Biocomposites prepared by alkaline phosphatase mediated mineralization of alginate microbeads†
Minli
Xie
a,
Magnus Ø.
Olderøy
a,
Zhibing
Zhang
b,
Jens-Petter
Andreassen
c,
Berit L.
Strand
d and
Pawel
Sikorski
*a
aDepartment of Physics, Norwegian University of Science and Technology, Trondheim, NO-7491, Norway. E-mail: sikorski@ntnu.no
bSchool of Chemical Engineering, University of Birmingham, Edgbaston, Birmingham B15 2TT, UK
cDepartment of Chemical Engineering, Norwegian University of Science and Technology, Trondheim NO-7491, Norway
dDepartment of Biotechnology, Norwegian University of Science and Technology, Trondheim NO-7491, Norway.
First published on 19th December 2011
Abstract
This study set out to develop a biomimetic scaffold by incorporating osteoinductive hydroxyapatite (HA) particles into a porous alginate gel matrix via a cell-friendly pathway. Two types of alginate/calcium phosphate (Alg/CP) composites were prepared through alkaline phosphatase (ALP) mediated mineralization and counter-diffusion precipitation. Structural characteristics were analyzed by scanning electron microscopy (SEM) and scanning transmission electron microscopy (STEM). Thermal stability and mineral content were studied by means of thermogravimetric (TG) analysis. X-ray diffraction (XRD) and Rietveld refinement showed the presence of bone-like hydroxyapatite. Our studies suggested that the gradual nature of enzymatic process together with alginate matrices provide regulation of nanocrystalline hydroxyapatite formation. The ALP-mediated mineralization process has great advantages over counter-diffusion precipitation, providing homogenous mineral distributions, smaller crystal sizes, and increased apparent Young's moduli, which creates a better structure for bone defect repair scaffolds.
Introduction
Bone tissue exhibits a complex, hierarchical structure over several length scales. It differs from the other connective tissues because of its greater stiffness and strength. These properties stem from it being a two-phase composite material composed of a collagen matrix reinforced with 50–60 wt% hydroxyapatite (HA) crystals.1 The HA crystals are nanoscale, plate-like, and elongated with a c-axis preferred orientation in directions of principal stress. Thus, bone tissue exhibits anisotropic mechanical properties with elastic modulus in the range of 0.05–23 GPa, depending on the bone type and tissue density.2Extracellular matrix (ECM) plays critical roles not only by providing a space to deposit mineral crystals but also by regulating crystal growth rates, nanoscale organization and the crystallographic orientation of HA nanocrystals.3–4 In the past few years, several attempts have been made to mimic bone structure and properties by using tailored biopolymers to mimic ECMs. Among the biopolymers, polysaccharides such as alginate, carrageenans, chitin and its derivative chitosan, are widely used due to their biocompatibility and a variety of chemical structures.Alginate is a linear copolymer consisting of β-D-mannuronic acid (M) and α-L-guluronic acid (G) in widely varying sequences and composition. The relative amount, length, and sequential distribution of homogeneous M segments (M-blocks), homogeneous G segments (G-blocks) and alternating M–G segments (MG-blocks), depend on marine sources and seasonal geographical variations.5 The gel properties including biocompatibility, stability, permeability, mechanical resistance and swelling behavior are significantly affected by the alginate compositions and sequential structures.6 In addition, highly homogenous sequential structures can be achieved by enzymatic modification on polyM derived from bacterial origin, offering significant advantages over native alginates.7–8Alginate can be crosslinked with divalent cations under cell-friendly conditions. Its easy gelling property is valuable for applications in tissue engineering. Therefore, calcium crosslinked alginate hydrogels have been extensively studied for their significant clinical applications, such as drug delivery,9–10cell encapsulation and immobilization,11–13 and scaffolds for cartilage and bone regeneration.14–15 A number of cell types including osteoblasts, chondrocytes, and mesenchymal stem cells have been encapsulated in alginate hydrogels for the purpose of rapid regeneration of bone tissue, potentially at a rate faster than natural healing.16–17 However, a potential limitation in using alginate gels is their weak mechanical properties, lack of osteoconductivity and cellular interaction.18 A desirable modification is mineralization with HA, creating a bone-like environment for cells. M. Gelinsky et al.19–20 reported a HA mineralized alginate hydrogel scaffold with orientated tube-like pore structures and tested the scaffold with human bone marrow stromal cells seeding and cultivation. Their studies demonstrated that the incorporation of HA was advantageous in terms of increasing scaffold stability, facilitating cell attachment, speeding up cell proliferation and inducing osteogenic differentiation by the addition of osteogenic supplements. Therefore, the formation of bone-like HA is essential to achieve direct interactions with cells and thereby enhance osteoblastic cell differentiation, biological affinity as well as mechanical strength of these scaffolds.21 HA reinforced structures have shown improved strength and higher Young's modulus proportional to increased HA content. However, the scaffold mechanical properties often become variable at high reinforcement levels.22–23 Therefore, composites with discrete regions of HA which have interactions with the polymer matrix are desirable for bone tissue interfaces.
Formation of minerals in organic matrices is regarded as one of the mineralization methods that mimic hard tissue. In general, there are many techniques available to fabricate calcium phosphate-based alginate biocomposites such as mechanical blending24 or in situprecipitation in a gel diffusion system.25–28 The shortcoming of direct blending is lack of physicochemical homogeneity and often leads to the aggregation of HA particles. It may exert neither any morphological control nor any chemical interaction between the mineral and polymer phases.29–30 These drawbacks will limit the applications of these composite materials. Gel diffusion systems are often used, since they provide a one-step strategy for the formation of calcium phosphates with well-controlled crystal size. The aggregation of crystals can be avoided efficiently due to the homogeneous nucleation and growth of nano crystals within matrix. Studies have also shown that calcium phosphates with different compositions can be obtained by varying pH and concentrations of calcium and phosphate solutions.27 In our previous work, we have described a porous Alg/CP composite made by counter-diffusion precipitation in a gel diffusion system.31 It showed that alginate played an active role in controlling CP crystal size, morphology and polymorphism. Nanocrystalline HA were achieved within porous alginate gel networks.
However, in biological systems, many inorganic, organic, and biological compounds are present and the concentrations of all ions as well as pH are well balanced during biomineralization. Simulated body fluid (SBF) is widely used as a biomimetic solution inducing formation of HA in polymer matrices.32Alginate fibers treated with calcium hydroxide solution have the ability to form bone-like HA on their surfaces in SBF.33 It takes up to a few weeks for a sufficient amount of HA to deposit. To better mimic biological HA, Banks34 first used the enzyme alkaline phosphatase (ALP) to prepare HA/collagen composites. ALP is one particularly important enzyme involved in the bone formation process which liberates phosphates necessary for mineralization. The use of ALP in biomineralization has considerable advantages.35 The enzyme acts as a “controllable precipitate supplier” and offers better control of precipitation compared to the gel diffusion method. HA is fabricated effectively in close contact with the matrix. Films and multilayer assemblies of HA/collagen composites have been prepared using ALP.36–37 The composites promoted differentiation of preosteoblast cell lines and resorbed with no evidence of cytotoxicity when implanted in muscle tissues.38 Furthermore, enzymatically assisted routes offer the possibility to synthesize materials with excellent control of the structural organization. A spatially selective ALP-directed HA mineralization has been conducted in a 3D nanofiber peptide amphiphile gel matrix.39
With the aim of preparing a bone-mimicking composite, ALP-directed crystallization of calcium phosphate in alginate hydrogels was attempted, and compared to the counter-diffusion approach, in this work. Enzymatic mineralization can be carried out under cell-friendly conditions to assure a high level of cell viability by co-encapsulation of cells and enzymes, which is a prerequisite for cell transplantation. It provides a way to develop new bone regeneration biomaterials.
Materials and methods
2.1. Materials
Sodium alginate with a high content of G (FG = 0.67, FGG = 0.56, FGGG = 0.52) and high viscosity ([η] = 1018 ml g−1) from Laminaria hyperborea stipe (Protanal SF60) was used in this study. Alkaline phosphatase (ALP, EC3.1.3.1, calf intestinal mucosa, 15 units/mg), CaCl2·2H2O, and β-glycerophosphate were purchased from Sigma Aldrich. Mesenchymal stem cell growth medium (MSCGM) was made by adding 50 mL mesenchymal cell growth supplement (MCGS, PT-4106E, Lonza), 10 mL L-glutamine (PT-4107E, Lonza) and 0.5 mL gentamicin sulfate and amphotericin-B (GA-1000, PT-4504E, Lonza) to 500 mL mesenchymal stem cell basal medium (MSCBM, Lonza). All reagents and solvents were of analytical grade and used as received.2.2. Alg/CP composite beads made by counter-diffusion precipitation
Composite beads were made using an electrostatically driven bead generator operated at electrostatic potential of 7 kV as previously described.31 Briefly, NaH2PO4/Na2HPO4 dissolved in alginate solution (1.8% (w/v) alginate, 100 mM PO43−, pH 7.4), which was then dripped into a calcium containing gelling bath (300 mM CaCl2, 0.9% NaCl, 10 mM tris-buffer, pH 7.4) through a 0.4 mm diameter needle at a flow rate of 8 mL h−1. The beads were left for 30 min in the gelling bath with stirring at room temperature (RT) and then taken out and rinsed with distilled water. Sample was named as Alg/CP–CD for short.2.3. Alg/CP composite beads made using ALP
Using the same method of gel formation, 1.8% (w/v) alginate solutions with various concentrations of ALP was employed to make composite beads with encapsulated ALP in a gelling bath consisting of 50 mM CaCl2, 0.9% NaCl, pH 7.4. The beads were subsequently incubated in a mineralization bath (CaCl2 and β-glycerophosphate in tris-buffer) for 24 h with stirring. To optimize mineralization conditions, different concentrations of ALP (0.1 mg mL−1 and 0.5 mg mL−1), β-glycerophosphate (2.5 mM and 5 mM), CaCl2 (25 mM and 50 mM), tris-buffer (10 mM, 50 mM and 100 mM), pH (9.0 and 7.4) and temperature (RT and 37 °C) were tested. The sample made with 0.1 mg mL−1 ALP, 5 mM β-glycerophosphate, 10 mM tris-buffer at pH 7.4 and RT for 24 h was fully characterized and named as Alg/CP–ALP for short.To make the reaction condition compatible with cell culture applications, Alg/CP beads (0.5 mg mL−1 ALP) were mineralized in MSCGM (in addition containing 15 mM CaCl2 and 10 mM β-glycerophosphate) at 37 °C. In this experiment, cell medium was changed every 3 h during day time and 12 h while overnight and mineralization process was allowed to continue for 48 h. Sample was named Alg/CP–ALP Cell for short.
2.4 Alginate controls and CP precipitates
Non-mineralized Ca–alginate beads were made as controls with neither mineral precursor nor ALP added to the alginate solution prior to gel formation in 50 mM CaCl2. The beads were left 15 min in the gelling bath with stirring at RT and then rinsed with distilled water.CP precipitates were made using MQ-grade water containing 300 mM Na2HPO4 in the absence of alginate. The phosphate solution was added dropwise into 1 M CaCl2 gelling bath according to the same procedure as making beads.
2.5. Fluorescent labelling of ALP and Ca-minerals
ALP was fluorescently labeled with Alexa Fluor 633 protein labeling kit (A20170, Molecular Probes, Invitrogen). Ca-minerals were fluorescently labeled with Calcein-high purity (C481, Invitrogen).2.6. Characterization of Alg/CP composite beads
Thin cross sections were freeze-cut by microtome (Leica CM3050 S, thickness 10 μm) from frozen beads which were embedded in Tissue-tek (Qiagen), and frozen in acetone pre-cooled with liquid nitrogen. All synthesized beads and cross-sections were critical point dried (CPD, Emitech K850) before electron microscopic characterization using Zeiss Ultra 55 LE and Hitachi S-5500 scanning electron microscopes. Hitachi S-5500 electron microscope in scanning transmission mode (STEM) was used to characterize size of mineral particles after calcination of beads at 300 °C for 2 h. The distributions of ALP, CP and alginate through the beads were visualized using Leica TCS SP5 confocal laser scanning microscope (CLSM) with a HCX PL FLUOTAR 10 ×/0.15 objective using settings suitable for used fluorescent dyes. Thermal stability and mineral content of beads were evaluated by thermogravimetry (NETZSCH STA 449C Jupiter). Powder XRD (Bruker XS D8 Focus, 40 kV and 40 mA, Cu K) was conducted on calcined samples after heat treatment at 1000 °C. Phase composition and quantification was studied using Rietveld refinement analysis with the TOPAS 4.2 software (Bruker AXS, Germany). Mechanical properties of beads in water were measured on a modified micromanipulation technique as described by Zhang et al.40–41 Single beads with a diameter around 420–480 μm, were compressed at a speed of 240 μm s−1 up to a deformation of 50% and then held for 10 s. The force being imposed on the beads was recorded by a transducer. For each type of sample, ten beads were measured and force-time data and force-displacement data were obtained. Hertz theory was used to describe the relation between the force F, and displacement Δ (up to 30% deformation), according to the following equation:where R is the radius of the bead, E is the apparent Young's modulus and υ is the Poisson's ratio. υ = 0.5 was used according to previous reports.40–41
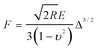
Results
3.1 Enzyme-mediated mineralization in alginate beads
In this work, Alg/CP composite beads were prepared through encapsulation of ALP in alginate gel beads, followed by enzymatic hydrolysis of β-glycerophosphate to supply phosphate selectively within the gel beads, and finally precipitation of CP within the beads. The distributions of ALP and Ca-minerals were visualized by CLSM. Presence of ALP within gel beads was confirmed by fluorescently labeled ALP after 24 h (Fig. 1, A). Produced CP was visualized with calcium-calcein staining of mineral phase.42Alg/CP–ALP showed a relatively uniform distribution of Ca-minerals throughout the beads (Fig. 1, B). By comparison, the counter-diffusion precipitation method (see 2.2) resulted in an inhomogenous distribution of CP (Fig. 1, C). Analysis of confocal z-stacks showed a similar distribution of ALP and Ca-minerals in the z-direction as in the xy-plane. | ||
| Fig. 1 CLSM micrographs showing the distribution of ALP (A) and Ca-minerals (B, C) in alginate beads; (A, B) Alg/CP–ALP; (C) Alg/CP–CD. | ||
3.2 Morphology and microstructure of Alg/CP composite beads
The morphology of beads was characterised by light microscopy (LM) and SEM. From LM studies, mineralized beads appear dark under transmitted light, while non-mineralized beads are transparent (see Fig. 1 in ESI). The darker the bead is, the more minerals have been produced. Alg/CP–ALP cell beads appeared the darkest under LM. Unlike Alg/CP–CD, Alg/CP–ALP and Alg/CP–ALP cell beads were less mineralized on the surfaces. The dark areas were more concentrated in the centre, indicating a different distribution of minerals compared to Alg/CP–CD.Microstructures of non-mineralized Ca–alginate beads have been studied before, showing smooth surfaces and porous internal gel networks.31 In the previous work, mineralized alginate beads were made by counter-diffusion precipitation. It has been found that large crystals are more likely to form on beads surface with increased ratio of phosphate precursor concentration to CaCl2 concentration. Deformed beads with unstable networks were obtained if CaCl2 concentration was too low to provide calcium ions for a rapid gel network formation in addition to calcium phosphate precipitation. In this work, 100 mM mineral precursor and 300 mM CaCl2 were chosen to produce stable gel beads. Alg/CP–CD beads showed relatively rough surfaces due to the big crystals on their surfaces (Fig. 2, A left). In comparison, Alg/CP–ALP beads presented smooth surfaces with fewer and smaller crystals on surfaces (Fig. 2, B left). More crystals on the surface can be observed once the concentration of ALP increased from 0.1 mg mL−1 to 0.5 mg mL−1 (see Fig. 2 in ESI). Beads made with 0.5 mg mL−1 ALP in hMSC medium had wrinkled surfaces which were full of CP nanogranules (Fig. 2, C left). The cross-sections of Alg/CP–CD beads showed CP minerals in plate-like morphology which were dispersed in the gel networks (Fig. 2, A right). Alg/CP–ALP bead sections, unlike Alg/CP–CD, exhibited mineral domains in bundles with needle-like morphology (Fig. 2, B right). When hMSC medium was employed, round shaped nanogranules were observed within gel networks (Fig. 2, C right) which looked similar to those observed on the beads surfaces.
 | ||
| Fig. 2 SEM micrographs of Alg/CP composite beads, surfaces (left) and internal structures (right): (A) Alg/CP–CD; (B) Alg/CP–ALP; and (C) Alg/CP–ALP Cell. | ||
Because specimens placed in the SEM are examined under vacuum, they must first be thoroughly dried. Direct air-drying can result in considerable distortion of specimen shape due to the adverse effects of surface tension forces. Water has an extremely high surface tension (72.75 N m−2 at 20 °C) and the receding liquid boundary results in unacceptably high levels of drying artifact. To avoid the problem of drying artifacts, Critical Point Dryer was utilized as a drying technique. It achieves a phase change from liquid to dry gas without the effects of surface tension and is therefore suitable for delicate alginate gel beads. To examine the internal microstructures, thin sections from gel beads were freezing cut by microtome. Pre-cooled acetone was used as a media to freeze beads before cutting. It was not possible to avoid all artifacts, like change in observed pore-size and network density. However, these were minimized by optimizing the preparation procedures and drying techniques.
Critical point dried Alg/CP beads were heat-treated at 300 °C, to partly degrade the alginate matrices and obtain dispersed CP particles. CP minerals were retrieved and their morphology was characterized by bright-field STEM, as shown in Fig. 3. After heat treatment at 300 °C, Alg/CP–CD beads showed nanocrystals in granules with the grain size of about 50 nm or larger, mostly agglomerated (Fig. 3, A). Alg/CP–ALP beads presented irregular agglomerated crystals in needle-like morphology (Fig. 3, B, inset). While nano-rods with length of 50–100 nm and width of 10–20 nm were observed for Alg/CP–ALP Cell beads (Fig. 3, C).
 | ||
| Fig. 3 Bright-field STEM micrographs of Alg/CP composite beads after heat treatment at 300 °C: (A) Alg/CP–CD; (B) Alg/CP–ALP; (C) Alg/CP–ALP Cell. | ||
3.3 Thermal stability and CP content
Thermal stability and mineral content was studied by means of thermogravimetric (TG) analysis. Fig. 4 shows the TG curves of pure alginate and Alg/CP composites, as well as the curve for pure calcium phosphates prepared in absence of alginate. Ca–alginate beads exhibit three decomposition steps as described below.43 The mass loss in the first stage is due to the evaporation of adsorbed water up to 250 °C. The alginate decomposition begins around 250 °C and is completed below 450 °C. At 450 °C, the calcination of the Ca–alginate gels results in CaCO3, which decomposes to CaO above 600 °C. When heated up to 900 °C, pure calcium phosphates also show a weight loss in three stages. The first stage corresponds to the vaporization of surface adsorbed water, while the second is due to the vaporization of crystal water and the third stage is due to loss of carbonate incorporated in the calcium phosphate during synthesis.44 The determined final mass of prepared CP precipitates in absence of alginate was 87% of the starting dry mass. | ||
| Fig. 4 Thermogravimetric curves showing thermal decomposition of pure alginate gel beads, CP precipitates and Alg/CP composites: (A) was made with 0.1 mg mL−1 ALP and 5 mM β-glycerophosphate at RT with varying pH, reaction time and buffer strength; (B) was made through counter-diffusion precipitation with and without tris-buffer. Estimated mineral content are plotted with dotted lines (see Table 1 and 2 for quantitative results). | ||
The CP mineral content presented in as-prepared beads was estimated by combining TG curves of pure Ca–alginate and CP precipitates, according to the following formula. A fraction (N) of the curve for CP mineral f(CP) was added to a fraction (1–N) of curve for pure Ca–alginate f(Alg), and the resulting curve was plotted to match the curves recorded for mineralized beads by varying N. Then the fraction N, was accepted to be percentage of CP mineral in the dry mass (data given in Table 1 and 2).
| f(Alg/CP) = Nf(CP) + (1 − N)f(Alg) |
| pH | Ca2− (mM) | Time (h) | Temp. (°C) | Buffer Conc. (mM) | CP Content (% of dry mass) | HA (% of dry mass) | TCP (% of dry mass) | HA/TCP ratio |
|---|---|---|---|---|---|---|---|---|
| a Italic font indicates the sample Alg/CP–ALP and Alg/CP–ALP Cell. | ||||||||
| 9.0 | 25 | 6 | RT | 50 | 10.0 | 9.3 | 0.7 | 14.2 |
| 9.0 | 50 | 6 | RT | 50 | 10.8 | 10.2 | 0.6 | 15.8 |
| 9.0 | 50 | 24 | RT | 10 | 15.2 | — | — | — |
| 9.0 | 50 | 24 | RT | 50 | 30.4 | 28.8 | 1.6 | 18.0 |
| 9.0 | 50 | 24 | RT | 100 | 31.8 | — | — | — |
| 7.4 | 50 | 24 | RT | 10 | 15.0 | 13.2 | 1.8 | 7.3 |
| 7.4 | 50 | 24 | 37 | 10 | 17.0 | 15.4 | 1.6 | 9.4 |
| 7.4 | 15 | 48 | 37 | MSCGM | 27.4 | 26.2 | 1.2 | 24.6 |
The effect of pH, reaction time, temperature and buffer concentrations on the CP mineral content was studied. For ALP mineralization, CP content increased with increasing pH, reaction time and temperature (Fig. 4(A) and Table 1). High buffer concentration resulted in increased mineral content both for ALP mediated mineralization (CP content up to 31.8% of the dry mass) and counter-diffusion precipitation (CP content up to 33.4% of the dry mass, Fig. 4(B) and Table 2). ALP/CP–ALP Cell beads showed mineral content of 27.4% in the dry mass after 48 h of mineralization.
3.4 Mineral phase composition in Alg/CP composite beads
X-ray diffraction and Rietveld refinement analysis were used to identify the resulting mineral phases of CP in alginate beads.31XRD patterns of the as-prepared samples for Alg/CP–CD, Alg/CP–ALP and Alg/CP–ALP Cell are shown in Fig. 5(A). As-prepared Alg/CP composites exhibited broad and overlaping diffraction peaks in the characteristic range of 2θ = 25–35° which can be indexed as HA phase (ICDD Card No.9–432). The peak broadening reflected the nanocrystalline nature and poor crystallinity of HA. Mineralized beads were heat-treated in air to improve the crystallinity. After heat treatment at 300 °C, the XRD patterns showed similar broad diffraction peaks for Alg/CP–CD, Alg/CP–ALP and Alg/CP–ALP Cell beads, indicating the presences of nanocrystalline HA. This is consistent with STEM results, showing nano-sized particles in the alginate matrices. | ||
| Fig. 5 XRD patterns recorded for alginate/CP composites: (A) mineralized beads before (as-prepared) and after heat treatment at 300 °C: a, Alg/CP–CD; b, Alg/CP–ALP Cell; c, Alg/CP–ALP. (B) Alg/CP–ALP, after heat treatment at 1000 °C. Black and red curves are raw- and refined data, respectively, while the blue curves show the difference between raw- and calculated data, Rwp (%) is 4.77, and GOF is 1.89. | ||
As the heat treatment temperature increased to 1000 °C, the intensity of HA characteristic peaks increased. The XRD patterns exhibited an increase in peak height and a decrease in peak width, thus indicating an increase in crystallinity and crystallite size. The most prominent characteristic peaks for HA, which corresponds to the plane (211), (300), and (202) were observed. However, heat treatment at 1000 °C resulted in partial decomposition of the HA phase to form tricalcium phosphate (β-TCP). Calcium oxide (CaO) was formed as a calcination product of Ca2+ ions cross-linking the alginate networks, which was not present in the as-prepared beads. Hence HA, β-TCP and CaO phases were identified as present phases in the calcined beads (Fig. 5, B). As can be seen, the most characteristic peaks of calcined Alg/CP–ALP corresponded to those of HA (JCPDS data file no.9–432). The β-TCP characteristic peaks could also be observed, but the intensities were far lower than those of HA. Rietveld refinement analysis further showed that calcined Alg/CP–ALP mainly composed of HA. The change of HA/β–TCP ratio is noticeable between different calcined mineralized beads which were made at different conditions. Alg/CP–ALP Cell beads yielded the highest HA/TCP ratio. For Alg/CP–ALP beads, HA/TCP ratio increased with pH value and temperature (Table 1). For Alg/CP–CD beads, higher HA/TCP ratio was obtained when tris-buffer was applied (Table 2).
3.5 Mechanical performance of Alg/CP composite beads
A micromanipulation technique40–41 was used for the mechanical characterization of selected alginate/CP composites, including Alg/CP–CD, Alg/CP–ALP and non-mineralized beads as a control. A single bead was compressed to approximately 50% deformation and held for 10 s, and the resulting force was recorded.Fig. 6 shows the typical force-time and force-displacement curves from the micromanipulation measurements. It has previously been shown that Hertz theory can describe the force-deformation behavior for deformations up to 30% for alginate hydrogel beads.41 Therefore, the Hertz approximation45 was applied to determine the apparent Young's moduli using data in the compressive deformation range of 30% (Table 3). The force-displacement curve will to a great extent depend on the speed of compression, because alginate hydrogels are viscoelastic. This can clearly be seen as a force-relaxation when the probe is held at constant position (Fig. 6A). Nevertheless, the calculated apparent Young's moduli can be used to compare different samples. Modeling of the viscoelastic behavior will be done for these beads elsewhere. The mineralized composite beads were found to be stiffer than the alginate hydrogel controls. Fig. 6 (B) shows that about twice the force was measured for the alginate/CP composites compared to the alginate controls at 30% deformation. In addition, ALP-mediated composites had steeper force-displacement curves than composites from counter-diffusion precipitation, which indicates that they have higher apparent Young's moduli. Neither the Alg/CP composites nor alginate controls ruptured with the applied force.
 | ||
| Fig. 6 (A) Typical curves showing force versus time for compression of beads at a speed of 240 μm s−1 with subsequent hold for 10 s. (B) Force-displacement data from alginate beads and Alg/CP composite beads (dot) fitted with model curves (line) based on the Hertz model (line) up to 30% deformation. These results are based on the mean value of ten beads for each sample. Sample types refer to Table 3. The error bars represent the standard deviation. | ||
| Samples | Mean diameter (μm) | Calculated apparent Young's modulus (kPa) |
|---|---|---|
| Alginate control | 420 ± 9 | 239 ± 27 |
| Alg/CP–CD | 487 ± 9 | 326 ± 49 |
| Alg/CP–ALP | 437 ± 4 | 475 ± 17 |
Discussion
The aim of this study was to develop biomimetic scaffolds by incorporating osteoinductive HA particles into highly porous and biocompatible alginate hydrogels. The combination of reinforcing and osteoinductive HA phase with biocompatible alginate hydrogels would produce a composite structure which possesses biological, morphological and mechanical characteristics required in tissue engineering. Previously our laboratory has developed a one-step method by counter-diffusion precipitation to fabricate nanostructured Alg/CP composites. It needs high concentration of CaCl2 to produce a stable gel network in addition to form a certain amount of CP minerals inside gel beads. This is due to the fact that calcium is consumed both by alginate gelation and mineral formation. For cell encapsulation, mineralization using the counter-diffusion method requires short (15 min) exposure of the cells to relatively high calcium and phosphate concentrations (∼300 mM), comparing to physiological concentration in a range of 1–2.5 mM. Some cell lines, like C2C12 (mouse myoblast cell line) and hMSC (human mesenchymal stem cell) can survive under these conditions. However, for some cell lines, like osteoblasts this can negatively influence cell viability and as a consequence methods for HA mineralization without the use of high Ca2+ concentration could be beneficial. In this study, ALP was utilized to direct enzymatic HA formation in 3D alginate gel matrices. Concentrations of Ca2+ applied in ALP-mediated mineralization were 50 mM or lower. ALP gradually hydrolyzes β-glycerophosphate and the kinetic of hydrolysis can be controlled by choosing appropriate reaction conditions, such as pH, buffer and temperature etc. The key role of ALP is to provide regulation of free phosphate concentration, preventing rapid, uncontrolled mineralization in the alginate matrices. This process allows calcium, phosphate precursor and ALP (Fig. 1, A) to be uniformly distributed throughout the alginate matrix, prior to mineralization. As a consequence, more uniform distribution of CP was achieved through an enzymatic mineralization process (Fig. 1, B) comparing to inhomogenous CP distribution resulting from counter-diffusion precipitation (Fig. 1, C). Moreover, counter-diffusion precipitation generally creates a concentration gradient due to the nature of diffusion in a gel system. It is more likely to form crystals on the bead surface rather than inside, which have been observed both in the LM and SEM images (Fig. 2, A, also see Fig. 1 in the ESI†). Although magnified SEM images in some cases have been used to reveal distributions of HA,46 however mineral distributions can hardly be described when composites have low mineral content. It is an advantage of using CLSM as a tool to visualize the inorganic/organic distributions throughout whole composite beads.47 Studies have shown that concentration of HA affects differentiation and fate of human MSC, where high HA contents show significantly higher extracellular matrix deposition and direct the stem cells towards an osteogenic differentiation pathway.48 A higher CP content was obtained by counter-diffusion precipitation (33.4% of dry mass) than by ALP-mediated mineralization (15.2% of dry mass); however, the more internal CP content and homogenous distribution make the minerals more available for encapsulated cells and makes cell-mineral interactions more likely.As the name suggests, ALP is most effective in an alkaline environment. High pH values have been found necessary for optimal activity of ALP in vitro. However, the ALP also works with a decreased activity in a broad range of pH-values. The pH-optimum for the ALP used in this study is 10.0 at RT and 9.8 at 37 °C49 which is far from physiological pH. This definite difference between the pH required for optimal activity and pH of living cells indicates that ALP is not optimally active in vivo. In addition to the pH, variation of other parameters such as temperature, ionic concentrations, the amount of enzyme, and the buffer employed directly influence the rate of phosphate release.49–50 Therefore, in this work all these parameters were studied systematically to optimize and control the fabrication process. It was found that the resulting minerals depend on the concentrations of ALP, β-glycerophosphate, Ca2+, as well as reaction time, pH, buffer concentration, and temperature. Higher concentration of ALP and β-glycerophosphate accelerated the precipitation and growth of CP minerals. Alkaline pH resulted in higher CP content compared with physiological pH. However, the efficiency of ALP can be enhanced by increasing reaction temperature and buffer concentration. Due to the enzymatic hydrolysis of the phosphate ester bond in β-glycerophosphate, a decrease of the pH-value appears in the case of calcium phosphate precipitation.49 Thus, a buffer was employed to prevent the decrease in pH during CP precipitation and resulted in more CP content in the beads.
S(T)EM revealed that nano-sized mineral particles were obtained within the alginate matrices. The nanocrystalline nature of as-prepared minerals was also reflected by broadening peaks in the XRD patterns. Our previous studies have shown that the alginate matrices play an important role in controlling crystal size and polymorphism.31,51–52 However, ALP-directed crystallization in solution without the present of organic matrices generally results in μm-sized crystals.49 Crystal morphological differences were obtained by using different mineralization methods. CP crystals shown as three different morphologies in plate-like, needle-like and spherical granules were observed by SEM. After heat treatment at 300 °C, alginate matrices were depolymerized and CP minerals were detached from gel networks. Nanocrystalline HA were still present after being heat-treated at 300 °C. XRD patterns showed broad diffraction peaks corresponding to HA phase. Nano-sized crystal particles were observed by STEM. After calcination at 1000 °C, the XRD patterns of calcined beads exhibited an increase in the intensity of HA characteristic peaks with sharp and narrow peaks, indicating increased crystallinity and crystallite size. HA was identified as the major mineral phase. A minor amount of β-TCP at 1000 °C may be associated with the partial decomposition of HA phase. It has been reported that the thermal stability of HA strongly depends on the Ca/P molar ratio. Stoichiometric HA has a Ca/P ratio of 1.67, which is thermally stable up to 1000 °C. HA with a Ca/P ratio of 1.67-1.5, called calcium deficient hydroxyapatite (CDHA), has the same crystal structure as HA but its decomposition temperature is lower than that of stoichiometric HA. Zhou et al.53 reported that synthetic HA with a Ca/P ratio near to 1.67 was stable below 1200 °C and decomposed to form TCP when heat-treated at temperatures above 1200 °C. Aoki54 heated nine kinds of vertebrate animal bones to 800 °C and found that calcined bones were mainly composed of HA. At temperatures above 1000 °C, bones were observed to decompose into β-TCP. Sintered bovine bone contained about 93 wt% of HA and 7 wt% of β-TCP.55
The Ca/P molar ratio of resulting minerals can be controlled by adjusting pH, temperature, initial [Ca2+]as well as buffer medium. Various Ca/P molar ratios were obtained at different mineralization conditions. The difference in Ca/P is dependent on the composition change between the two phases from [Ca/P]HA =1.67 to [Ca/P]TCP =1.5. The HA/TCP ratio at 1000 °C apparently increased with increasing pH, temperature and initial [Ca2+], indicating higher Ca/P ratio of HA was obtained. Applied buffer played a role in maintaining the pH value. Soluble phosphate ions concentration was highly dependent on pH-value and Ca concentration. The available phosphate ions decreased at higher pH and Ca concentration, and hence favored the formation of HA with high Ca/P ratio. Calcined Alg/CP–ALP Cell beads at 1000 °C resulted in mineral phases consisting of about 96 wt% of HA and 4 wt% of β-TCP, which is similar to natural bones.
Mechanical properties of tissue engineering scaffolds are vital to ensure long-term structural and functional stability in vivo. Mechanical performance of Alg/CP composites has been enhanced by the CP minerals inside these beads. Apparent Young's moduli increased to almost twice that of alginate controls. Tested Alg/CP–ALP beads contained less mineral phase (CP content 15.2% of dry mass) than Alg/CP–CD beads (CP content 33.4% of dry mass). However, their apparent Young's moduli were 45% larger (475 kPa) than those of Alg/CP–CD beads (326 kPa). This can be explained by reinforcing effect from homogenous distribution of CP crystals within the composite material. Closer interaction between CP and alginate gel network might also have a contribution to the improved stiffness of the ALP mineralized samples. Thus, the enhanced stiffness can be attributed to a better microscopic organization of this biocomposite. It has been reported that 3D bone-like constructs from the osteogenic differentiation of encapsulated mESCs in alginate hydrogels displayed an over 10 fold higher Young's modulus (666.2 kPa) than the typical 3D porous PLGA scaffold (55.4 kPa).56 The apparent Young's moduli of Alg/CP composites fabricated in our study are close to the values of 3D bone-like constructs reported in that study.
The incorporation of HA into alginate-based scaffolds is advantageous not only in terms of reinforcing scaffold structures but also in facilitating cell adhesion and promoting osteogenesis. Our preliminary results show that the methods described in this work have good cell compatibility. Human mesenchymal stem cells derived from bone marrow (hBMSCs) were encapsulated successfully in mineralized alginate gel beads at a density of 2 × 106cells ml−1 and maintained high cell viability post encapsulation for 21 days. The enzyme mediated mineralization provided more cell-friendly conditions, compared to the counter diffusion approach. By using ALP, the beads were rapidly and heavily mineralized appearing dark in the light microscope at day 2 post encapsulation. After the first 48 h of cultivation in mineralization medium, the cells in beads were cultured in regular medium for up to 21 days. An addition of CaCl2 (15 mM) to the regular growth medium is needed to preserve the beads stability.
Conclusions
In conclusion, a biomimetic Alg/CP scaffold has been developed through a cell-friendly pathway. Enzymatic CP mineralization was directed by ALP in alginate gel matrices, presenting advantages over the counter-diffusion precipitation method. We have demonstrated that gradually released phosphates combined with alginate gel networks can lead to formation of nano-sized HA in a 3D environment. Mineral morphology, mineral content, and Ca/P molar ratio of HA can be controlled through reaction conditions, which are compatible with cell culture. The ALP-mediated composite scaffolds have a homogenous CP mineral distribution. They also exhibit increased stiffness due to the addition of an osteoinductive HA phase and a well-defined microstructural organization. This scaffold is comprised of bone's natural constituent minerals, ensuring non-toxic degradation by-products, biocompatibility and the potential to facilitate and promote osteogenesisin vivo. Future work that incorporates stem cells into Alg/CP composite beads may offer interesting strategies in the design of advanced bone regeneration materials.Acknowledgements
We acknowledge the financial support by The Research Council of Norway (NANOMAT program, NO. 182047) and Helse Midt-Norge (BLS). We would also like to thank Doctor Therese Standal (Department of Cancer Research and Molecular Biology, NTNU) for her knowledge and guidance in working with mesenchymal stem cells and Henriette Elisabeth Myhr Sætrang who contributed to parts of this paper with her project work.References
- S. C. Cowin and S. B. Doty, Tissue mechanics, Springer, New York, 2007 Search PubMed.
- R. K. Roeder, G. L. Converse, R. J. Kane and W. M. Yue, JOM, 2008, 60, 38–45 CrossRef CAS.
- F. Nudelman, K. Pieterse, A. George, P. H. H. Bomans, H. Friedrich, L. J. Brylka, P. A. J. Hilbers, G. de With and N. A. J. M. Sommerdijk, Nat. Mater., 2010, 9, 1004–1009 CrossRef CAS.
- H. Colfen, Nat. Mater., 2010, 9, 960–961 CrossRef.
- G. G. d'Ayala, M. Malinconico and P. Laurienzo, Molecules, 2008, 13, 2069–2106 Search PubMed.
- J. M. Guisan, Immobilization of enzymes and cells, 2nd edn, Humana Press, Totowa, N.J., 2006 Search PubMed.
- Y. A. Morch, I. Donati, B. L. Strand and G. Skjak-Braek, Biomacromolecules, 2007, 8, 2809–2814 Search PubMed.
- Y. A. Morch, S. Holtan, I. Donati, B. L. Strand and G. Skjak-Braek, Biomacromolecules, 2008, 9, 2360–2368 CrossRef CAS.
- D. W. Green, S. Mann and R. O. C. Oreffo, Soft Matter, 2006, 2, 732–737 RSC.
- A. M. Smith, M. R. Jaime-Fonseca, L. M. Grover and S. Bakalis, J. Agric. Food Chem., 2010, 58, 4719–4724 Search PubMed.
- C. M. Bunger, B. Tiefenbach, A. Jahnke, C. Gerlach, T. Freier, K. P. Schmitz, U. T. Hopt, W. Schareck, E. Klar and P. de Vos, Biomaterials, 2005, 26, 2353–2360 Search PubMed.
- J. S. Yu, K. T. Du, Q. Z. Fang, Y. P. Gu, S. S. Mihardja, R. E. Sievers, J. C. Wu and R. J. Lee, Biomaterials, 2010, 31, 7012–7020 CrossRef CAS.
- A. Goren, N. Dahan, E. Goren, L. Baruch and M. Machluf, FASEB J., 2010, 24, 22–31 Search PubMed.
- I. Ghidoni, T. Chlapanidas, M. Bucco, F. Crovato, M. Marazzi, D. Vigo, M. L. Torre and M. Faustini, Cytotechnology, 2008, 58, 49–56 Search PubMed.
- S. J. Bidarra, C. C. Barrias, M. A. Barbosa, R. Soares and P. L. Granja, Biomacromolecules, 2010, 11, 1956–1964 CrossRef CAS.
- R. M. Hernandez, G. Orive, A. Murua and J. L. Pedraz, Adv. Drug Delivery Rev., 2010, 62, 711–730 CrossRef CAS.
- M. Endres, N. Wenda, H. Woehlecke, K. Neumann, J. Ringe, C. Erggelet, D. Lerche and C. Kaps, Acta Biomater., 2010, 6, 436–444 Search PubMed.
- P. X. Ma and J. H. Elisseeff, Scaffolding in tissue engineering, Taylor&Francis, Boca Raton, 2005 Search PubMed.
- R. Dittrich, G. Tomandl, F. Despang, A. Bernhardt, T. Hanke, W. Pompe and M. Gelinsky, J. Am. Ceram. Soc., 2007, 90, 1703–1708 Search PubMed.
- A. Bernhardt, F. Despang, A. Lode, A. Demmler, T. Hanke and M. Gelinsky, J. Tissue Eng. Regener. Med., 2009, 3, 54–62 Search PubMed.
- G. P. Chen, T. Ushida and T. Tateishi, Macromol. Biosci., 2002, 2, 67–77 CrossRef CAS.
- G. L. Converse, T. L. Conrad and R. K. Roeder, J. Mech. Behav. Biomed. Mater., 2009, 2, 627–635 Search PubMed.
- P. K. D. V. Yarlagadda, M. Chandrasekharan and J. Y. M. Shyan, Bio-Med Mater Eng, 2005, 15, 159–177 Search PubMed.
- H. R. Lin and Y. J. Yeh, J. Biomed. Mater. Res., 2004, 71B, 52–65 Search PubMed.
- L. Wang, Y. Li and C. Z. Li, J. Nanopart. Res., 2009, 11, 691–699 CrossRef CAS.
- S. H. Teng, L. J. Chen, Y. C. Guo and J. J. Shi, J. Inorg. Biochem., 2007, 101, 686–691 Search PubMed.
- S. H. Teng, J. J. Shi and L. J. Chen, Colloids Surf., B, 2006, 49, 87–92 Search PubMed.
- I. Leveque, K. H. Rhodes and S. Mann, J. Mater. Chem., 2002, 12, 2178–2180 RSC.
- I. Yamaguchi, K. Tokuchi, H. Fukuzaki, Y. Koyama, K. Takakuda, H. Monma and T. Tanaka, J. Biomed. Mater. Res., 2001, 55, 20–27 CrossRef CAS.
- M. Sivakumar and K. P. Rao, Biomaterials, 2002, 23, 3175–3181 CrossRef CAS.
- M. L. Xie, M. O. Olderoy, J. P. Andreassen, S. M. Selbach, B. L. Strand and P. Sikorski, Acta Biomater., 2010, 6, 3665–3675 Search PubMed.
- X. Liu, L. A. Smith, J. Hu and P. X. Ma, Biomaterials, 2009, 30, 2252–2258 CrossRef CAS.
- T. Kokubo, M. Hanakawa, M. Kawashita, M. Minoda, T. Beppu, T. Miyamoto and T. Nakamura, J. Mater. Sci.: Mater. Med., 2004, 15, 1007–1012 Search PubMed.
- E. Banks, S. Nakajima, I. C. Shapiro, O. Tilevitz, J. R. Alonzo and R. R. Chianelli, Science, 1977, 198, 1164–1166 Search PubMed.
- H. Unuma, Int. J. Appl. Ceram. Technol., 2007, 4, 14–21 Search PubMed.
- K. Yamauchi, T. Goda, N. Takeuchi, H. Einaga and T. Tanabe, Biomaterials, 2004, 25, 5481–5489 Search PubMed.
- O. Tomomatsu, A. Tachibana, K. Yamauchi and T. Tanabe, J. Ceram. Soc. Jpn., 2008, 116, 10–13 Search PubMed.
- Y. Doi, T. Horiguchi, Y. Moriwaki, H. Kitago, T. Kajimoto and Y. Iwayama, J. Biomed. Mater. Res., 1996, 31, 43–49 CrossRef CAS.
- E. D. Spoerke, S. G. Anthony and S. I. Stupp, Adv. Mater., 2009, 21, 425 CrossRef CAS.
- Z. Zhang, R. Saunders and C. R. Thomas, J. Microencapsulation, 1999, 16, 117–124 CrossRef CAS.
- C. Wang, C. Cowen, Z. Zhang and C. R. Thomas, Chem. Eng. Sci., 2005, 60, 6649–6657 Search PubMed.
- J. f Bandrows and C. L. Benson, Clin Chem, 1972, 18, 1411 Search PubMed.
- R. Horga, F. Di Renzo and F. Quignard, Appl. Catal., A, 2007, 325, 251–255 CrossRef CAS.
- A. L. Daniel-da-Silva, A. B. Lopes, A. M. Gil and R. N. Correia, J. Mater. Sci., 2007, 42, 8581–8591 Search PubMed.
- Y. Yan, Z. B. Zhang, J. R. Stokes, Q. Z. Zhou, G. H. Ma and M. J. Adams, Powder Technol., 2009, 192, 122–130 CrossRef CAS.
- H. G. Zhao, L. Ma, C. Y. Gao and J. C. Shen, Polym. Adv. Technol., 2008, 19, 1590–1596 Search PubMed.
- B. L. Strand, Y. A. Morch, T. Espevik and G. Skjak-Braek, Biotechnol. Bioeng., 2003, 82, 386–394 CrossRef CAS.
- S. P. Kwei, K. L. Moffat, S. Doty and H. H. Lu, Bioengineering Conference, Proceedings of the 2010 IEEE 36th Annual Northeast, 2010 Search PubMed.
- C. Hoffmann, C. Zollfrank and G. Ziegler, J. Mater. Sci.: Mater. Med., 2008, 19, 907–915 CrossRef CAS.
- M. H. Ross, J. O. Ely and J. G. Archer, J Biol Chem, 1951, 192, 561–568 Search PubMed.
- M. O. Olderoy, M. L. Xie, B. L. Strand, K. I. Draget, P. Sikorski and J. P. Andreassen, Cryst. Growth Des., 2011, 11, 520–529 Search PubMed.
- M. O. Olderoy, M. L. Xie, B. L. Strand, E. M. Flaten, P. Sikorski and J. P. Andreassen, Cryst. Growth Des., 2009, 9, 5176–5183 Search PubMed.
- J. M. Zhou, X. D. Zhang, J. Y. Chen, S. X. Zeng and K. Degroot, J. Mater. Sci.: Mater. Med., 1993, 4, 83–85 Search PubMed.
- F. Tavangarian, R. Emadi and S. I. R. Esfahani, Mater. Lett., 2010, 64, 993–996 Search PubMed.
- S. Ramesh, C. Y. Ooi and M. Hamdi, Ceram. Int., 2007, 33, 1171–1177 Search PubMed.
- Y. S. Hwang, J. Cho, F. Tay, J. Y. Y. Heng, R. Ho, S. G. Kazarian, D. R. Williams, A. R. Boccaccini, J. M. Polak and A. Mantalaris, Biomaterials, 2009, 30, 499–507 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: LM, SEM images and XRD refinement data for Alg/CP composite beads. See DOI: 10.1039/c1ra00750e/ |
| This journal is © The Royal Society of Chemistry 2012 |