulfophthalocyanines (MPcS) and monopersulfate or hydrogen peroxide as oxidants for the DBT ODS. Among the various MPcS catalysts examined (MFe, Co and Ru), the ruthenium derivative exhibited the best performance with persulfate and iron derivative and hydrogen peroxide. Gutierrez et al.125 developed and evaluated Mo/γ–Al2O3 catalysts for the ODS of diesel fuel using H2O2 as the oxidizing reagent. The results showed that the activity for sulfur elimination depended mainly on the presence of hepta-and octamolybdates species in the catalyst support and the use of a polar aprotic solvent. Likewise, the presence of phosphate markedly increased the sulfur elimination. Using this catalyst, it was possible to reduce sulfur levels in diesel fuel from about 320 to less than 10 parts per million by weight (ppmw) at 333 K and atmospheric pressure. Caero et al.126 evaluated ODS activities of DBTs in hexadecane for a series of V2O5 catalysts supported on alumina, titania, ceria, niobia and silica. It was observed that the sulfone yield was not proportional to textural properties or V content. Total S-removal was close to 99% using vanadia on titania as catalyst, and this decreased according to the support used in the order: alumina > titania > niobia > Al–Ti mixed oxide > SBA-15. The oxidation activity of DBTs for V catalyst supported on niobia or alumina presented higher catalytic activity than all the other catalysts (niobia > alumina > SBA-15 > titania > ceria > Al–Ti mixed oxide). However, in the presence of an N-compound such as indole the best catalytic performance was obtained with titania-supported catalysts.
Collins121 used tetra-amido macrocyclic ligand (TAML) activators to enhance the oxidizing ability of hydrogen peroxide at low catalyst concentration and mild reaction conditions. TAML activators are used in many different areas including the pulp and paper, textile and laundry industries; mineralization of organohalogens, and others.127,128 They are capable of rapidly oxidizing the DBT derivatives that are of concern to the petroleum industry. Fe–TAML activators were used under different reaction conditions including variable pH, temperature and solvent composition. Micromolar concentrations of the FeF2B activator were reacted with H2O2 to convert greater than 99% of millimolar solutions (>7000:1 substrate: catalyst concentrations) of DBT derivatives to the corresponding sulfones under mild conditions. Yang et al.100 reported oxidation of DBT into sulfone using polyoxometalates with a Keggin structure such as H3PM12O40 [M, Mo(VI), W(VI)]. H3PW12O40 (HPWA) was supported on mesoporous molecular sieves SBA-15 to obtain HPWA-SBA-15. HPWA-SBA-15 has both catalytic oxidation ability and adsorption ability. The non-polar DBT were converted into polar DBT sulfones that were easily absorbed on HPWA-SBA-15.
5.3. ODS by other methods
Accumulation of hydrogen hampers mercaptan conversion, but controlled access of ozone-containing air mixed into the reactor allows one to regulate the degree of mercaptan conversion. The application of ozone as an oxidizer for oil mixtures was proposed earlier.130 One of the virtues of this technology is the possibility of achieving any desired degree of sulfur oxidation at minimum processing time and maximum process simplicity. This approach is technically feasible; however, it requires the development of special equipment and use of a multi-step process.
Nadirov et al.131 and Zaykin et al.132 studied the processing of heavy fuel oil by two irradiation methods under two different modes with the purpose of producing light oil fractions from the feedstock and, simultaneously, transforming sulfur into harmless and easily extractable forms. The feedstock was irradiated by 2 MeV electrons in the temperature range 300–400 °C using different values of other operational parameters (dose rate, P; dose, D). Mode 1 (P = 6 kGy/s, D = 30 kGy) used severe irradiation conditions and resulted in high yields of motor fuels. Mode 2 (P = 2 kGy/s, D = 70 kGy) was milder and caused lesser changes in hydrocarbon contents and appeared to be more favorable for conversion of sulfur compounds. 80% mercaptan conversion was reached in this milder mode and more than 90% of the total sulfur was concentrated in the heavy liquid fraction with boiling temperature higher than 350 °C.
Catalytic ODS process under phase transfer conditions and ultrasonication is termed as ultrasound assisted ODS (UAOD). This photo-induced oxidation process uses the absorption of short wavelength UV light for the direct photo-excitation of DBTs. This process relies on shock waves to agitate sulfur molecules, and allows for their extraction from oil as sulfones.138
The conceptual model of the oxidation step in the UAOD process may be depicted as a catalytic cycle, as depicted in Fig. 2.139–141 It consists of four basic steps: First, the metal precursor (simply represented as W(O)n), is peroxidized and disaggregated to form anionic peroxometal complex as W(O2)n in the presence of excess H2O2, phosphotungstic acid; second, quaternary ammonium salts such as Oc4N+Br−with large lipophilic cation function as PTA, and transfer the peroxometal anion into organic phase; third, organic sulfur compounds such as DBT get oxidized by the peroxometal complex with high efficiency and high selectivity; lastly, the reduced oxo species, which dissociate with PTA, returns to aqueous phase and restores the catalytic cycle.102
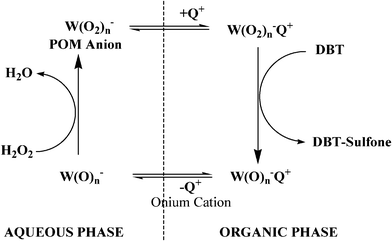
A high pressure mercury lamp (λ > 280 nm) has been used for desulfurization by direct photo-oxidation,143,144,146 gasoline147 and gas oil.148–149 Hirai et al.143 investigated photodecomposition of DBTs dissolved in tetradecane by the use of a high-pressure mercury lamp (λ > 280 nm). The decomposed products were removed to the water phase as SO42− at conditions of room temperature and atmospheric pressure. The order of reactivity for the DBTs was DBT < 4-MDBT < 4,6-DMDBT which is just opposite to that for the HDS method. The desulfurization yield of commercial light oil, however, was only 22% following 30 h of irradiation. This low yield was due to the depression of photoreaction of the DBT by the presence of aromatic compounds such as naphthalene and its derivatives in the light oil.143 Hirai et al.144 used same method for photodecomposition of benzothiophenes and alkyl sulfides.
Photosensitized oxidation has been also investigated for light oil to utilize light in the visible region (λ > 400 nm) more efficiently.145,150–151 Hirai et al.142 found that the addition of benzophenone (BZP), a triplet photosensitizer, enhanced the removal of DBT from tetradecane. However, this reaction didn't proceed in the presence of naphthalene (NP), because of triplet energy transfer from photoexcited DBT or BZP to ground-state NP. The addition of H2O2 enhanced the desulfurization of commercial light oil as well as the removal of DBT from tetradecane. This was due to the fact that H2O2 acted as a weak oxidizing agent for photoexcited DBT and interrupted the energy transfer from excited DBT to NP to some extent. The desulfurization yield, with the use of 30% H2O2 solution, of commercial light oil was 75% following 24 h of photo-irradiation and the sulfur content in the light oil was reduced from 0.2 wt% to less than 0.05 wt%.145 In these studies,143–145 desulfurization was basically done by photodecomposition of sulfur-containing compounds in the light oil phase, followed by the transfer of the resultant decomposed compounds into the aqueous phase. Thus, deactivation of the photoexcited DBTs by naphthalene was predictable. However, H2O2 is known to be photodecomposed by the absorption of the short-wavelength UV light (λ < 280 nm), which is necessary for the direct photoexcitation of DBT.
In earlier studies, photochemical reactions suffered from the quenching effect caused by associated aromatic hydrocarbons in fuels; and the desulfurization yield for real fuels was significantly lower than those for model solvents such as hexane and tetradecane.152 Extractive photo-oxidation, which combines the extraction of organosulfur compounds in polar solvents and subsequent photo-oxidation, was investigated to reduce the quenching effect by aromatic hydrocarbons.8,146,150,153 Shiraishi et al.8 found that DBT gets converted to DBT–O and then to DBT–O2, DBT sultine, and aromatic sulfonate or sulfinate anion in presence of acetonitrile by the UV irradiation. These products are highly polarized and are therefore not distributed into the nonpolar light oil phase. This process reduced the sulfur content of commercial light oil (CLO) from 0.2 to 0.05 wt% after 2 h of irradiation and that of straight-run light gas oil (LGO) from 1.4 to 0.05 wt% after 10 h of irradiation. The separation of the coextracted aromatics from acetonitrile was carried out successfully by using light paraffinic hydrocarbon stripping agents. Shiraishi et al.153 identified in detail the acidic products of BT and DBT in acetonitrile. This was achieved by methylation of the compounds with diazomethane and analysis using gas chromatography with atomic emission detection (GC-AED) analysis and Gas chromatography with mass spectra (GC-MS) analysis.
Studies on indirect photo-oxidation that generates oxidizing agents in situ by UV irradiation (400 nm > λ > 300 nm)154 and photocatalytic oxidation using TiO2 (λ > 290 nm)155–157 have also been reported. Abdel-Wahab and Gaber158 used anatase-type TiO2 and studied photocatalytic oxidation of DBT in acetonitrile. Matsuzawa et al.159 reported photo-oxidation of DBT and 4,6DMDBT in acetonitrile using titanium dioxide (TiO2) including P25 as catalysts. DBT was found to be more stable than 4,6DMDBT and that the rate of photo-oxidation differs depending on the kind of TiO2.
UV light below 280 nm has been used for fundamental studies on the mechanism of photo-oxidation of aliphatic disulfides, sulfides and thiols160–163 as well as the mechanism of photodissociation of aliphatic disulfides.164 Robertson and Bandosz157 used a multilayer TiO2–hectorite nanofilm photocatalyst for the photo-oxidation (λ < 280 nm) of dibenzothiophene (DBT) in nonpolar organic solution (tetradecane), as a model for diesel fuel. Photo-oxidation of DBT was performed with and without catalyst, at 254 and 300 nm and comparison was made with a commercially available TiO2 catalyst, Degussa P25. At 300 nm in the presence of catalyst, photo-oxidation preceded primarily by indirect photolysis, according to a zero-order rate law. At 254 nm, the rate was determined primarily by first-order direct photolysis, though the presence of the catalyst increased the overall rate of removal. Further adsorption of oxidation products by silica reduced the sulfur concentrations below 10 ppm. Tao et al.152 investigated selective photooxidation and adsorptive desulfurization of kerosene using a 5W low-pressure mercury lamp at 25 °C in the presence of O2. There is a group of sulfur compounds that are easily photo-oxidized with UV light below 280 nm in the presence of molecular oxygen without using any catalysts. The reactivity of these sulfur compounds was at least 100 times higher than those of DBTs and the photooxidation of these highly reactive sulfur compounds was completed within 30 min. The easily oxidized sulfur compounds can be removed from kerosene with adsorbents, such as molecular sieve, silica gel and activated alumina. On the other hand, non-reactive sulfur compounds such as DBTs were removed by adsorbents such as activated carbon by hydrophobic interaction. As a result, total sulfur content can be reduced from 7 ppm to less than 0.1 ppm.
However, at the present stage, there are a number of problems that need to be solved to make the extractive photo-oxidation desulfurization process technically and economically feasible. Better solvents need to chosen so as to increase sulfur compounds solubility and aromatic rejection. Recovery of solvent needs special attention. Combination of a solvent and a photosensitizer has to be optimized to increase the rate of the organosulfur compounds photo-transformation. Photosensitizer may be stabilized on the surface of a solid carrier so without losing its ability to accelerate photo-oxidation of the sulfur compounds.88
Table 4 summarizes recent researches conducted for optimization of ODS processes with and without use of any oxidants.
| Process | Sample | Model Oil | Reagent | System | S Conc. Co (ppm) | Optimum Conditions | % S Removal | Reference | |
|---|---|---|---|---|---|---|---|---|---|
| Temp. (°C) | Pressure (atm) | ||||||||
| Oxidation-Adsorption | DBT | n-Octane | H2O2 + Activated C | Batch | 800 | 60 | 1 | 99% | 103 |
| Oxidation-Adsorption | DBT | n-Octane | H2O2 | Batch | 800 | 60 | 1 | 60% | 103 |
| Oxidation-Extraction | DBT | Toluene (40%) Hexane (60%) | H2O2 | Batch | 5000 | 50 | 1 | 92% | 93 |
| Oxidation | BTs | Decalin | H2O2 + Amphiphilic Catalyst | Batch | 500 | 40 | 1 | 98% | 104 |
| Oxidation | Tetrahydrothiophene | cyclohexane | H2SO4 | Batch | 2000 | 22 | 1 | 99% | 5 |
| Oxidation | Thiophene + 3-methylthiophene | n-Heptane | H2O2 + Formic Acid | Batch | 500 | 50 | 1 | 80% | 119 |
| Oxidation | DBT + 4,6-DMDBT | Octane | Na2WO4 + 30% H2O2 + CH3CO2H | Batch | 500 | 70 | 1 | 100% | 120 |
| Oxidation-Adsorption | DBT | Iso-Octane | t-BuOOH + HPWA-SBA-15 | Batch | 174 | 70 | 1 | 97.43% | 105 |
| Oxidation | Alkylated DBTs | Diesel | H2O2 + Mo/γ–Al2O3 | Batch | 320 | 60 | 1 | 97% | 125 |
| Oxidation | DBTs | Hexadecane | H2O2 + V2O5 based catalysts | Batch | 500 | 60 | 1 | 99% | 126 |
| Oxidation | BT + DBT | Petroleum ether | K2FeO4 | Batch | 457 | 35 | 1 | 98% | 108 |
| Radiation Oxidation | — | Crude Oil | — | — | 3000 | 350 | — | 90% | 129 |
| Ultrasound Oxidation | DBT | Toluene | H2O2 | Batch | 3000 | 75 | 1 | 98% | 102 |
| Electrochemical Oxidation | — | Gasoline | CeO2/C based anode | Electrolysis Cell | 310 | 25 | — | 83% | 165 |
5.4. Extraction in ODS
The sulfoxides and sulfones produced after ODS have increased relative polarity, and are preferentially extracted from light oil using a non-miscible solvent.88,92,96,167 The extraction efficiency depends on the solvent's polarity. Polarity, however, is not the only criteria for the selection of suitable solvents. Methanol, for example has sufficient polarity, but its density, 0.79 kg m−3, is about the same as that of typical light diesel oil and is therefore not preferred extraction solvent. Other properties such as boiling point, freezing point, and surface tension need to be considered carefully to evaluate the potential for separation and recovery of the solvent for recycling and reuse.92,96Depending on the solvents used for extraction, the oxidized compounds and solvent are separated from the light oil by gravity separation or centrifugation. Any unused oxidant that remains in the light oil is removed by water washing and extracting. The light oil is water washed to recover any traces of dissolved extraction solvent and unused oxidant. It is then polished using other methods such as absorption using silica gel and aluminum oxide. The solvent is separated from the mixture of solvent and oxidized compounds by a simple distillation for recycling and re-use.
Some common water-soluble polar solvents used are dimethyl sulfoxide (DMSO), dimethylformamide (DMF), acetonitrile, etc.88,99,114,168–169 The former two solvents have a high extractability for sulfones but also have a high boiling point (573 K). This is close to the boiling point of the sulfones, thus creates difficult in separation and reuse for further extraction.
The earliest application for desulfurization by IL was reported in 2001.176 They found that multi-step extraction using AlCl3 ILs reduced the sulfur concentration from 500 mg l−1 down to 235 mg l−1. Phillips210 also used AlCl3 and AlCl4 based ILs for the removal of S-containing compounds. Eber et al.211 proposed design of industrial equipment for deep desulfurization of oil refinery streams by extraction with ILs. Imidazolium-based ILs with PF6− and BF4− anions;192–193,212 and N-alkyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide ILs213–216 have also been used by several investigators. However with these ILs, a change in the content of aromatics was observed and suitability for real industrial fuels was not well established. Imidazolium-based phosphoric ILs were also tested184–186,217–218 which showed encouraging results. Gao et al.179–180 and Domanska et al.219 performed several extraction experiments with alkyl-and alkylmethyl-pyridinium-based ILs with ethanoate and thiocyanate anions as alternative to the perfluorinated and tetrafluoborate ones giving the good extraction performance. Instability and regeneration of the Ils, corrosion problem, negative effects on fuel quality and high total costs of deep fuel desulfurization make the use of Ils in still a challenging task for researchers.172,212 A number of excellent reviews have already been published which critically evaluate desulfurization of liquid fuels by Ils and these are referred for further study.170,209,212,220–223
5.5. Future challenges to ODS
There are two major problems associated with ODS. First, the oxidants chosen do not always perform selectively. Some oxidants cause unwanted side reactions that reduce the quantity and quality of the light oil. The second problem is the selection of a suitable solvent for the extraction of the sulfur compounds. Using the wrong solvent may result in removing desirable aromatic/olefinic compounds from the fuel or extracting less than desired amount of the sulfur compounds from the fuel.93 Reaction selectivity, safety and cost are important concerns for the selection of oxidant, catalyst and operating conditions for ODS processing.The catalytic systems reported are mostly toxic and expensive. There are issues relating to ultimate fuel quality and economy of the process. Thus, there is a need for a new technology that can perform the oxidation reaction under mild conditions, and one that can selectively oxidize the sulfur compounds. However, despite all the disadvantages, greatest advantage of the ODS process is low reaction temperature and pressure. It removes the need for expensive hydrogen that is used in the conventional HDS process. Another feature of ODS is that the refracted sulfur compounds in ODS are easily converted by oxidation.115 ODS avoids the aromatic and olefin saturation, and thus low octane number. Therefore, ODS has great potential to be complementary process to traditional HDS for achieving deep desulfurization. The development and application of oxidation/extraction method are considered among the most desired options because they can lower the sulfur without negative impact on capital investment.
6. Adsorptive desulfurization
6.1. Introduction
Adsorption has been applied variously for removal of sulfur compounds from liquid hydrocarbon fuels. Removal of DBT and other sulfur compounds has been studied over zeolites, aluminosilicates, activated carbon (AC), alumina, zinc oxide, etc.224–239 However, only few adsorbents have shown high selectivity for difficult to hydrotreat sulfur compounds such as 4,6-dimethylDBT. In the following sections, recent studies which utilized various adsorbants have been discussed and research gaps have been identified.6.2. Adsorbents
Sano et al.226 proposed an integrated process for deep desulfurization where conventionally hydrodesulfurized straight run gas oil (HDS-SRGO) was desulfurized to less than 10 ppm S over an ACF. The adsorption bed used in the desulfurization of HDS-SRGO was used again to denitrogenate and desulfurised SRGO as a pre-treatment step of conventional HDS process. As shown in Fig. 3, twice use of ACF in sequence as an adsorbent for HDS-SRGO and SRGO reduced the frequency of regeneration and oil loss in the adsorption bed. Fully saturated adsorption bed was regenerated by conventional solvent, such as toluene. ACF showed low pressure drop and high performance among the AC materials examined.
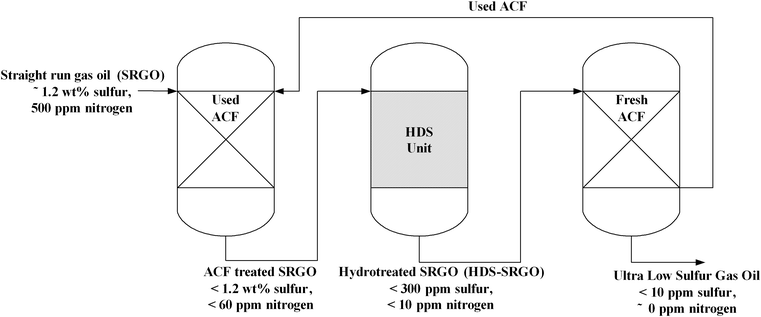 | ||
| Fig. 3 Schematic diagram of consecutive adsorption process.226 | ||
Modifications of carbon surfaces by incorporation of metals and oxidation of carbon surface can have a positive effect on the adsorption of DBTs.241–264 This happens due to π-electron interactions of sulfur-containing aromatic compounds with metals on the activated carbon257 and due to oxidation of carbon surface.246,248,256
Incorporation of metals such as copper and iron increases the capacity and selectivity for adsorption and the catalytic transformations on the surface.249,256 The selectivity increase is due to acid–base interactions of slightly basic thiophenes with acidic oxygen containing groups246,248,254 or due to increased polarity and redox reactions.249,258 Ania and coworkers,248–249 Zhou and co-workers250 and Yang and co-workers253 have reported on the positive role of acidic groups for desulfurization.
It has been reported that both microporosity and the surface chemistry are important for efficient desulfurization.254 The increase in the adsorption capacity has been linked to the development of pores with diameters less than 10 Å as a result of various modifications of carbon materials.248,259–261 Pores similar in size to DBT or DMDBT molecules, are important at low surface coverage where dispersive forces are the strongest.249,254,258–262 The amount adsorbed is also governed by the volume in pores smaller than 10 Å. The small pores, which are too small to accommodate functional groups, govern the physical nonspecific adsorption process that is based on dispersive interactions.
With the progress of adsorption, oxygen containing functional groups248,254,262–263 and/or the introduced catalytic inorganic phase258 start to affect/enhance the amount adsorbed via specific interactions. Oxidation is an important factor since on the surface of unmodified activated carbons other aromatic components of liquid fuels are also adsorbed in significant quantities. Oxidation of carbon surface leads to the formation of oxidized species of DBT and 4,6-DMDBT, which are strongly adsorbed via interactions with polar groups of the carbon surface. Thus, oxidation increases the selectivity of DBT adsorption via an increase in the number of specific adsorption centers, whereas the adsorption of aromatic hydrocarbons decreases with an increase in the extent of oxidation.246,248,254,258,262–263
Activated alumina has good adsorptive properties and has been used for the removal of organic compound from aqueous solutions. Unlike silica gel, which is amorphous, activated alumina is crystalline. Oxygen vacancies (defects) are easily formed on its surfaces, thus alumina has both Lewis and Brønsted acid sites.265 The surface chemistry, as well as the pore structure of activated alumina, can be modified, for example, by treatment with acid (HCl or HF) or alkaline (to alter the acidity) and controlled thermal treatment (to tailor the pore structure). As a result, activated alumina is more versatile than silica gel and has been applied more often as a sorbent.266 Larrubia et al.227 studied the adsorption of BT, DBT and 4,6-DMDBT on alumina, zirconia and magnesia. It was found that adsorption on alumina was the strongest, forever, 4,6-DMDBT adsorption was limited due to steric effect. Etemadi and Yen228 studied the surface properties of two different phases of alumina (amorphous acidic alumina and nanopowder alumina) through SEM images. The effect of calcining on alumina particles was also investigated through SEM images. Adsorption capacity of amorphous acidic alumina was found to be 1.6 times lower than the nanopowder sample, though the surface area was 2.3 to 4.6 times smaller. Crystallinity of the adsorbent decreased its adsorption capacity. Higher disorder provided more topological traps, irregularities, and hidden grooves for higher adsorption capacity. Therefore, the amorphous phase provided more sites for adsorption. Srivastav and Srivastava267 recently reported the usage of commercial grade activated alumina (aluminum oxide) as adsorbent for the removal of DBT dissolved in n-hexane. Optimum adsorbent dose was found to be 20 g l−1. The adsorption of DBT on alumina was found to be gradual process, and quasi-equilibrium reached in 24 h. Baeza et al.239 used copper supported on zirconia to separate low TH concentration from a mixture of 2000 ppmw of TH in n-octane at room temperature and atmospheric pressure. The results showed that the capacity of copper supported zirconia to adsorb TH increased as the copper content increased, reaching a maximum at a concentration of 3% of copper. Kumar et al.268 also studied various laboratory prepared zirconia based adsorbents for removal of DBT from iso-octane.
Zeolites are crystalline aluminosilicates of alkali or alkali earth elements, such as sodium, potassium, and calcium. The primary structural units of zeolites are the tetrahedra of silicon and aluminum, SiO4 and A1O4. These units are assembled into secondary polyhedral building units such as cubes, hexagonal prisms, octahedra, and truncated octahedra. The silicon and aluminum atoms, located at the corners of the polyhedra, are joined by shared oxygen. The final zeolite structure consists of assemblages of the secondary units in a regular three-dimensional crystalline framework.266 Weitkamp et al.229 found that TH is adsorbed more selectively than benzene when passed (in vapor phase) over a fixed-bed adsorber packed with ZSM-5 zeolite as a sorbent. Yang et al.230 and Takahishi et al.231 compared the vapor phase adsorption isotherm for the different sorbents, it was found that CuY and AgY zeolites had the best TH adsorption capacity. The adsorption capacities followed the order Cu–Y and AgY ≫ NanZSM–S > AC > Na–Y > modified alumina. Hernandez and Yang232 used the combination of CuY zeolite with AC as sorbent. This sorbent's total sulfur uptake (saturation loading) was 18.9 mg S g−1 sorbent for commercial diesel fuel (430 ppmw S) fed to the column at room temperature. The breakthrough loading was calculated to be 10.9 mg S g−1 sorbent.
Several works have developed new adsorbents to remove the thiophenic compounds from commercial fuels via π-complexation. Gongshin233 prepared sorbents using ion-exchange techniques to introduce d-block metals like Ag+, Cu+, Ni2+ and Zn2+ into zeolites. These ion exchanged materials were capable of producing fuels with a total sulfur concentration of less than 1 ppm. Ngamcharussrivichai et al.234 synthesized zeolites using coal fly ash. The zeolites were used for adsorption of TH and BTH in n-hexane solution. It was shown that the introduction of different heteroatoms into the framework of zeolites leads to different catalytic and adsorption properties. Tang et al.236 introduced gallium atoms into the framework of Y zeolite by treating the zeolite with an aqueous solution of ammonium hexafluoro gallate. Desulfurization of various model fuels containing about 500 μg sulfur/g were studied over the synthesized Y zeolite ([Ga]AlY) with a liquid hourly space velocity of 7.2 h−1 at ambient conditions. At ambient conditions, the breakthrough capacity for the adsorption of TH, THT and 4,6-DMDBT were found to be 7.0, 17.4 and 14.5 mg of sulfur/g of adsorbent, respectively. Ma et al.269 explored various transition metal-based adsorbents to find an adsorbent in which the metal interacts with the sulfur atom and not the CC double bond of the thiophenic compounds. The proprietary metal compound was supported on porous silica gel with 5% loading. The adsorbent was tested for removal of DBT and 4,6-DMDBT from gasoline, diesel and jet fuel. It was found that the adsorbent had significant selectivity toward the sulfur compounds over naphthalene and 2-methylnaphthalene. Wang et al.270 achieved desulfurization of a JP-5 light fraction (841 ppmw S) for fuel cell applications by Pi-complexation adsorption with CuCl and PdCl2 supported on the MCM-41 and SBA-15 mesoporous materials. PdCl2/SBA-15 exhibited the highest sulfur selectivity and capacity for the desulfurization of jet fuel among all known sorbents. The total sulfur content of JP-5 light fraction was reduced from 841 ppmw S to below 50.0 ppmw S. The spent PdCl2/SBA-15, regenerated by purging with benzene at 70 °C, could recover about 44% and 48% of the adsorption capacity at breakthrough and saturation, respectively, compared with the fresh adsorbent. Lu271 developed and used intermetallic crystalline powder made of tin and antimony for the destructive adsorption of sulfur compounds from heavy oil. This bimetallic powder had a highly porous structure with discrete crystals. This adsorbent was prepared by melting an equi-atomic mixture of tin and antimony in a graphite crucible. The melt was held at 500 °C with a hydrogen gas cover to avoid oxidation. It was then pored into an atomization nozzle that was operated at high nitrogen pressure. The adiabatic expansion of gas rapidly cools the droplets and freezes into a uniform size of about 10–15 μm. A specific organic chemical was added during the sputtering process at 375 °C to produce the proper size of cavities and pits on the surface of powders. Lu271 showed in his studies that intermetallic powder had the ability to lower the contents of sulfur both in organic and inorganic form.
Metal–organic frameworks (MOF) consist of metal cations linked by polyfunctional organic linkers. Organic linkers act as bridging ligands between the metal ions to form highly ordered porous three-dimensional networks with large pore volumes and high inner surface areas.272–275 MOF have been used to adsorb selectively organo-sulfur compounds.273–276 Gaseous sulfur compounds have also been removed using MOFs.277 A few important factors, such as open metal sites compounds and pore functionality, have been suggested for the efficient removal of S-compounds.273–276
Cychosz et al.273 also studied different MOF materials and their adsorption characteristics for organosulfur compounds in model oil. Blanco-Brieva et al.275 demonstrated that the adsorption of DBT at ambient temperature (304 K) is much higher on MOF systems than on the benchmarked Y-type zeolite and activated carbons. It was also found that the extent of adsorption on the Cu–(C300) and Al-containing (A100) MOF systems than on the Fe-containing (F300) MOF. The higher adsorption capacity of DBT observed on C300 was due to the stronger interaction of S–atom of DBT with surface Cu2+ ions. Achmann et al.276 observed that a special copper-containing MOF (copper benzene-1,3,5-tricarboxylate, Cu–BTC-MOF) was able to remove 78 wt% and 86 wt% of the sulfur content from thiophene containing model oils (initial sulfur content = 30 mg kg−1) and tetrahydrothiophene (THT)-based model oils (initial sulfur content = 9 mg kg−1). The sulfur content of low-sulfur gasoline was reduced by more than 22% to 6.5 mg kg−1. The sulfur level in diesel fuel was reduced by an extent of 13 wt%. It was also demonstrated that sulfur adsorption occurred in the first hour after addition of the sorption-material. Khan et al.278 studied the adsorption kinetics and thermodynamics in the adsorption of BT over MOFs. The study has focused on the adsorption over isotypic MOFs12 such as MIL-53(Cr, Al)279–280 and MIL-47(V)281 to understand the effect of central metal ions on the adsorption of S-compounds. MIL-47 showed the highest BT adsorption capacity among the isotypic Me–BDCs because of high acidity. The driving force of BT adsorption over the adsorbents was due to entropy effect. It was concluded that for efficient S–compounds removal, a MOF-type material should have a specific adsorption site like an acidic site. Shi et al.282 functionalized MOF-5 by decomposing Mo(CO)6 onto its surface and evaluated it for the selective adsorption of dibenzothiophene from solutions containing i-octane, naphthalene and/or benzene. The resulting materials had Mo loadings up to 20 wt%, surface areas in excess of 1800 m2 g−1. Sulfur uptakes at breakthrough approached 0.5 mmol S g−1. The presence of relatively low concentrations of aromatics like naphthalene and benzene did not interfere with the adsorption of DBT, however, at the high aromatics concentrations the sulfur adsorption capacities decreased. The results indicate that this new type of sorbent could be used for removing organosulfur compounds typically left in gasoline, diesel and JP-8.282
Table 5 summarizes some of the researches conducted for the sulfur removal with various types of adsorbents at optimized conditions.269–274
| Process | Sample | Model Oil | Adsorbent | System | S Conc. Co (ppm) | Optimum Conditions | % S Removal | Reference | |
|---|---|---|---|---|---|---|---|---|---|
| Temp. (°C) | Pressure (atm) | ||||||||
| Adsorption | DBT | — | Activated Carbon | Batch | 178 | 25 | 1 | 95% | 225 |
| 2 step Adsorption | Thiophenes | Gas Oil | Activated Carbon | Packed Bed | 300 | 70 | 1.5 | 88% | 226 |
| Adsorption | DBT sulfone | — | Alumina | Packed Column | 700 | 200 | — | 30% | 227 |
| Adsorption | Thiophenes | Diesel | CuCl/γ–Al2O3 | Fixed Bed | 140 | 25 | 1 | 99% | 233 |
| Adsorption | Thiophene + BT | n-Hexane | Zeolites from coal fly ash | Batch | 500 | 30 | 1 | 63% | 234 |
| Adsorption | 4,6-DMDBT | cyclohexane | NiMoP/Al2O3 catalyst + NaY zeolites | Fixed Batch | 450 | 340 | 40 | 56% | 235 |
| Adsroption | 4,6-DMDBT | n-Nonane | Gallium + Y zeolite | Fixed bed flow reactor | 500 | 60 | — | 97% | 236 |
| Selective Adsorption | — | Commercial diesel | Ni nanoparticles supported on silica | Fixed bed | 11.7 | 200 | — | 99% | 237 |
| Reactive Adsorption | Thiophene | Ni/SiO2 and Ni/ZnO | Fixed Bed | 300 | 0.02 | 238 | |||
| Adsorption | Thiophene | n-Octane | Cu–zirconia | Fixed bed | 2000 | 180 | — | 99% | 239 |
| Pi-complexation adsorption | — | JP-5 light fraction | CuCl and PdCl2 supported on the MCM-41 and SBA-15 | Fixed bed | 841 | 350 | — | 94% | 270 |
| Adsorption | DBT | Crude Oil | Intermetallic Powder | Packed Bed | 250 | 25 | 1 | 55% | 271 |
| Adsorption | DBT | Toluene (45%) Hexane (55%) | Ruthenium Complexes | Batch | 40 | 25 | 1 | 50% | 283 |
| Adsorption | 4,6Me2DBT | Toluene (45%) Hexane (55%) | Ruthenium Complexes | Batch | 40 | 25 | 1 | 40% | 283 |
| Bond cleavage | Thiophenes | n-Hexane | (C5Me5)Rh(PMe3)(Ph)H | Batch | — | 64 | 1 | — | 284 |
| Alkylation | 3-methylthiophene | n-Heptane | Silica-supported 12-phosphotungstic zeolite | Slurry tank reactor | 340 | 85 | 1 | 60% | 285 |
6.3. Reactive adsorption
Reactive adsorption desulfurization (RADS) combines the advantages of both the catalytic HDS and adsorption desulfurization.286–291 In the RADS, sulfur-containing molecules react with a solid adsorbent in the presence of hydrogen.286 Conoco Phillips Petroleum Co. developed a new S–Zorb process for the production of low-sulfur gasoline by reactive adsorption of sulfur compounds over a solid sorbent at elevated temperatures under a low hydrogen pressure.243,292 Transition metals supported on base oxides are usually used as the adsorbents for the RADS. Ni/ZnO is an ideal adsorbent, in which the ZnO acts not only as an acceptor of sulfur released during the regeneration of sulfided Ni species, but also acts as a co-catalyst for the hydrogenation of sulfur-containing compounds over the surface of Ni species.88,238,243,286–296Tawara et al.288 used Ni/ZnO for bringing down sulfur in kerosene to <0.1 μg g−1 in the temperature range 270–300 °C under H2 atmosphere at a pressure of 0.60 Mpa. The sulfur-containing compounds were first decomposed through the catalytic hydrogenation. The H2S–like species formed were then adsorbed in the adsorbent. It was supposed that ZnO acts as an acceptor for sulfur produced from sulfur-containing molecules on Ni particles which were suggested to be “continuously regenerated” during reaction.286
During the reaction, the active phase of the adsorbent consisting of metallic particles supported on ZnO, is transformed into a mixture of sulfides. After complete transformation (saturation), the adsorbent is regenerated in a two-stage process: sulfides are firstly calcined to obtain oxides which are than treated in H2 in order to reduce the supported metal. RADS with Ni/ZnO requires less hydrogen as compared to HDS and also due to absence of side hydrogenation reactions, decrease in octane number is not observed.297
Many efforts have been made to reveal the mechanism of RADS. Babich and Moulijn proposed a reaction scheme in which thiophene is decomposed on nickel surface that was then sulfidized followed by hydrogenation of NiS site and transfer of H2S to ZnO.88 Bezverkhyy et al.238 studied the kinetics of thiophene RADS on Ni/SiO2 and Ni/ZnO by thermal gravimetric analysis at 280–360 °C under 5–40 mbar of thiophene in H2. In the case of Ni/SiO2, the interaction proceeded in two steps: a rapid surface reaction followed by a slower bulk transformation into Ni3S2. Maximum Ni conversion depended on reaction conditions and observed conversion profiles were described by an exponential equation corresponding to a reaction of first order relatively to both sulfidable Ni amount and thiophene. In case of the interaction between Ni/ZnO and thiophene, a rapid increase of weight, similar to the first stage observed on Ni/SiO2, was not followed by bulk Ni sulfidation, but instead by a nucleation-controlled ZnO surface transformation. After formation of the surface ZnS layer, a complete particles sulfidation had with kinetics being strongly dependent on the reaction conditions. Zhang et al.298 also reported that the decomposition of thiophene on Ni/ZnO may occur, while S is trapped by ZnO and converted into ZnS in the reactive adsorption process. ZnO with favorable textural structures has good activity of taking up the S, which may be the main reason that the Ni/ZnO adsorbents with ZnO calcined at different temperatures display different desulfurization activities.
Huang et al.287 carried out sulfur K-edge X-ray absorption near-edge structure (XANES) to investigate the transfer of sulfur species during the RADS process with Ni/ZnO under nitrogen and hydrogen. The results indicated that the desulfurization processes under nitrogen and hydrogen are different in the reaction mechanism (Fig. 4). In nitrogen, the desulfurization over Ni/ZnO is achieved through physical and chemical adsorption; a severe decrease in the desulfurization activity of Ni/ZnO is observed with the time on stream and the desulfurization capacity is very low. Hydrogen facilitates the decomposition of DBT on active Ni species, the formation of Ni3S2, and thereafter the transfer of sulfur to ZnO. Metallic Ni as the active nickel species is preserved until most of ZnO is converted to ZnS. On the basis of these observations, a possible sulfur transfer mechanism for the RADS is then proposed. Thus in hydrogen, the desulfurization turns to be a reactive adsorption process and Ni/ZnO exhibits a high desulfurization activity and capacity.287
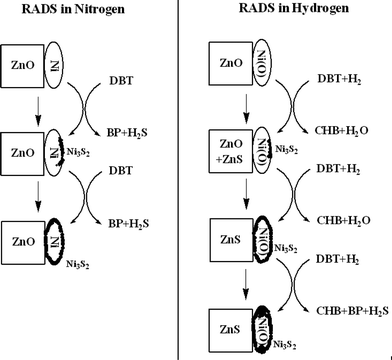
6.4. Future challenges for desulfurization by adsorption
One of the advantages of desulfurization of fuels by adsorption over HDS is operation at low temperature. Another advantage is that while H2 is abundant in refineries for use in the HDS process, it is a precious reactant for generating power for fuel cell applications. In addition, recycling the H2 exiting the reformer back to the pre-reformer HDS unit adds complexity to the fuel processing system. On another note, HDS cannot be easily applied to internal reforming fuel cell systems, such as molten carbonate and solid oxide fuel cells, since there is no H2 rich stream to feed to the HDS reactor. Furthermore, the sulfur content of the fuel can theoretically be reduced to a very low level due to removal of the refractory sulfur compounds that cannot be removed easily by HDS.Desulfurization by adsorption faces two major challenges. The first is to develop easy remunerable adsorbents with a high adsorption capacity for sulfur compounds. The second challenge is to find adsorbents that selectively adsorb the sulfur compounds, which are mainly aromatic sulfur compounds that have not been removed in the HDS process in refinery, over the other aromatic and olefinic compounds present in tests hydrocarbon fuel. While the adsorption process is highly effective, the adsorbents are difficult to regenerate, often requiring calcinations or solvent washing.232 The adsorption capacity of many adsorbents has been on low. Economical clay materials displayed capacities of 1–4 mg S compound per g clay, thus requiring huge amounts of adsorbent. Pressure swing adsorption is not effective due to the strong interaction of sulfur with the adsorbent. Large adsorbent beds are, therefore, required to minimize the number of turnovers, and multiple beds are needed to keep a refinery on-stream. Repeated calcinations can also lead to a loss of surface area due to sintering; reducing the amount of sulfur a bed can remove. Most research efforts, therefore, focus on creating less expensive and higher surface area materials. In developing the selective adsorbent, the key consideration is to design the adsorbent materials which selectively interact with sulfur in the presence of large excess of aromatic compounds, which exist in concentrations of >20% in comparison with less than 1 wt% sulfur compounds.
Investigations into the structure–activity relations of adsorbents as well as the active sites in these systems are needed. It may also be informative to establish the relationship between desulfurization activity and the chemisorption of model hydrocarbon compounds which are commonly found in fossil-based fuels. The mechanisms and kinetics of desulfurization reactions over various adsorbents also deserve research attention.
7. Biodesulfurization
7.1. Introduction
Biodesulfurization (BDS) has drawn wide attention recently because of its green processing of fossil fuels for removal of sulphur compounds. During initial research, many bacterial species that had the ability to consume DBTs as their energy source were isolated from their natural habitats. However, these isolated microbial species could not specifically remove sulfur from DBTs. Some of the isolated microorganisms used thiophenic compounds as carbon and sulfur sources. Some other used metabolized DBTs as carbon source and, in a series of oxidizing steps, converted them into several water-soluble compounds. The accumulation of these water-soluble end products significantly inhibited microbial growth and DBT oxidation.288 Several bacterial species which are capable of either bio-transforming DBT or growing on it as a sole sulfur source have now been identified. These include Arthrobacter, Brevibacterium, Pseudomonas, Gordona, and Rhodococcus spp.7.2. Mechanism of BDS
Most of the work over the last decade has focused on the metabolism of sulfur heterocycles like DBT by rhodococcus strains, pseudomonas bacteria and other related species.168,300Rhodococcus seems particularly well suited for hydrocarbon metabolism. Sulfur-metabolizing bacteria have been shown to reduce the sulfur content of diesel fuel from 535 ppm to 75 ppm in 24 h. Several species of bacteria metabolize organosulfur compounds in one of three ways: reductive C–S bond cleavage, oxidative C–S bond cleavage, and oxidative C–C bond cleavage.Generally, there are two primary pathways for BDS of alkyl-DBTs. One pathway is that in which the initial attack is directed against one of the carbon atoms (the Kodama pathway) (Fig. 5) and one in which initial catalysis is directed against the sulfur center (the 4S pathway) (Fig. 6).
 | ||
| Fig. 5 Kodama enzymatic pathway on dibenzothiophene.299 | ||
 | ||
| Fig. 6 The ‘4s’ pathway for the biological desulfurization of DBT and its derivatives.102,307 | ||
In the destructive BDS (oxidative C–C bond cleavage) mechanism, the initial attack is directed against one of the carbon atoms, preferentialy one of the DBT phenyl rings. This results in the breakage of one bond or a fragment from the phenyl ring. The oxidative and carbon-destructive series of enzymatic actions that attack carbon atoms in DBT phenyl ring is known as Kodama pathway.301 This pathway for DBT is shown in Fig. 5. It consists of three main steps including hydroxylation, ring cleavage, and hydrolysis.302 The oxidation in most cases produces 3-hydroxy-2-formyl-benzothiophene (HFBT) and pyruvic acid (PA) are produced. The steps after production of HFBT are not fully known. HFBT is chemically unstable and it is probably mineralized in nature. In cases where methyl-DBT was subjected to Kodama desulfurization, it was found that the carbon–carbon cleavage occurred on the benzene ring that had no substituting group to form corresponding methyl-HFBT.299 Several different genera, majority of which are pseudomonas cultures have been reported to carry out desulfurization through DBT carbon-destructive pathway.303–307
Enzymatic attack at a carbon atom, typical of many aromatic hydrocarbon degradative pathways, is undesirable for a process designed to selectively remove organic sulfur compounds without oxidation of other aromatics found in petroleum products. Due to the undesired breakage of carbon-carbon bonds in benzene rings, this type of desulfurization is considered to be a destructive process.
Several anaerobic strains have demonstrated the ability to remove organic sulfur from petroleum feedstocks by reductive C–S bond cleavage (anaerobic BDS). For example, desulfovibrio desulfuricans metabolizes DBT, releasing sulfur as H2S in the presence of a reducing agent.299
Kim et al.308 investigated the specific desulfurization by Desulfovibrio desulfuricans M6. This anaerobic strain could degrade 96% and 42% of BT and DBT, respectively. Metabolite analyses proved that this strain could convert DBT to biphenyl and H2S.
Some anaerobic microorganisms, such as Desulfomicrobium scambium and Desulfovibrio longreachii, have been reported to have the ability to desulfurize only about 10% of DBT dissolved in kerosene. Under anaerobic conditions, oxidation of hydrocarbons to undesired compounds such as colored and gum forming products is minimal.301 This advantage can be counted as an incentive to continue research on reductive BDS. However, maintaining an anaerobic process is extremely difficult and the specific activity of most of the isolated strains have been reported to be insignificant for DBTs.309
The specific oxidative BDS (oxidative C–S bond cleavage) pathway is often referred to as the ‘4s pathway’ because of the four sulfur-containing intermediates (sulfoxide–sulfone–sulfinate–sulfate). It involves consecutive biocatalytic oxidation of DBT sulfur to sulfoxide (DBTO), sulfone (DBTO2), sulfinate (HPBS) and hydroxybiphenyl (HBP). An example is shown in Fig. 6 for converting DBT to sulfoxides, sulfones and finally to hydroxyl biphenyl.226,310–315
This mechanism involves four enzymes that converts DBT-like molecule into a phenolic product and SO32−. Flavin-dependent DBT mono-oxygenase (DszC or DBT-MO (a tetramer encoded by the desulfurizationc gene)) and DBT-sulfone mono-oxygenase (DszA or DBTO2-MO (a dimer encoded by the desulfurizationa gene)) are first two of these enzymes that require a third enzyme (the flavin reductase DszD) for their activity. The fourth enzyme, HPBS desulfinase (DszB), completes the reaction sequence.307 There are a number of potentially rate limiting steps including enzyme kinetics involved in the desulfurization of CX-DBTs, and other molecules dissolved in diesel. The first, and sometimes rate-limiting, step in CX-DBT metabolism is the transfer of the polyaromatic sulfur heterocycles from the oil phase into the cell. In rhodococcus, the desulfurization enzymes are soluble and presumably found in the cytoplasm.307 Once the molecules reach the cell, the CX-DBT molecules are subjected to a series of oxidations. The first two are catalyzed by the same enzyme, DBT-MO. This enzyme requires flavin mononucleotide for activity, which is provided by flavin mononucleotide oxidoreductase. The oxidation of dibenzothiophene sulfone (DBTO2) to hydroxyl-phenyl benzene sulfonate is catalyzed by the next monooxygenase (DBTO2–MO). This reaction also requires reduced flavin mononucleotide (FMNH2) and molecular oxygen. Hydroxy-phenyl benzene sulfonate is readily soluble in water. In the final step, the inorganic sulfur is released by a HPBS desulfinase. This reaction yields a hydrophobic aromatic molecule, hydroxy biphenyl and sulfite. The hydroxy biphenyl is very soluble in oil, and finds its way back to the petroleum fraction, thus, conserving the fuel value of the oil. It is also unclear how Cx-hydroxy biphenyl or Cx-hydroxyphenyl benzene sulfonate exit the cells. Hydroxy biphenyl (also known as orthophenyl phenol or ‘dowicide number 1’) is a potent industrial biocide.307 Experimental results suggest that mass transfer is not limited by intermediate oil-to-water, water-to-cell steps.
The throughput of substrates in this pathway is hindered at several steps, including substrate acquisition, the supply of reducing equivalents and enzyme turnover rates for specific substrates.316 These slow the overall rate at which whole bacterial cells can remove sulfur. In general, DBT and C1-DBT are attacked preferentially followed by the more highly alkylated molecules. The position of the methyl group also influences the reaction rate. Alkylation near the sulfur leads to slower rate of desulfurization. Although, sphingomonas strain shows the opposite trend.307
The genes responsible for the “4S” metabolic pathway (oxidative C–S cleavage) have been cloned, sequenced and engineered from a variety of microorganisms, and have been transferred to several bacterial species after its initial discovery in Rhodococcus erythropolis strain IGTS8. In the past few years several new flavin reductases, including thermo-tolerant enzymes, have been discovered. In addition, the least well understood enzyme in the pathway, HPBS desulfinase, is becoming better characterized.316
7.3. Recent studies in BDS
BDS using microorganisms and/or enzymes has been studied since last few decades. Soleimani et al.299 recently published a review paper on BDS of refractory organic sulfur compounds in fossil fuels. This section presents the results of very recent studies on BDS.Conventional refining processes have been performed at much higher temperature; therefore thermophilic biodesulfurizaiton is desirable and could be easily integrated into the refining process without cooling the stock to 30 °C.317 Moreover, thermophilic biodesulfurization also reduces the viscosity of crude oil, which makes the development of crude oil biodesulfurization more practicable.318 Li et al.319 investigated the desulfurization for DBT in tetradecane by a facultative thermophilic bacterium Mycobacterium goodii X7B. The total sulfur level of DBT in tetradecane was reduced by 99%, from 200 to 2 ppm within 24 h at 40 °C. After 72 h treatment, 59% of the total sulfur content (from 3600 to 1478 ppm) in Liaoning crude oil was removed.
Mohebali et al.320 investigated the ability of a newly isolated bacterium, Gordonia lkanivorans RIPI90A (GenBank accession number DQ321498) to desulfurize both DBT and DBT-containing hexadecane during both the growth and resting stages. The highest specific activity, in terms of DBT-utilization occurred in cells harvested from the late exponential growth phase. TH or BTH was completely degraded by a DBT desulfurizing bacterium Mycobacterium sp. ZD-19 within 10 h or 42 h, and 100% DBT or 4,6-DMDBT was removed within 50 h or 56 h, respectively.321 Diphenylsulfide (DPS) possessed the lowest desulfurization efficiencies with 60% being transformed within 50 h and 80% at 90 h. The desulfurization activities of five substrates were found to be in order of TH > BTH > DPS > DBT > 4,6-DMDBT. The desulfurization rate of DBT or 4,6-DMDBT in mixture was found to be lower than they are desulfurized separately, indicating that the substrate competitive inhibition existent when DBT and 4,6-DMDBT are mixed.
Caro et al.322 reported the BDS of DBT in oil–water emulsions by aerobic Rhodococcus erythropolis IGTS8 strain. Addition of β-cyclodextrins increased the diffusion of DBT into the aqueous phase and avoided the accumulation of HBP, thus improving the BDS yield. Higher biocatalyst cell concentrations decreased the HBP production rates indicating combination of both inhibition effects and mass transfer limitations. Alves et al.323 tested enzymatic hydrolyzates of recycled paper sludge as suitable feedstock for BDS by Gordonia alkanivorans strain 1B. Only the hydrolyzate obtained after enzymatic mixture dialysis (dialyzed hydrolyzate) allowed DBT desulfurization, in spite of faster bacterial growth occurring on non-dialyzed hydrolyzate. For dialyzed hydrolyzate, 250 M DBT was consumed after 96 h displaying a maximum specific productivity of 2-hydroxybiphenyl of 1.1 mol g−1(dry cell weight) h. Complete consumption of DBT was observed upon the addition of only phosphates and ammonia. Addition of zinc further increased the 2-hydroxybiphenyl production by 14%. Madeira et al.324 performed a stepwise evaluation of the enzymatic oxidation of DBT by horseradish peroxidase (HRP). Reactions were carried out in monophasic organic media containing 25% (v/v) acetonitrile. Best results were observed in a reaction medium at pH 8.0 presenting HRP 0.06 IU/ml, DBT 0.267 mM, DBT:H2O2 molar ratio of 1:20 (stepwise hydrogen peroxide addition) and incubated at 45 °C for 60 min. Under these conditions 60% of DBT was converted into DBT sulfoxide (12%) and DBT sulfone (46%).
A summary of various researches conducted for the sulfur removal using BDS at optimized conditions is given in Table 6.319–335
| Process | Sample | Model Oil | Microorganism | System | S Conc. Co (ppm) | Optimum Conditions | % S Removal | Reference | |
|---|---|---|---|---|---|---|---|---|---|
| Temp. (°C) | Pressure (atm) | ||||||||
| 4S | DBT | hexadecane | Gordonia alkanivorans RIPI90A | Batch | 100[320] | 30 | — | 90% | |
| 4S | DBT + 4,6-DMDBT | — | Mycobacterium sp. ZD-19 | Batch | 92 | 30 | — | 100% | 321 |
| 4S | DBT | Tetradecane | Mycobacterium goodii X7B | Fed Batch | 200 | 40 | — | 99% | 319 |
| 4S | DBT | Hexadecane | Rhodococcus erythropolis IGTS8 | Batch | 100 | 30 | — | 80% | 322 |
| 4S | DBTs | n-heptane | Gordonia alkanivorans strain 1B | Batch | 100 | 35 | — | 63% | 323 |
| Biocatalytic oxidation | Organosulfides and thiophenes, | Straight-run diesel fuel | Caldariomyces fumago | Batch | 1600 | 25 | 1 | 99% | 325 |
| 4S | DBTs | n-tridecane | Bacillus subtilis WU-S2B | Batch | 100 | 50 | — | 50% | 326 |
| BDS | DBTs | n-tridecane | Mycobacterium phlei WU-F1 | Batch | 150 | 50 | — | 99% | 327 |
| BDS | DBT | n-Hexadecane | Rhodococcus sp. strain P32C1 | Batch | 1000 | 30 | — | 75% | 328 |
| BDS | DBT | Decane | Rhodococcus erythropolis ATCC 53968 | Interface Bioreactor | 184 | 30 | — | 90% | 329 |
| 4S | DBT | Hydrodesulfurized diesel | Mycobacterium sp. X7B | Batch | 535 | 45 | — | 86% | 330 |
| BDS | DBT | Ethanol | Microbacterium strain ZD-M2 | Batch | 36 | 30 | — | 94% | 331 |
| BDS | DBT | n-Hexadecane | Pseudomonas stutzeri UP-1 | Batch | 500 | 31 | — | 74% | 332 |
| BDS | DBT + 4,6DMDBT | Light gas oil | Sphingomonas subarctica T7b | Fermentor | 280 | 27 | — | 94% | 333 |
| 4S | DBT | n-Hexadecane | Bacterium, strain RIPI-22 | Batch | 100 | 30 | — | 77% | 334 |
| BDS | — | Hydrodesulfurized diesel oil | Pseudomonas delafieldii R-8 | Fermenter | 591 | 30 | — | 47% | 335 |
7.4. Future challenges of BDS
Lower capital and operating costs, ability to produce substantially less greenhouse gases and high valuable byproducts are the potential benefits of BDS. Moreover, BDS takes advantage of the specificity of enzymes, especially for DBT and substituted DBTs. Therefore, BDS offers an alternative way to obtain ‘zero sulfur’ products and has more promising prospects.Development of biocatalytic desulfurization for the selective removal of polyaromatic sulfur heterocycles from petroleum products has focused on the 4S pathway which can remove sulfur from substituted and un-substituted DBTs by attacking the sulfur site and converting it to sulfone. Biological removal of sulfur has several limitations that prevent it from being applied today. The metabolism of sulfur compounds is typically slow compared to chemical reactions.
Although, there is a lot of research going on in this area, the application of BDS approach is limited by the slow biodegradation process.302 The rate of metabolism is rate limiting in the process, though mass transfer resistance from the oil/water interface to the microbe is also slow compared to the rate of transfer of the sulfur compound to the oil–water interface. Large amounts of biomass are needed (typically 2.5 g biomass per g sulfur), and biological systems must be kept alive to function under variable input conditions found in refineries. This is difficult since the rate of desulfurization strongly depends on pH, temperature, and dissolved oxygen concentration. Separation of the cells from the oil can also be difficult, and immobilized cells often have lower activity and limited lifetimes. Despite considerable progress in BDS, there are still some difficulties in commercial application in the industry. Critical aspects of the process include cost of biocatalyst, reactor design and oil–water separation. Mass production of biocatalyst with high desulfurization activity is an important technique to decrease the cost of biocatalysts, which is carried out by high cell density cultivation.335
Despite considerable progress in understanding of BDS, there are several bottlenecks limiting commercialization of this process, such as biocatalyst's desulfurization rate and ability of organic sulfur compounds.336 There have been few reports on the microbial treatment mechanism of different sulfur compounds when they are coexisting. Additionally, little is known about which compound would be degraded preferentially or whether the desulfurization rate is different between single and their mixture. An understanding of the metabolic mechanism is still expected and this will be the focus of the majority of research.337
8. Conclusions
Most researchers in the field of desulfurization have focused their research into three categories: ODS, metabolism of sulfur compounds using microbes, and selective adsorption. Despite these research areas, the primary method for sulfur removal is still HDS, which is a conventional, heterogeneous hydrogenation reaction. Studies regarding reaction mechanisms and catalytic centers involved in the hydrogenation of olefins and HDS of thiophene and related compounds are still important. This issue has gained a renewal of interest in recent years with the need of deep desulfurization coupled to the necessity of saving a maximum of octane rating in the fractions composing commercial gasoline.The extensive use of catalytic hydrogenation for sulfur removal suggests that better techniques for desulfurization may be found in a typical hydrogenation reactor. Before they can be applied to desulfurization, however, the operation of different types of hydrogenation reactors must be understood. The oxidation-extraction technique can be used as an additional process to the HDS to enable the refiners to meet the future environmental sulfur regulations. The conventional HDS can be used to lower the sulfur content to few hundreds ppm. Then, the oxidation/extraction approach is applied to go for ultra-deep desulfurization as it provides better mean and cost effective way in order to meet the future sulfur environmental requirements. Given the economic limitations of peroxide-based ODS and the need for an alternate technology for refineries where a supply of hydrogen peroxide is not readily available, oxidation methods besides H2O2 are needed. There is also not many detailed works to define the appropriate conditions in terms of the optimum temperature, oxidants, catalysts, solvents/fuel ratio for extraction, and the impact of such solvents extraction on fuel quality. So these approaches still need further research, especially in the area of designing the appropriate selective catalysts. Much work still remains to be done to address the suitability of future approaches to meet the sulfur limit requirements.
Abbreviations
| 4E6MDBT | 4-ethyl,6-methyl-DBT |
| AC | Activated Carbon |
| ACF | Activated Carbon Fiber |
| BT | Benzothiophene |
| BDS | Biodesulfurization |
| BiCh | Bicyclohexyl |
| CGO | Coker Gas Oil |
| CHB | Cyclohexylbenzene |
| CUS | Coordinately unsaturated sites |
| DBT | Dibenzothiophene |
| DBT-MO | Dibenzothiophene-monooxygenase |
| DBTO | Dibenzothiophene-5-oxide, DBT sulfoxide |
| DBTO2 | Dibenzothiophene-5,5-dioxide, DBT sulfone |
| DBTO2-MO | Dibenzothiophene-5,5-dioxide-monooxygenase |
| DDS | Direct desulfurization |
| DMBPh | Dimethylbiphenyl |
| DMDBT | DiMethyl Dibenzothiophene |
| DMSO | Dimethyl sulfoxide |
| DPS | Diphenylsulfide |
| EPA | Environmental protection agency |
| EDF | Equilibrium deposition filtration |
| FCC | Fluid catalytic cracking |
| FMNH2 | Reduced flavin mononucleotide |
| FTIR | Fourier Transform Infrared Spectroscopy |
| GC | Gas Chromatography |
| HBP | Hydroxybiphenyl |
| HBPS | 2′-hydroxybiphenyl-2-sulfinate |
| HBPSi | 2′-hydroxybiphenyl-2-sulfinate, sulfinate |
| HBPSo | 2′-hydroxybiphenyl-2-sulfonate, sulfonate |
| HDEBP | 2-hydroxy-3,3′-diethylbiphenyl |
| HAD | Hydrodearomatization |
| HDS | Hydrodesulfurization |
| HHDBT | Hexahydrodibenzothiophene |
| HFBT | 3-hydroxy-2-formyl-benzothiophene |
| HPBS | Hydroxyphenyl benzene sulfonate |
| HRP | Horseradish peroxidase |
| HYD | Hydrogenation |
| IL | Ionic liquid |
| MDBT | Methyl Dibenzothiophene |
| MT | Methylthiophene |
| NA | Naphthalene |
| NOx | Oxides of nitrogen |
| ODS | Oxidative Desulfurization |
| PA | Pyruvic acid |
| PBS | 2-phenylbenzene sulfinate |
| PM | Particulate matter |
| ppmw | Parts per million by weight |
| SCANfining | Selective cat naphtha hydrofining process developed by ExxonMobil |
| SEM | Scanning electron micrograph |
| SOx | Oxides of sulfur |
| SRGO | Straight (distillation) run gas oil |
| TH | Thiophene |
| THDBT | Tetrahydrodibenzothiophene |
| TPR | Temperature-programmed reduction |
| XRD | X-ray diffraction |
Acknowledgements
Author is thankful to Prof. I. D. Mall and Prof. I. M. Mishra for their suggestions, continuous help and motivation. Author is also thankful to all his students particularly Mr. Ankur Srivasatav for the help during preparation of the manuscript.References
- Annual Energy Review. Energy Dept, Energy Information Administration, Office of Energy Markets and End Use. DOE/EIA-0384, 2004.
- G. H. James, G. E. Handewerk, Petroleum Refining: Technology and Economics, Second Edition, Marcel Dekker, New York, 1984 Search PubMed.
- W. L. Fang, Inventory of U. S. Greenhouse Gas Emissions and Sinks, 1990-2003. Clean Air Markets Division, 2004 Search PubMed.
- G. A. Emison, United States Environmental Protection Agency, Office of Air Quality Planning and Standards, 2000 Search PubMed.
- J. P. Nehlsen, Ph.D. Dissertation, Princeton University, 2006 Search PubMed.
- T. A. Koch, K. R. Krause, L. E. Manzer, M. Mehdizadeh, J. M. Odom and S. K. Sengupta, New J. Chem., 1996, 20, 163–173 Search PubMed.
- D. D. Whitehurst, I. Isoda and I. Mochida, Adv. Catal., 1998, 42, 345–357 CAS.
- Y. Shiraishi, T. Hirai and I. Komasawa, Ind. Eng. Chem. Res., 1998, 37, 203–211 CrossRef CAS.
- G. Parkinson, Chem. Eng., 2000, 107, 45–48 Search PubMed USEPA. http://www..
- USEPA. http://www..
- F. M. Collins, A. R. Lucy and C. J. Sharp, J. Mol. Catal. A: Chem., 1997, 117, 397–403 CrossRef CAS.
- P. S. Tam, J. R. Kittrell and J. W. Eldridge, Ind. Eng. Chem. Res., 1990, 29, 321–324 CrossRef CAS.
- CA-Asia, Summary of Country/City Synthesis Reports Across Asia, 2006.
- A. Avidan, B. Klein and R. Ragsdale, HydroCarbon, Process., 2001, 80, 47–53 Search PubMed EC-Environment Canada, http://www., 2006.
- EC-Environment Canada, http://www., 2006.
- J. J. Kilbane, Curr. Opin. Biotechnol., 2006, 17, 305–14 CrossRef European Standard for Gasoline EN 228/1999 and for diesel EN 228/1999 and for diesel EN 590/1999, 1999.
- European Standard for Gasoline EN 228/1999 and for diesel EN 590/1999, 1999.
- Auto Fuel Policy, Ministry of petroleum and Natural gas, Government of India, 2003, http://petroleum..
- C. N. Saterfield, Heterogenous catalysis in industrial practice, 2nd Ed, McGraw-Hill: New York, 1993 Search PubMedAuto Fuel Policy, Ministry of petroleum and Natural gas, Government of India, 2003, http://petroleum..
- K. Chan, J. Jung, J. Lee, B. Sang, C. Kyungil and H. M. Sang, Appl. Catal., A, 2000, 200, 233–242 CrossRef CAS.
- H. Nikkolaj, M. Brosrson and T. Henrik, Catal. Lett., 2000, 65, 196–174 Search PubMed.
- G. T. Austin, Shreve's Chemical Process Industries, McGraw-Hill: New York, 1984 Search PubMed.
- R. Shafi and G. J. Hutchings, Catal. Today, 2000, 59, 423–442 CrossRef CAS.
- C. Song, Catal. Today, 2002, 77, 17–49 CrossRef CAS.
- S. Katikaneni, C. Yuh, S. Abens and M. Farooque, Catal. Today, 2002, 77, 99–106 Search PubMed.
- R. Bartsch and C. Taniellian, J. Catal., 1974, 35, 353–358 CrossRef CAS.
- M. Houalla, D. H. Broderick, A. V. Sapre, N. K. Nag and V. H. J. D. Beer, J. Catal., 1980, 61, 523–527 CrossRef CAS.
- M. Houalla, N. K. Nag, A. V. Sapre, D. H. Broderick and B. C. Gates, AIChE J., 1978, 24, 1015–1021 Search PubMed.
- S. Haji, Ph.D. Dissertation, University of Connecticut, 2005 Search PubMed.
- I. March, Advanced Organic Chemistry, John Wiley and Sons: New York, 1985 Search PubMed.
- N. K. Nag, A. V. Sapre, D. H. Broderick and B. C. Gates, J. Catal., 1979, 57, 509–512 CrossRef CAS.
- D. R. Kilanowski, H. Teeuwen, B. C. Gates, V. H. J. D. Beer, G. C. A Schuit and H. Kwart, J. Catal., 1978, 55, 129–137 CrossRef CAS.
- T. Isoda, S. Nagao, X. L. Ma, Y. Korai and I. Mochida, Energy Fuels, 1996, 10, 1078–1082 Search PubMed.
- T. Isoda, S. Nagao, X. L. Ma, Y. Korai and I. Mochida, Energy Fuels, 1996, 10, 482–486 Search PubMed.
- M. Egorova and R. Prins, J. Catal., 2004, 224, 278–287 CrossRef CAS.
- M. Nagai and T. Kabe, J. Catal., 1983, 81, 440–449 Search PubMed.
- J. Chen and Z. Ring, Fuel, 2004, 83, 305–313 Search PubMed.
- A. Ishihara, J. Lee, F. Dumeignil, M. Yamaguchi, S. Hirao, E. W. Qian and T. Kabe, J. Catal., 2004, 224, 243–251 CrossRef CAS.
- T. Kabe, Y. Aoyama, D. Wang, A. Ishihara, W. Qian, M. Hosoya and Q. Zhang, Appl. Catal., A, 2001, 209, 209–237 Search PubMed.
- W. Y. Liu, Z. L. Lei and I. K. Wang, Energy Fuels, 2001, 15, 38–43 Search PubMed.
- I. Funakoshi and T. Aida, US Patent, 5753102, 1998 Search PubMed.
- S. Otsuki, T. Nonaka, N. Takashima, W. Qian, A. Ishihara, T. Imai and T. Kabe, Energy Fuels, 2000, 14, 1232–1239 CrossRef CAS.
- T. Kabe, W. H. Qian, S. Ogawa and A. Ishihara, J. Catal., 1993, 143, 239–248 CrossRef CAS.
- X. Ma, K. Sakanishi and I. Mochida, Fuel, 1994, 73, 1667–1671 Search PubMed.
- D. Yitzhaki, M. V. Landau, D. Berger and M. Herskowitz, Appl. Catal., A, 1995, 122, 99–110 Search PubMed.
- M. K. Andari, F. Abu-Seedo, A. Stanislaus and H. M. Qabazard, Fuel, 1996, 75, 1664–1670 Search PubMed.
- L. Vradman, M. V. Landau and M. Herskowitz, Catal. Today, 1999, 48, 41–48 Search PubMed.
- F. T. T. Ng and I. K. Milad, Appl. Catal., A, 2000, 200, 243–254 Search PubMed.
- S. Shin, H. Yang, K. Sakanishi, I. Mochida, D. A. Grudoski and J. H. Shinn, Appl. Catal., A, 2001, 205, 101–108 Search PubMed.
- U. T. Turaga and C. Song, Catal. Today, 2003, 86, 129–140 Search PubMed.
- E. Pedernera, R. Reimert, N. L. Nguyen and V. van. Buren, Catal. Today, 2003, 79-80, 371–381 Search PubMed.
- K. H. Choi, N. Kunisada, Y. Korai, I. Mochida and K. Nakano, Catal. Today, 2003, 86, 277–286 Search PubMed.
- N. Kunisada, K. H. Choi, Y. Korai, I. Mochida and K. Nakano, Appl. Catal., A, 2004, 269, 43–51 Search PubMed.
- N. Kunisada, K. H. Choi, Y. Korai and I. Mochida, Appl. Catal., A, 2004, 260, 185–190 Search PubMed.
- C. Schmitz, L. Datsevitch and A. Jess, Chem. Eng. Sci., 2004, 59, 2821–2829 CrossRef CAS.
- P. D. Costa, J. M. Manoli, C. Potvin and G. Djéga-Mariadassou, Catal. Today, 2005, 107-108, 520–530 Search PubMed.
- R. Hubaut, J. Altafulla, A. Rives and C. Scott, Fuel, 2007, 86, 743–749 Search PubMed.
- A. M. Venezia, V. L. Parola, G. Deganello, D. Cauzzi, G. Leonardi and G. Predieri, Appl. Catal., A, 2002, 229, 261–271 Search PubMed.
- C. Papadopoulou, J. Vakros, H. K. Matralis, C. Kordulis and A. Lycourghiotis, J. Colloid Interface Sci., 2003, 261, 146–153 CrossRef CAS.
- T. A. Pecoraro and R. R. Chianelli, J. Catal., 1981, 67, 430–445 CrossRef CAS.
- M. J. Ledoux, O. Michaux, G. Agostini and P. Panissod, J. Catal., 1986, 102, 275–288 CrossRef CAS.
- P. Grange and X. Vanhaeren, Catal. Today, 1997, 36, 375–392 CrossRef CAS.
- W. R. A. M. Robinson, J. A. R. Veen van, V. H. J. de Beer and R. A. van Santen, Fuel Process. Technol., 1999, 61, 103–116 CrossRef CAS.
- W. Qian, Y. Yoda, Y. Hirai, A. Ishihara and T. Kabe, Appl. Catal., A, 1991, 184, 81–88 Search PubMed.
- E. Lecrenay, K. Sakanishi and I. Mochida, Catal. Today, 1997, 39, 13–20 Search PubMed.
- S. K. Bej, S. K. Maity and U. T. Turaga, Energy Fuels, 2004, 18, 1227–1237 CrossRef CAS.
- X. Li, A. Wang, M. Egorova and R. Prins, J. Catal., 2007, 250, 283–293 Search PubMed.
- E. Rodriguez-Castellon, A. Jimenez-Lopez and D. Eliche-Quesada, Fuel, 2008, 87, 1195–1206 Search PubMed.
- B. Yoosuk, J. H. Kim, C. Song, C. Ngamcharussrivichai and P. Prasassarakich, Catal. Today, 2008, 130, 14–23 Search PubMed.
- K. Sakanishi, T. Nagamatsu, I. Mochida and D. D. Whitehurst, J. Mol. Catal. A: Chem., 2000, 155, 101–109 Search PubMed.
- R. Nav, R. A. Ortega, G. Alonso, C. Ornelas, B. Pawelec and J. L. G. Fierro, Catal. Today, 2007, 127, 70–84 Search PubMed.
- R. Navarro, B. Pawelec, J. L. G. Fierro, P. T. Vasudevan, J. F. Cambra and P. L. Arias, Appl. Catal., A, 1996, 137, 269–286 CrossRef CAS.
- W. R. A. M. Robinson, van. J. A. R. Veen, V. H. J. de Beer and R. A. van Santen, Fuel Process. Technol., 1999, 61, 89–101 Search PubMed.
- H. R. Reinhoudt, R. Troost, van. S. Schalkwijk, van. A. D. Langeveld, S. T. Sie, van. J. A. R. Veen and J. A. Moulijn, Fuel Process. Technol., 1999, 61, 117–131 CrossRef.
- V. G. Baldovino-Medrano, S. A. Giraldo and A. Centeno, Fuel, 2008, 87, 1917–1926 Search PubMed.
- Y. Okamoto, K. Ochiai, M. Kawano, K. Kobayashi and T. Kubota, Appl. Catal., A, 2002, 226, 115–127 CrossRef CAS.
- T. Klimova, J. Reyes, O. Gutierrez and L. Lizama, Appl. Catal., A, 2008, 335, 159–171 Search PubMed.
- T. A. Zepeda, B. Pawelec, J. L. G. Fierro and T. Halachev, Appl. Catal., B, 2007, 71, 223–236 CrossRef CAS.
- E. Furimsky and F. E. Massoth, Catal. Today, 1992, 52, 381–495 Search PubMed.
- B. M. Vogelaar, P. Steiner, van. A. D. Langeveld, S. Eijsbouts and J. A. Moulijn, Appl. Catal., A, 2003, 251, 85–92 Search PubMed.
- D. H. Broderick, G. C. A. Schuit and B. C. Gates, J. Catal., 1978, 54, 94–97 Search PubMed.
- H. Farag, K. Sakanishi, M. Kouzu, A. Matsumura, Y. Sugimoto and I. Saito, Catal. Commun., 2003, 4, 321–326 Search PubMed.
- H. Farag and K. Sakanishi, J. Catal., 2004, 225, 531–535 CrossRef CAS.
- T. Isoda, Y. Takase, K. Kusakabe and S. Morooka, Energy Fuels, 2000, 14, 585–590 Search PubMed.
- J. G. Michael and C. C Bruce, Ind. Eng. Chem. Res., 1991, 30, 2021–2058 CrossRef CAS.
- K. Heeyeon, J. Jung, S. Lee and M. Heup, Appl. Catal., B, 2003, 44, 287–299 Search PubMed.
- A. S. Rappas, V. P. Nero and S. J. Decanio, US Patent 6402940, B1, 2002 Search PubMed.
- I. V. Babich and J. A. Moulijn, Fuel, 2003, 82, 607–631 CrossRef CAS.
- B. W. Higgins, Chem. Innovation, 2001, 31, 39–40 Search PubMed.
- G. E. Dolbear and E. R. Skov, Am. Chem. Soc, Div. Pet. Chem., 2000, 45, 375–378 Search PubMed.
- S. E. Bonde and G. E. Dolbear, Am. Chem. Soc, Div. Pet. Chem., 2001, 46, 69–73 Search PubMed.
- G. M. K. Abotsi and A. W. Scaroni, Fuel Process. Technol., 1989, 22, 107–133 Search PubMed.
- M. H. Ali, A. Al-Maliki, B. El-Ali, G. Martinie and M. N. Siddiqui, Fuel, 2006, 85, 1354–1363 CrossRef CAS.
- F. Zannikos, E. Lois and S. Stournas, Fuel Process. Technol., 1995, 42, 35–45 CrossRef CAS.
- T. Aida, D. Yamamoto and K. Sakata, Adv. Mater., 1993, 5, 391–395 Search PubMed.
- W. Gore, US Patent, 6274785, 2001 Search PubMed.
- Y. Shiraishi, K. Tachibana, T. Hirai and I. Komasawa, Ind. Eng. Chem. Res., 2002, 41, 4362–4375 CrossRef CAS.
- Y. Shiraishi, T. Hirai and I. Komasawa, J. Chem. Eng. Jpn., 2002, 35, 1305–1311 CrossRef CAS.
- H. Vasile, F. Francois and B. Jacques, J. Catal., 2000, 198, 179–186 Search PubMed.
- R. T. Yang, F. H. Yang, A. Takahashi and A. J. H. Maldonado, US Patent, 7029574, 2002 Search PubMed.
- R. F. Zaykina, Y. A. Zaykin, T. B. Mamaonava and N. K. Nadirov, Radiat. Phys. Chem., 2002, 63, 621–624 Search PubMed.
- H. Mei, B. W. Mei and T. F. Yen, Fuel, 2003, 82, 405–414 CrossRef CAS.
- G. Yu, S. Lu, H. Chen and Z. Zhu, Carbon, 2005, 43, 2285–2294 CrossRef CAS.
- H. Lu, J. Gao, Z. Jiang, F. Jing, Y. Yang, G. Wang and C. Li, J. Catal., 2006, 239, 369–375 CrossRef.
- L. Yang, J. Li, X. Yuan, J. Shen and Y. Qi, J. Mol. Catal. A: Chem., 2007, 262, 114–118 CrossRef CAS.
- A. Attar and W. H. Corcoran, Ind. Eng. Chem. Prod. Res. Dev., 1978, 17, 102–109 CrossRef CAS.
- S. E. Bonde and W. E. D. G. Gore, Am. Chem. Soc. Div. Fuel. Chem., 1999, 199–201 Search PubMed.
- S. Liu, B. Wang, B. Cui and L. Sun, Fuel, 2008, 87, 422–428 CrossRef CAS.
- Y. Shiraishi, Y. Taki, T. Hirai and I. Komasawa, Ind. Eng. Chem. Res., 2001, 40, 1213–1224 CrossRef CAS.
- Y. Shiraishi, K. Tachibana, Y. Taki, T. Hirai and I. Komasawa, Ind. Eng. Chem. Res., 2001, 40, 1225–1233 CrossRef CAS.
- Y. Shiraishi, K. Tachibana, T. Hirai and I. Komasawa, Ind. Eng. Chem. Res., 2001, 40, 4919–4924 Search PubMed.
- Y. Shiraishi, T. Naito, T. Hirai and I. Komasawa, Ind. Eng. Chem. Res., 2002, 41, 4376–4382 CrossRef CAS.
- S. T. Patrick, R. K. James and W. E. John, Ind. Eng. Chem. Res., 1990, 29, 321–324 CrossRef CAS.
- S. Kumar, V. C. Srivastava and R. P. Badoni, Oxidative desulfurization by chromium promoted sulfated zirconia, Fuel Process. Technol., 2011 DOI:10. 1016/j.fuproc.2011.08.017.
- C. A. Audeh, US Patent, 5310479 1994 Search PubMed.
- M. C. Frances and R. L. Anderw, J. Mol. Catal. A: Chem., 1997, 117, 397–403 CrossRef CAS.
- M. J. Grassman, M. Siskin, D. T. Ferrughelli and M. K. Lee, US Patent, 5910440, 1999 Search PubMed.
- T. F. Yen, S. H. Lu, US Patent, 6402939, 2002 Search PubMed.
- C. Lanju, G. U. O. Shaohui and Z. H. A. O. Dishun, Chin. J. Chem. Eng., 2007, 15, 520–523 Search PubMed.
- F. Al-Shahrani, T. Xiao, A. Simon, S. Barri, Z. Jiang, H. Shi, G. Martinie and L. H. M. Green, Appl. Catal., B, 2007, 73, 311–316 CrossRef CAS.
- T. J. Collins, Acc. Chem. Res., 1994, 27, 279–285 CrossRef CAS.
- K. Yazu, Y. Yamamoto, T. Furuya, K. Miki and K. Ukegawa, Energy Fuels, 2001, 15, 1535–1536 CrossRef CAS.
- S. Herbstman and J. Patel, In Symposium on Upgrading of Synthetic Crudes, American Chemical Society: Kansas City, 1982 Search PubMed.
- N. D'Alessandro, T. Lucia, B. Monica, D. Milena, B. Mario and M. Antonino, New J. Chem., 2003, 27, 989–993 RSC.
- J. L. G. Gutierrez, A. G. Fuentes, M. E. Hernandez-Teran, P. Garcıa, F. Murrieta-Guevara and F. Jimenez-Cruz, Appl. Catal., A, 2008, 334, 366–373 CrossRef CAS.
- L. C. Caero, H. Gomez-Bernal, A. Fraustro-Cuevas, D. H. Guerra-Gomez and R. Cuevas-Garcia, Catal. Today, 2008, 133-135, 244–254 Search PubMed.
- T. I. Collins, Science, 2001, 291, 48–49 CrossRef CAS.
- T. J. Collins, Nature, 2001, 414, 161–163 Search PubMed.
- R. F. Zaykina, Y. A. Zaykin, G. Mirkin and N. K. Nadirov, Radiat. Phys. Chem., 2002, 63, 617–620 Search PubMed.
- V. F. Kamyanov, A. K. Lebedev, P. P. Sivirilov and T. A. Filimonova, Neftekchimiya (Oil Chem.), 1991, 31, 255–263 Search PubMed.
- N. K. Nadirov, R. F. Zaykina, Y. A. Zaykin, T. B. Mamonova and C. F. Bakirova, Oil Gas Kazakhstan, 1998, 3, 129–134 Search PubMed.
- A. Y. Zaykin, R. F. Zaykina, N. K. Nadirov and T. B. Mamonova, Oil Gas Kazakhstan, 1998, 4, 91–96 Search PubMed.
- L. H. Thompson and L. K. Doraiswamy, Ind. Eng. Chem. Res., 1999, 38, 1215–1249 CrossRef CAS.
- J. L. Luche, Synthetic Organic Sonochemistry, New York: Plenum Press, 1998 Search PubMed.
- Y. T. Shah, A. B. Pandit and V. S. Moholkar, Cavitation Reaction Engineering, New York: Kluwer academic/Plenum publishers, 1999 Search PubMed.
- T. F. Yen, R. D. Gilbert and J. H. Fendler, Membrane mimetic chemistry and its applications. New York: Plenum Press, 1994 Search PubMed.
- S. P. Tu and T. F. Yen, Energy Fuels, 2000, 14, 1168–1175 Search PubMed.
- Y. Shiraishi, Y. Taki, T. Hirai and I. Komasawa, Ind. Eng. Chem. Res., 1999, 38, 3310–3318 Search PubMed.
- G. Strukul, editor.Catalytic oxidations with hydrogen peroxide as oxidant. Dordrecht: Kluwer, 1992 Search PubMed.
- C. Venturello, E. Alneri and M. Ricci, J. Org. Chem., 1983, 48, 3831–3833 CrossRef CAS.
- S. Campestrini, V. Conte, F. D. Furia, G. Modena and O. Bortolini, J. Org. Chem., 1988, 53, 5721–5724 CrossRef CAS.
- S. Yasuhiro, T. Kenya, H. Takayuki and K. Isao, Ind. Eng. Chem. Res., 2000, 39, 2826–2836 Search PubMed.
- T. Hirai, K. Ogawa and I. Komasawa, Ind. Eng. Chem. Res., 1996, 35, 586–589 CrossRef CAS.
- T. Hirai, Y. Shiraishi and I. Komasawa, J. Chem. Eng. Jpn., 1997, 30, 173–175 Search PubMed.
- T. Hirai, Y. Shiraishi, K. Ogawa and I. Komasawa, Ind. Eng. Chem. Res., 1997, 36, 530–533 Search PubMed.
- Y. Shiraishi, T. Hirai and I. Komasawa, Ind. Eng. Chem. Res., 1999, 38, 3300–3309 Search PubMed.
- A. Ibrahim, S. B. Xian and Z. Wei, Pet. Sci. Technol., 2003, 21, 1555–1573 Search PubMed.
- Y. Shiraishi, T. Hirai and I. Komasawa, Ind. Eng. Chem. Res., 2001, 40, 293–303 CrossRef CAS.
- A. Ibrahim, S. B. Xian and Z. Wei, Pet. Sci. Technol., 2004, 22, 287–301 Search PubMed.
- Y. Shiraishi, H. Hara, T. Hirai and I. Komasawa, Ind. Eng. Chem. Res., 1999, 38, 1589–1595 CrossRef CAS.
- K. Yazu, Y. Yamamoto, T. Furuya, K. Miki and K. Ukegawa, Energy Fuels, 2001, 15, 1535–1536 CrossRef CAS.
- H. Tao, T. Nakazato and S. Sato, Fuel, 2009, 88, 1961–1969 Search PubMed.
- Y. Shiraishi, Y. Taki, T. Hirai and I. Komasawa, Chem. Commun., 1998, 23, 2601–2602 Search PubMed.
- A. Paybarah, R. L. Bone and W. H. Corcoran, Ind. Eng. Chem. Process Des. Dev., 1982, 21, 426–431 CrossRef CAS.
- S. Matsuzawa, J. Tanaka, S. Sato and T. Ibusuki, J. Photochem. Photobiol., A, 2003, 149, 183–189 Search PubMed.
- L. Wang, B. X. Shen and S. Z. Li, Energy Fuels, 2006, 20, 1287–1293 Search PubMed.
- J. Robertson and T. J. Bandosz, J. Colloid Interface Sci., 2006, 299, 125–135 Search PubMed.
- A. M. A. Abdel-Wahab and A. E. A. M. Gaber, J. Photochem. Photobiol., A, 1998, 114, 213–218 Search PubMed.
- S. Matsuzawa, J. Tanaka, S. Sato and T. Ibusuki, J. Photochem. Photobiol., A, 2002, 149, 183–189 CrossRef CAS.
- E. Robert-Banchereau, S. Lacombe and J. Ollivier, Tetrahedron, 1997, 53, 2087–2102 CrossRef CAS.
- T. Akasaka, A. Yabe and W. Ando, J. Am. Chem. Soc., 1987, 109, 8085–8087 Search PubMed.
- D. Sinnreich, H. Lind and H. Batzer, Tetrahedron Lett., 1976, 39, 3541–3542 Search PubMed.
- T. Tezuka, H. Miyazaki and H. Suzuki, Tetrahedron Lett., 1978, 22, 1959–1960 Search PubMed.
- C. W. Bookwalter, D. L. Zoller, P. L. Ross and M. V. Johnston, J. Am. Soc. Mass Spectrom., 1995, 6, 872–876 CrossRef CAS.
- W. Wang, S. Wang, H. Liu and Z. Wang, Fuel, 2007, 86, 2747–2753 Search PubMed.
- W. Wang, S. Wang, Y. Wang, H. Liu and Z. Wang, Fuel Process. Technol., 2007, 88, 1002–1008 Search PubMed.
- R. B. Long and F. A. Caruso, US Patent, 4493765, 1985 Search PubMed.
- M. J. Grossman, M. Siskin, D. T. Ferrughelli, M. K. Lee and J. D. Senius, US Patent, 5910440, 1999 Search PubMed.
- J. L. Frank and H. Yuan, Production of ultra low sulfur fuels by selective hydroperoxide oxidation, NPRA, AM-03-23, Annual Meeting March 23–25, 2003 Search PubMed.
- H. Zhao, S. Q. Xia and P. S. Ma, J. Chem. Technol. Biotechnol., 2005, 80, 1089–1096 CrossRef CAS.
- G. Yu, X. Li, X. Liu, C. Asumana and X. Chen, Ind. Eng. Chem. Res., 2011, 50, 2236–2244 Search PubMed.
- K. Kendra-Krolik, M. Fabrice and J. -N. Jaubert, Ind. Eng. Chem. Res., 2011, 50, 2296–2306 CrossRef CAS.
- L. Alonso, A. Arce, M. Francisco, O. Rodriguez and A. Soto, AIChE J., 2007, 53, 3108 CrossRef CAS.
- L. Alonso, A. Arce, M. Francisco and A. Soto, Fluid Phase Equilib., 2008, 263, 176–181 Search PubMed.
- L. Alonso, A. Arce, M. Francisco and A. Soto, J. Chem. Thermodyn., 2008, 40, 966 CrossRef CAS.
- A. Bösmann, L. Datsevich, A. Jess, A. Lauter, C. Schmitz and P. Wasserscheid, Chem. Commun., 2001, 23, 2494–2495 RSC.
- X. M. Chu, Y. F. Hu, J. G. Li, Q. Q. Liang, Y. S. Liu, X. M. Zhang, X. M. Peng and W. J. Yue, Chin. J. Chem. Eng., 2008, 16, 881–884 Search PubMed.
- J. Esser, P. Wasserscheid and A. Jess, Green Chem., 2004, 6, 316–322 RSC.
- H. S. Gao, Y. G. Li, J. M. Wu, Y. Wu, M. F. Luo, Q. Li, J. M. Xing and H. Z. Liu, Energy Fuels, 2009, 23, 2690–2694 CrossRef CAS.
- H. S. Gao, M. F. Luo, J. M. Xing, Y. Wu, Y. G. Li, W. L. Li, Q. F. Liu and H. Z. Liu, Ind. Eng. Chem. Res., 2008, 47, 8384–8388 CrossRef CAS.
- Q. X. Guo and X. D. Tang, J. Southwest Pet. Univ. China, 2007, 29, 95 Search PubMed.
- J. D. Holbrey, I. Lopez-Martin, G. Rothenberg, K. R. Seddon, G. Silvero and X. Zheng, Green Chem., 2008, 10, 87–92 RSC.
- C. P. Huang, B. H. Chen, J. Zhang, Z. C. Liu and Y. X. Li, Energy Fuels, 2004, 18, 1862–1864 CrossRef CAS.
- X. C Jiang, Y. Nie, C. X. Li and Z. H. Wang, Fuel, 2008, 87, 79–84 CrossRef CAS.
- N. H. Ko, J. S. Le, E. S. Huh, H. Lee, K. D. Jung, H. S. Kim and M. Cheong, Energy Fuels, 2008, 22, 1687–1690 CrossRef CAS.
- Y. Nie, C. X. Li, H. Meng and Z. H. Wang, Fuel Process. Technol., 2008, 89, 978–983 CrossRef CAS.
- Y. Nie, C. X. Li, A. J. Sun, H. Meng and Z. H. Wang, Energy Fuels, 2006, 5, 2083–2087 Search PubMed.
- Y. Nie, C. X. Li and Z. H. Wang, Ind. Eng. Chem. Res., 2007, 46, 5108–5112 CrossRef CAS.
- J. Planeta, P. Karasek and M. Roth, Green Chem., 2006, 8, 70–77 RSC.
- J. L. Wang, D. S. Zhao, E. P. Zhou and D. Zhi, J. Fuel Chem. Technol., 2007, 35, 293–296 Search PubMed.
- Q. B. Wang, K. F Liu, X. P. Zhang and S. J. Zhang, Appl. Chem. Ind. China, 2008, 37, 7 Search PubMed.
- S. G. Zhang and Z. C. Zhang, Green Chem., 2002, 4, 376–379 RSC.
- S. G. Zhang, Q.L. Zhang and Z. C. Zhang, Ind. Eng. Chem. Res., 2004, 43, 614–622 CrossRef CAS.
- H. C. Zhou, N. Chen, F. Shi and Y. Q. Deng, J. Mol. Catal. A: Chem., 2005, 19, 95 Search PubMed.
- E. Lissner, W. de-Souza, B. Ferrera and J. Dupont, ChemSusChem, 2009, 2, 962–964 CrossRef.
- L. He, H. M. Li, W. S. Zhu, J. S. Guo, X. Jiang, J. D. Lu and Y. S. Yan, Ind. Eng. Chem. Res., 2008, 47, 6890–6895 CrossRef CAS.
- F. T. Li, R. H. Liu, J. H. Wen, D. S. Zhao, Z. M. Sun and Y. Liu, Green Chem., 2009, 11, 883–888 RSC.
- H. M. Li, X. Jiang, W. S. Zhu, J. D. Lu, H. M. Shu and Y. S. Yan, Ind. Eng. Chem. Res., 2009, 48, 9034–9039 CrossRef CAS.
- W. H. Lo, H. Y. Yang and G. T. Wei, Green Chem., 2003, 5, 639–642 RSC.
- L. Lu, S. Cheng, J. Gao, G. Gao and M. He, Energy Fuels, 2007, 21, 383–384 CrossRef CAS.
- J. L. Wang, D. S. Zhao and K. X. Li, Energy Fuels, 2009, 23, 3831–3834 CrossRef CAS.
- D. S. Zhao, R. Liu and J. L. Wang, Energy Fuels, 2008, 22, 1100–1103 CrossRef CAS.
- D. S. Zhao, Z. M. Sun, F. T. Li, R. Liu and H. D. Shan, Energy Fuels, 2008, 22, 3065–3069 CrossRef CAS.
- W. S. Zhu, H. M. Li, X. Jiang, Y. S. Yan, J. D. Lu, L. Hea and J. X. Xia, Green Chem., 2008, 10, 641–648 RSC.
- W. Zhu H. Li, X. Jiang, Y. Yan, J. Lu and J. Xia, Energy Fuels, 2007, 21, 2514–2516 CrossRef CAS.
- K. R Seddon., J. Chem. Technol. Biotechnol., 1997, 68, 351–356 CrossRef CAS.
- J. D. Holbrey and K. R. Seddon, Clean Prod. Proc., 1999, 1, 223–236 CrossRef.
- M. J. Earle and K. R. Seddon, Pure Appl. Chem., 2000, 72, 1391–1398 CrossRef CAS.
- M. Francisco, A. Arce and A. Soto, Fluid Phase Equilib., 2010, 294, 39–48 CrossRef CAS.
- C. Phillips, Energy Fuels, 2008, 22, 1774–1778 CrossRef CAS.
- J. Eber, P. Wasserscheid and A. Jess, Green Chem., 2004, 6(7), 316–322 RSC.
- C. Xuemei, H. Yufeng, L. Jiguang, L. Qianqing, L. Yansheng, Z. Xianming, P. Xiaomin and Y. Wenjia, Chin. J. Chem. Eng., 2008, 16, 881–884 Search PubMed.
- L. Alonso, A. Arce, M. Francisco and A. Soto, J. Chem. Eng. Data, 2007, 52, 2409–2412 Search PubMed.
- L. Alonso, A. Arce, M. Francisco and A. Soto, J. Chem. Eng. Data, 2008, 53, 1750–1755 Search PubMed.
- L. Alonso, A. Arce, M. Francisco and A. Soto, J. Solution Chem., 2008, 37, 1355–1363 Search PubMed.
- C. C. Cassol, A. P. Umpierre, G. Ebeling, B. Ferrera, S. S. X. Chiaro and J. Dupont, Int. J. Mol. Sci., 2007, 8, 593–605 CrossRef CAS.
- R. P. Swatloski, J. D. Holbrey and R. D. Rogers, Green Chem., 2003, 5, 361–363 RSC.
- J. Feng, C. X. Li, H. Meng and Z. H. Wang, Petrochem. Technol., 2006, 35, 272–275 Search PubMed.
- U. Domanska, M. Krolikowski and K. Slesinska, J. Chem. Thermodyn., 2009, 41, 1303–1311 Search PubMed.
- L. A. Aslanov and A. V. Anisimov, Petroleum Chem., 2004, 44, 65–69 Search PubMed.
- E. Ito and J. A. R. van-Veen, Catal. Today, 2006, 116, 446–460 CrossRef CAS.
- P. S. Kulkarni and C. A. M Afonso., Green Chem., 2010, 12, 1139–1149 RSC.
- H. -F. Fan, Z. B. Li and J. Ma, Oilfield Chem., 2007, 24, 283–286 Search PubMed.
- S. H. D. Lee, R. Kumar and M. Krumpelt, Sep. Purif. Technol., 2002, 26, 247–258 Search PubMed.
- C. O. Ania, J. B. Parra, A. Arenillas, F. Rubiera, T. J. Bandosz and J. J. Pis, Appl. Surf. Sci., 2007, 253, 5899–5903 Search PubMed.
- Y. Sano, K. Sugaraha, K. Choi, Y. Korai and I. Mochida, Fuel, 2005, 84, 903–910 CrossRef CAS.
- M. A. Larrubia, A. Gutierrez-Alejandre, J. Ramirez and G. Busca, Appl. Catal., A, 2002, 224, 167–178 Search PubMed.
- O. Etemadi and T. F. Yen, J. Colloid Interface Sci., 2007, 313, 18–25 Search PubMed.
- J. Weitkamp, M. Schwark and S. Ernst, J. Chem. Soc., Chem. Commun., 1991, 1133–1134 RSC.
- R. T. Yang, A. Takahashi and F. H. Yang, Ind. Eng. Chem. Res., 2001, 40, 6236–6239 Search PubMed.
- A. Takahashi, F. H. Yang and R. T. Yang, Ind. Eng. Chem. Res., 2002, 41, 2487–2496 Search PubMed.
- A. J. Hernandez and R. T. Yang, AIChE J., 2004, 50, 791–801 Search PubMed.
- Q. Gongshin, Ph.D. Dissertation, University of Michigan, 2006 Search PubMed.
- C. Ngamcharussrivichai, C. Chatratananon, S. Nuntang and P. Prasassarakich, Fuel, 2008, 87, 2347–2351 CrossRef CAS.
- F. Richard, T. Boita and G. Perot, Appl. Catal., A, 2007, 320, 69–79 Search PubMed.
- K. Tang, K. L. Song, L. Duan, X. Li, J. Gui and Z. Sun, Fuel Process. Technol., 2008, 89, 1–6 Search PubMed.
- J. G. Park, K. C. Hyun, K. B. Yi, J. H. Park, S. Han, S. H. Cho and J. N. Kim, Appl. Catal., B, 2008, 81, 244–250 CrossRef CAS.
- I. Bezverkhyy, A. Ryzhikov, G. Gadacz and J. P. Bellat, Catal. Today, 2008, 130, 199–205 Search PubMed.
- P. Baeza, G. Aguila, F. Gracia and P. Araya, Catal. Commun., 2008, 9, 751–755 CrossRef CAS.
- M. Suzuki, Adsorption Engineering, Kodansha Ltd., Tokyo, 1990 Search PubMed.
- A. B. S. H. Salem, Ind. Eng. Chem. Res., 1994, 33, 336–340 Search PubMed.
- A. B. S. H. Salem and H. S. Hamid, Chem. Eng. Technol., 1997, 20, 342–347 CAS.
- J. Gislason, Oil Gas J., 2001, 99, 72–76 Search PubMed.
- S. Haji and C. Erkey, Ind. Eng. Chem. Res., 2003, 42, 6933–6937 Search PubMed.
- C. Song, Catal. Today, 2003, 86, 211–263 CrossRef CAS.
- Z. Jiang, Y. Liu, X. Sun, F. Tian, F. Sun, C. Liang, W. You, C. Han and C. Li, Langmuir, 2003, 19, 731–736 Search PubMed.
- S. Velu, S. Watanabe, X. Ma and C. Song, Am. Chem. Soc. Proceed. Fuel Div., 2003, 48, 526–528 Search PubMed.
- C. O. Ania and T. J. Bandosz, Langmuir, 2005, 21, 7752–7759 CrossRef CAS.
- C. O. Ania and T. J. Bandosz, Carbon, 2006, 44, 2404–2412 CrossRef CAS.
- A. Zhou, X. Ma and C. Song, J. Phys. Chem., 2006, B110, 4699–4707 Search PubMed.
- J. H. Kim, X. Ma, A. Zhou and C. Song, Catal. Today, 2006, 111, 74–83 CrossRef CAS.
- O. Etemandi and T. F. Yen, Energy Fuels, 2007, 21, 1622–1627 Search PubMed.
- Y. Yang, H. Ku, P. Ying, Z. Jiang and C. Li, Carbon, 2007, 45, 3042–3059 CrossRef CAS.
- M. Seredych, J. Lison, U. Jans and T. J. Bandosz, Carbon, 2009, 47, 2491–2500 Search PubMed.
- E. Deliyanni, M. Seredych and T. J. Bandosz, Langmuir, 2009, 25(16), 9302–9312 CrossRef CAS.
- C. Shalaby, X. Ma, A. Zhou and C. Song, Energy Fuels, 2009, 23, 2620–2627 Search PubMed.
- A. J. Hernández-Maldonado, G. Qi and R. T. Yang, Appl. Catal., B, 2005, 61, 212–218 CrossRef CAS.
- M. Seredych and T. J. Bandosz, Fuel Process. Technol., 2010, 91, 693–701 Search PubMed.
- H. J. Jeon, C. H. Ko, S. H. Kim and J. N. Kim, Energy Fuels, 2009, 23, 2537–2543 CrossRef CAS.
- Y. A. Alhamed and H. S. Bamufleh, Fuel, 2009, 88, 87–94 Search PubMed.
- Q. Wang, X. Liang, W. Qiao, C. Liu, X. Liu, L. Zhan and L. Ling, Fuel Process. Technol., 2009, 90, 381–387 CrossRef CAS.
- M. Seredych and T. J. Bandosz, Energy Fuels, 2010, 24, 3352–3360 CrossRef CAS.
- M. Seredych and T. J. Bandosz, Carbon, 2011, 49, 1216–1224 Search PubMed.
- D. Rakesh-Kumar and V. C. Srivastava, CLEAN-Soil Air Water, 2011 DOI:10.1002/clen.201000368.
- F. Rouquerol, J. Rouquerol and K. Sing, Adsorption by Powders and Porous Solids, Academic Press, San Diego, CA, 1999 Search PubMed.
- R. T. Yang, Adsorbents: fundamentals and applications, John Wiley & Sons, Inc., Hoboken, New Jersey, 2003 Search PubMed.
- A. Srivastav and V. C. Srivastava, J. Hazard. Mater., 2009, 170, 1133–1140 Search PubMed.
- S. Kumar, V. C. Srivastava and R. P. Badoni, Fuel, 2011, 90, 3209–3216 Search PubMed.
- X. Ma, L. Sun and C. Song, Catal. Today, 2002, 77, 107–116 CrossRef CAS.
- Y. Wang, T. R. Yang and J. M. Heinzel, Chem. Eng. Sci., 2008, 63, 356–365 CrossRef CAS.
- S. H. Lu, Ph.D. Dissertation, University of Southern California, 2000 Search PubMed.
- U. Mueller, M. Schubert, F. Teich, H. Puetter, K. Schierle-Arndt and J. Pastre, J. Mater. Chem., 2006, 16, 626–636 RSC.
- K. A. Cychosz, A. G. Wong-Foy and A. J. Matzger, J. Am. Chem. Soc., 2008, 130, 6938–6939 CrossRef CAS.
- K. A. Cychosz, A. G. Wong-Foy and A. J. Matzger, J. Am. Chem. Soc., 2009, 131, 14538–14543 CrossRef CAS.
- G. Blanco-Brieva, J. M. Campos-Martin, S. M. Al-Zahrani and J.L.G. Fierro, Global NEST J., 2010, 12, 296–304 Search PubMed.
- S. Achmann, G. Hagen, M. Hammerle, I. Malkowsky, C. Kiener and R. Moos, Chem. Eng. Technol., 2010, 33, 275–280 CrossRef CAS.
- L. Hamon, C. Serre, T. Devic, T. Loiseau, F. Millange, G. Ferey and G. D. Weireld, J. Am. Chem. Soc., 2009, 131, 8775–8777 CrossRef CAS.
- N. A. Khan, J. W. Jun, J. H. Jeong and S. H. Jhung, Chem. Commun., 2011, 47, 1306–1308 RSC.
- C. Serre, F. Millange, C. Thouvenot, M. Nogues, G. Marsolier, D. Louer and G. Ferey, J. Am. Chem. Soc., 2002, 124, 13519–13526 CrossRef CAS.
- T. Loiseau, C. Serre, C. Huguenard, G. Fink, F. Taulelle, M. Henry, T. Bataille and G. Ferey, Chem.–Eur. J., 2004, 10, 1373–1382 CrossRef CAS.
- K. Barthelet, J. Marrot, D. Riou and G. Ferey, Angew. Chem., Int. Ed., 2002, 41, 281–284 CrossRef CAS.
- F. Shi, M. Hammoud and L. T. Thompson, Appl. Catal., B, 2011, 103, 261–265 Search PubMed.
- S. G. McKinley, Ph.D. Dissertation, Iowa State University, 2003 Search PubMed.
- A. W. Myers, L. Dong, T. A. Atesin, R. Skugrud, C. Laschenriem and W. D. Jones, Inorg. Chim. Acta, 2008, 361, 3263–3270 CrossRef CAS.
- M. Arias, D. Laurenti, C. Geantet, M. Vrinat, I. Hideyuki and Y. Yoshimura, Catal. Today, 2008, 130, 190–194 Search PubMed.
- K. Tawara, T. Nishimura and H. Iwanami, J. Jpn. Pet. Inst., 2000, 43, 114–120 Search PubMed.
- K. Tawara, T. Nishimura, H. Iwanami, T. Nishimoto and T. Hasuike, J. Jpn. Pet. Inst., 2001, 44, 43 Search PubMed.
- K. Tawara, T. Nishimura, H. Iwanami, T. Nishimoto and T. Hasuike, Ind. Eng. Chem. Res., 2001, 40, 2367–2370 CrossRef CAS.
- L. Huang, G. Wang, Z. Qin, M. Du, M. Dong, H. Ge, Z. Wu, Y. Zhao, C. Ma, T. Hu and J. Wang, Catal. Commun., 2010, 11, 592–596 Search PubMed.
- L. C. Huang, Z. F. Qin, G. F. Wang, M. X. Du, H. Ge, X. K. Li, Z. W. Wu and J. G. Wang, Ind. Eng. Chem. Res., 2010, 49, 4670–4675 Search PubMed.
- L. Huang, G. Wang, Z. Qin, M. Dong, M. Du, H. Ge, X. Li, Y. Zhao, J. Zhang, T. Hu and J. Wang, Appl. Catal. B: Environmental, 2011, 106, 26–38 Search PubMed.
- G. P. Khare, US Patent, 6274533, 2001 Search PubMed.
- Y. X. Yang, Y. L. Zhang, L. Wang, Y. N. Zhang, Z. X. Jiang and C. Li, Shiyou Huagong, 2008, 37, 243–246 Search PubMed.
- A. Ryzhikov, I. Bezverkhyy and J. -P. Bellat, Appl. Catal., B, 2008, B84, 766–772 Search PubMed.
- I. Bezverkhyy, O. V. Safonova, P. Afanasiev and J. -P. Bellat, J. Phys. Chem., 2009, C113, 17064–17069 Search PubMed.
- J. X. Fan, G. Wang, Y. Sun, C. M. Xu, H. J. Zhou, G. L. Zhou and J. S. Gao, Ind. Eng. Chem. Res., 2010, 49, 8450–8460 Search PubMed.
- G. Germana, D. Abbott and U. Turuga, Hydrocarbon Eng., 2004, 9, 35–38 Search PubMed.
- J. Zhang, Y. Liu, S. Tian, Y. Chai and C. Liu, J. Nat. Gas Chem., 2010, 19, 327–332 Search PubMed.
- M. Soleimani, A. Bassi and A. Margaritis, Biotechnol. Adv., 2007, 25, 570–596 CrossRef CAS.
- I. Konishi, I. Yoshitaka, O. Kouichi and M. Suzuki, US Patent, 5925560, 1999 Search PubMed.
- B. L. McFarland, D. J. Boron, W. Deever, J. A. Meyer, A. R. Johnson and R. M. Atlas, Crit. Rev. Microbiol., 1998, 24, 99–147 CAS.
- N. Gupta and P. K. Roychoudhury, Appl. Microbiol. Biotechnol., 2005, 66, 356–366 Search PubMed.
- K. Kodama, S. Nakatani, K. Umehara, K. Shimizu, Y. Minoda and K. Yamada, Agric. Biol. Chem., 1970, 34, 1320–1324 Search PubMed.
- K. Kodama, K. Umehara, K. Shimuza, S. Nakatani, Y. Minoda and K. Yamada, Agric. Biol. Chem., 1973, 37, 45–50 CAS.
- K. O. Yamada, M. Morimoto and Y. Tani, J. Biosci. Bioeng., 2001, 91, 91–93 Search PubMed.
- F. J. Hartdegen, J. M. Coburn and R. L. Roberts, Chem. Eng. Prog., 1984, 80, 63–67 Search PubMed.
- D. J. Monticello, D. Bakker and W. R. Finnerty, Appl. Environ. Microbiol., 1985, 49, 756–760 Search PubMed.
- H. Y. Kim, T. S. Kim and B. H. Kim, Biotechnol. Lett., 1990, 12, 761–764 Search PubMed.
- S. M. Armstrong, B. M. Sankey and G. Voordouw, Biotechnol. Lett., 1995, 17, 1133–1136 Search PubMed.
- H. M. Lizama, T. C. Scott and C. D. Scott, US Patent, 5458752, 1995 Search PubMed.
- M. R. Sol and T. G. David, Appl. Environ. Microbiol., 1996, 62, 4073–4080 Search PubMed.
- W. Ping and K. Steven, Appl. Environ. Microbiol., 1996, 62, 1670–1675 Search PubMed.
- W. Kimilko, N. Kenichi, K. Jin and M. Kenji, Biotechnol. Lett., 2003, 25, 1451–1456 Search PubMed.
- G. Magdalena, A. Sergey and J. Peter, Appl. Environ. Microbiol., 1995, 61, 3490–3493 Search PubMed.
- B. R. Folsom, D. R. Schieche, D. P. M. Grazia, J. Werner and S. Palmer, Appl. Environ. Microbiol., 1999, 65, 4967–4972 CAS.
- K. A. Gray, G. T. Machkoyz and C. H. Squires, Curr. Opin. Microbiol., 2003, 6, 229–235 Search PubMed.
- J. Konishi, Y. Ishii and T. Onaka, Appl. Environ. Microbiol., 1997, 63, 3164–3169 Search PubMed.
- S. L. Borgne and R. Quintero, Fuel Process. Technol., 2003, 81, 155–169 Search PubMed.
- F. Li, Z. Zhang, J. Feng, X. Cai and X. Ping, J. Biotechnol., 2007, 127, 222–228 Search PubMed.
- G. Mohebali, A. S. Ball, B. Rasekh and A. Kaytash, Enzyme Microb. Technol., 2007, 40, 578–584 Search PubMed.
- H. Chen, H. W. J. Zhang, J. M. Chen, Y. B. Cai and W. Li, Bioresour. Technol., 2008, 99, 6928–6933 Search PubMed.
- A. Caro, P. Leton, E. G. Calvo and L. Setti, Fuel, 2007, 86, 2632–2636 Search PubMed.
- L. Alves, S. Marque, J. Matos, R. Tenreiro and F. M. Gırio, Chemosphere, 2008, 70, 967–973 Search PubMed.
- L. Madeira, V. Santana and E. Pinto, Chemosphere, 2008, 71, 189–194 Search PubMed.
- M. Ayala, R. Tinoco, V. Hernandez, P. Bremauntz and R. V. Duhalt, Fuel Process. Technol., 1998, 57, 101–111 Search PubMed.
- K. Kirimura, T. Furuya, Y. Nishii, Y. Ishii, K. Kino and S. Usami, J. Biosci. Bioeng., 2001, 91, 262–266 Search PubMed.
- T. Furuya, K. Kirimura, K. Kino and S. Usami, FEMS Microbiol. Lett., 2001, 204, 129–133 Search PubMed.
- S. Maghsoudi, M. Vossoughi, A. Kheirolomoom, E. Tanaka and S. Katoh, Biochem. Eng. J., 2001, 8, 151–156 CrossRef CAS.
- S. Oda and H. Ohta, J. Biosci. Bioeng., 2002, 94, 474–477 Search PubMed.
- F. L. Li, P. Xu, C. Q. L. L. Ma and X. S. Luo, FEMS Microbiol. Lett., 2003, 223, 301–307 Search PubMed.
- W. Li, Y. Zhang, M. D. Wang and Y. Shi, FEMS Microbiol. Lett., 2005, 247, 45–50 Search PubMed.
- Y. Hou, Y. Kong, J. Yang, J. Zhang, D. Shi and W. Xin, Fuel, 2005, 84, 1975–1979 Search PubMed.
- I. B. W. Gunam, Y. Yaku, M. Hirano, K. Yamamura, F. Tomita, T. Sone and K. Asano, J. Biosci. Bioeng., 2006, 101, 322–327 Search PubMed.
- M. M. Akbarnejad, J. Towfighi, B. Rasekh and A. Keytash, Biochem. Eng. J., 2006, 29, 169–173 Search PubMed.
- S. Guobin, Z. Huaiying, X. Jianmin, C. Guo, L. Wangliang and L. Huizhou, Biochem. Eng. J., 2006, 27, 305–309 Search PubMed.
- D. J. Monticello, Curr. Opin. Biotechnol., 2000, 11, 540–546 CrossRef CAS.
- P. Xu, B. Yu, F. L. Li, X. F. Cai and C. Q. Ma, Trends Microbiol., 2006, 14, 338–405 Search PubMed.
| This journal is © The Royal Society of Chemistry 2012 |