Application of stable fused dienolates for diversity oriented synthesis of 2,5-dioxo-5,6,7,8-tetrahydro-2H-chromene-3-carboxamides†
Maria A.
Vodolazhenko
,
Nikolay Yu.
Gorobets
*,
Sergey A.
Yermolayev
,
Vladimir V.
Musatov
,
Valentin A.
Chebanov
* and
Sergey M.
Desenko
Department of Chemistry of Heterocyclic Compounds, SSI “Institute for Single Crystals” of National Academy of Sciences of Ukraine, Lenin Avenue 60, Kharkiv, 61001, Ukraine. E-mail: gorobets@isc.kharkov.com; chebanov@isc.kharkov.com; Fax: +38 (057) 340 9343; Tel: +38 057 341 0256
First published on 2nd December 2011
Abstract
A novel one-pot three-step method for the synthesis of 2,5-dioxo-5,6,7,8-tetrahydro-2H-chromene-3-carboxamide derivatives via hydrolysis of 4-cyanobuta-1,3-dienolate salts was developed. These chromenes contain a 2-pyrone scaffold which is present in many well-known natural and synthetic biologically active compounds. Employing our protocol, a 55 compounds library was quickly generated from 1,3-cyclohexanediones, dimethylformamide dimethylacetal, and various N-substituted cyanacetamides.
Introduction
Multi-component and one-pot reactions are widely applied in modern strategy towards diversity of drug-like small molecules.1–4 Among such methods the application of highly-functionalized reactants in this approach is a challenging but attractive task. Since several undefended functional groups are present in one reacting molecule, a simultaneous formation of different products can be generally expected. Fortunately in some cases it is possible to control the pathways and selectivity of the polyfunctionalized molecule transformations using optimized reaction conditions.5–15 In this way starting from one set of reactants it becomes possible to obtain several chemotypes of individual reaction products that may also contain functionalities opened for additional transformations.Previously our group introduced16 an efficient synthesis of polyfunctionalized fused dienolates 1 by a one-pot three component reactions of 1,3-cyclohexanediones with DMFDMA and active methylene nitriles. The salts 1 are reactive intermediates in the synthesis of various heterocyclic compounds16–20 where different functional groups are engaged in the heterocyclization depending on the reaction conditions applied (Scheme 1). In most cases where salts 1 are derived from cyclic 1,3-diketones they are stable enough to be isolated in individual states16,17 and used as starting materials, which is suitable for a preliminary optimization of the reaction conditions. However due to the high yields for the synthesis of these salts 1 it is possible to use them in the next reaction in one-pot fashion. Salts 1 are synthetic precursors of six member heterocyclic compounds because they contain an unsaturated chain of four carbon atoms with terminal nitrile and amide/hydrazide groups. Both terminal groups can react with enole functions to form a six-membered cycle. The nitrile group reacts under evaluated temperature resulting in the formation of different 2-pyridone derivatives e.g.519 and 6.17 Switching between the nitrile and the amide groups is controlled by the basicity of the reaction media and in the presence of piperidine excess the cyclization proceeds by the amide group. However, this competition is dependent also on the nucleophilicity of the R3-function. Thus, the type 6 compounds with hydrazide groups in the third position can be obtained only as admixture due to the high reactivity of the hydrazide function20 stipulating predominance of the reaction pathways (c) and (f). Also the earlier described17 2-benzimidazolyl compound 8 is formed under all types of reaction conditions indicated in Scheme 1. At the same time the reactions of salts 1 with N-nucleophiles generally proceed in acetic acid under mild conditions to give 9 and 10.16,19,20 In the case of hydrazide substituent for the salts 1 a new type of heterocyclic system 11 is formed in acetic acid.20
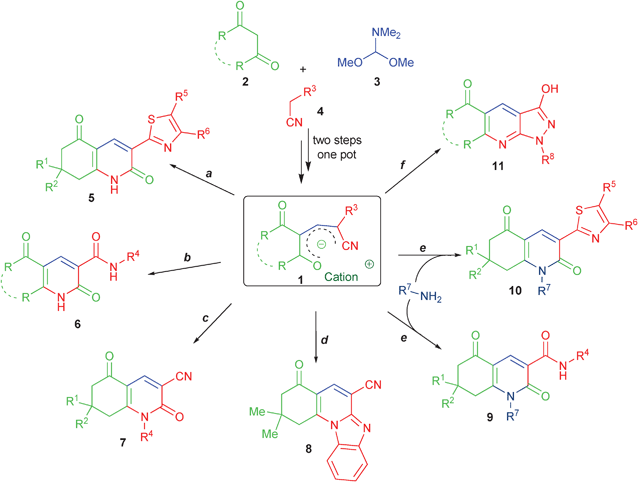 | ||
| Scheme 1 Reagents and conditions: (a) n-BuOH Δ, 10 min or i-PrOH, MW 100 °C/5 min; (b) i-PrOH, MW 100 °C/5 min; (c) for R4 = Ar, Het: piperidine, H2O, MW, 120 °C/10 min, for R4 = NH-Ar: H2O, Δ, 5 min; (d) i-PrOH, MW 100 °C/5 min or other (see text for details); (e) AcOH, rt, 5 min; (f) for R4 = NH-Ar: AcOH, rt, 10–15 min | ||
Interestingly, beside the electron withdrawing groups conjugated with the enolate,16 such salts are probably stabilized also by their cyclic chain fragment. Attempts to synthesize enolates starting from acyclic carbonyl compounds 12 were not successful.20 According to the data obtained by Mosti's group21 and the next following reports,22–24 the reaction in this case is accomplished by formation of amides 15 probably via a ring opening of the intermediate iminopyrane 14 (Scheme 2). The reason for the higher stability of the cyclic enolates lies probably in the higher acidity of the cyclic 1,3-dicarbonyl compounds as compared to their acyclic analogs.25,26
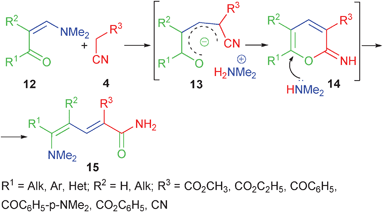 | ||
| Scheme 2 Transformation of enolates 13 without cyclic fragment. | ||
Here, we are intrigued by the possibility to apply the reactivity of the salts 1 in their acid catalyzed hydrolysis into the corresponding 2-pyrone derivatives.
One publication27 was found in the literature concerning the preparative synthesis of 2-pyrones 18 using similar enolates 17. Wolfbeis et al. described a stepwise synthesis of 5-oxo-5,6,7,8-tetrahydrocoumarins 18 using the condensation of a 1,3-cyclohexanediones 15 with triethoxymethane and urea derivatives to afford 2-ureidomethylenecyclohexane-1,3-diones 16 (Scheme 3). The former reacted with activated acetonitriles 4 in the presence of a strong base giving the desired 5-oxo-5,6,7,8-tetrahydrocoumarins 18 after acidic aqueous work-up. However this described approach can hardly be applied for an efficient diversity oriented synthesis of 5-oxo-5,6,7,8- tetrahydrocoumarins 18 because of its low molecular efficiency and the necessity to isolate the intermediate enamines 16. Mosti's group has also obtained a few of such 2-pyrone derivates as admixtures.28
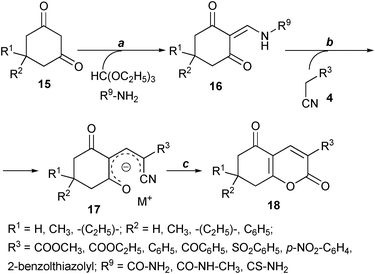 | ||
| Scheme 3 Reagents and conditions: (a) glacial CH3COOH, 90 °C, 3 h or i-PrOH, 80 °C, 0.5–3 h; (b) DMF, base; (c) HCl, H2O | ||
Here we aimed to combine our method for the synthesis of salts 1 with their acidic hydrolysis suggested by Wolfbeis et al. and develop a diversity oriented synthesis of 2,5-dioxo-5,6,7,8-tetrahydro-2H-chromene-3-carboxamide library 22.
The 2-pyrone ring is a fundamental structural motif of natural29–36 and synthetic31,37,38 biologically active compounds which action is varied in a broad range from toxins and pesticides,37 teratogens and carcinogens30 to those which shows analgesic, mild sedative, spasmolythic, muscle relaxant,37 antibacterial29,31,37 or antioxidant31 activity. Some of them can be applied in cancer29,30 or Alzheimer's disease29 treatments, known as HIV inhibitors,29 and can also induce apoptosis via caspase activation.32–36,38 However any data on biological activity of direct analogs of 2,5-dioxo-5,6,7,8-tetrahydro-2H-chromene-3-carboxamide library 22 is absent in the literature.
Results and discussion
Initially, the key intermediate enolate salts 21 are easily obtained by our previously described procedure16 by the one-pot subsequent reaction of 1,3-diones 15, dimethylformamide dimethylacetal (DMFDMA) 3 and various N-substituted cyanacetamides 20 in i-PrOH in the presence of excess base at room temperature (Scheme 4). At the same time, the following hydrolysis suggests application of aqueous HCl. Thus the optimal concentration of aqueous HCl had to be determined in our first optimization step. In the initial experiments we used dimedone 15{1} and unsubstituted cyanacetamide 20{1} as representatives of the CH-acid building blocks and active methylene nitriles correspondingly. The salt 21{1,1} was obtained by the stirring of equimolar amounts of neat dimedone 15{1} and DMFDMA 3 during 5 min at room temperature followed by the reaction with 1.0 equiv of nitrile 20{1} in different organic solvents (i-PrOH, EtOH, MeCN, DMF) at room temperature for 30 min. The obtained salt 21{1,1} was treated without isolation with aqueous HCl of different concentrations. The reactions were monitored by 1H NMR of the products isolated after an aqueous work-up. The best result was obtained applying MeCN as a reaction medium at room temperature for both steps of the synthesis of salt 21{1,1} and its hydrolysis using 18% HCl. The yield for the obtained product 22{1,1} was 60% (Method A).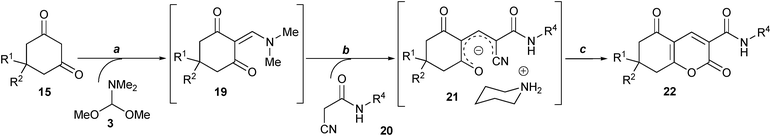 | ||
| Scheme 4 Reagents and conditions: (a) r.t., 5 min.; (b) 0 °C, CH3CN, 2 equiv. piperidine, 1 h; (c) 8 equiv. HCl (18%), 0.5 h; H2O, 0.5 h | ||
For the library generation we used 1,3-cyclohexandiones 15{1-6} bearing one diversity point (Table 1) and more diverse N-substituted cyanacetamides 20{1-36} derived from various benzyl- or (hetero)aromatic amines (Table 2) to obtain the piperidinium salts 21 which were used for further hydrolysis. For all the reactions three steps were done in a one pot parallel format.
| N° | R4 | N° | R4 | N° | R4 |
|---|---|---|---|---|---|
| 20{1} | H | 20{13} |
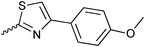
|
20{25} | 4-Cl-C6H4-CH2 |
| 20{2} | 2,5-di-Me-C6H3 | 20{14} |
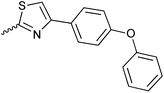
|
20{26} | 4-Me-C6H4-CH2 |
| 20{3} | 3-Me-C6H4 | 20{15} |
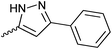
|
20{27} | 3-Br-2-F-C6H3 |
| 20{4} |
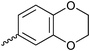
|
20{16} | 4-HO-C6H4 | 20{28} | 4-HOOC-CH2-C6H4 |
| 20{5} | 2-Et-C6H4 | 20{17} |
|
20{29} | 2,4-di-Me-C6H3 |
| 20{6} | 4-CF3O-C6H4 | 20{18} | 3-HOOC-C6H4 | 20{30} | 2,4-di-MeO-C6H3 |
| 20{7} | 2-EtO-C6H4 | 20{19} |
|
20{31} | 4-CF3-C6H4 |
| 20{8} | 3,5-di-Me-C6H3 | 20{20} | 4-EtOOC-C6H4 | 20{32} |
|
| 20{9} | 4-MeCO-C6H4 | 20{21} | 4-NH2CO-C6H4 | 20{33} | 2,5-di-Me-4-Cl-C6H2 |
| 20{10} | 2,5-di-MeO-4-Cl-C6H2 | 20{22} | 4-EtO-C6H4 | 20{34} | 3,4-di-MeO-C6H3 |
| 20{11} | 2,5-di-Cl-C6H3 | 20{23} | 4-MeO-C6H4-CH2 | 20{35} | 2,4-di-Me-C6H3 |
| 20{12} | 3-Me-4-F-C6H3 | 20{24} | 4-F-C6H4-CH2 | 20{36} | 4-Me-3-F-C6H3 |
However, application of the Method A for the library generation did not give a satisfactory result. More than 50% of the obtained products had the impurity level higher than 10% according to 1H NMR, and these final products needed additional purification steps to satisfy the purity requirements. And in worse cases the yields were very poor or the derivates of 2-pyrones were not isolated (Table 3, Method A, Entries 13, 16, 18, 25 and 26). The major impurity was found to be the corresponding 2-pyridone derivative of the type 6 (Scheme 1). In the cases of 22{5,4-5}, 22{5,8} the isolated product contained from 20% to 50% of this admixture. Previously we also noted17 that enolates 21 obtained from the parent cyclohexane-1,3-dione 15{5} are more inclined to the intermolecular cyclization into 2-pyridones 6 than the corresponding derivatives of dimedone 15{1}.
| Entry | Library Member | Isolated yield (%) | |
|---|---|---|---|
| Method A | Method B | ||
| 1 | 22{1,1} | 60 | 70 |
| 2 | 22{1,2} | 63 | 68 |
| 3 | 22{1,3} | 46 | 55 |
| 4 | 22{1,4} | 46 | 65 |
| 5 | 22{1,5} | 64 | 70 |
| 6 | 22{1,6} | 51 | 58 |
| 7 | 22{1,7} | 59 | 65 |
| 8 | 22{1,8} | 55 | 60 |
| 9 | 22{1,10} | 67 | 73 |
| 10 | 22{1,11} | 50 | 56 |
| 11 | 22{1,14} | 76 | 80 |
| 12 | 22{1,15} | 58 | 65 |
| 13 | 22{1,16} | 5 | 55 |
| 14 | 22{1,19} | 96 | 90 |
| 15 | 22{1,22} | 40 | 70 |
| 16 | 22{1,23} | 10 | 53 |
| 17 | 22{1,24} | 62 | |
| 18 | 22{1,26} | 17 | 46 |
| 19 | 22{1,28} | 45 | |
| 20 | 22{2,2} | 48 | 55 |
| 21 | 22{3,30} | 64 | 71 |
| 22 | 22{3,31} | 87 | 90 |
| 23 | 22{4,19} | 86 | 86 |
| 24 | 22{4,21} | 82 | 80 |
| 25 | 22{4,28} | 11 | 56 |
| 26 | 22{4,1} | 0 | 76 |
| 27 | 22{4,23} | 42 | |
| 28 | 22{4,16} | 65 | |
| 29 | 22{4,31} | 53 | |
| 30 | 22{5,21} | 61 | 65 |
| 31 | 22{5,4} | 46 | 45 |
| 32 | 22{5,8} | 50 | |
| 33 | 22{6,22} | 58 | |
| 34 | 22{6,24} | 50 | |
| 35 | 22{6,1} | 66 | |
To obtain purer products it was necessary to make an additional optimization step. Since the spontaneous formation of such 2-pyridones is promoted by higher temperature17 to avoid their formation in our final procedure we performed both the synthesis of salts 21 and their hydrolysis into 2-pyrones 22 at 0 °C. This allowed us to isolate the model product 22{1,1} in 70% yield without additional purification and any impurities according to 1H NMR data (see Scheme 4 for the final optimized conditions, Method B). Applying Method B we obtained the products 22 with higher yields and purity especially for cases where the yields were very poor using Method A (Table 3).
The purity of nearly 70% of the products isolated as precipitates directly for the reaction mixtures was higher than 90%, including the above mentioned difficult cases, and the other products had a purity higher than 80%. In some cases we found the corresponding 2-pyridone derivative of the type 6 (Scheme 1) as an impurity in products 22 but its amount was insignificant.
Carrying out the reactions using our final optimized conditions, we quickly generated a library of 55 representatives of chromenes 22{1-6,1-36} in parallel format with yields 42–96% (see a random sampling of the library members in Table 3). In those cases where the impurity level exceeded 10%, the compounds were recrystallized from a suitable solvent, EtOH or n-BuOH.
The structure and purity of all the library members were controlled by means of 1H NMR spectroscopy. The spectra do not contain exchangeable NH proton belonging to 2-pyridone cycle which is observed for compounds 6. In the cases when R1 and R2 are not equal, the appearance of a chiral center in the molecules leads to coupling of CH2 proton signals into complex multiplets. 13C NMR, mass spectra and elemental analysis measured for random representatives of the library do not contradict the proposed structures (see ESI for details)†.
Since the direct hydrolysis of a cyano group requires much tougher reaction conditions than the applied for the hydrolysis of salts 1, two reaction mechanisms can be considered for this process. Both pathways suggest sequential neutralization of the enolate, formation of an intermediate iminopyrane 23 and its protonation to form 2-aminopyrrilium cation 24. The former may undergo the attack of a water molecule resulting in the formation of tetrahydrocoumarin 22via sp3-hybridized intermediate 25. Such mechanism has been earlier determined for the hydrolysis of 2-iminocoumarin derivatives39 as direct analogs of our suggested intermediate 23. 2-Iminocoumarins are known to be quickly hydrolyzed in the presence of acids.40
Alternatively, water may attack the nodal carbon atom (same as HNMe2 in the Scheme 2) to give an open chain amide 26 that under acidic conditions may give the final product 22 (Scheme 5).
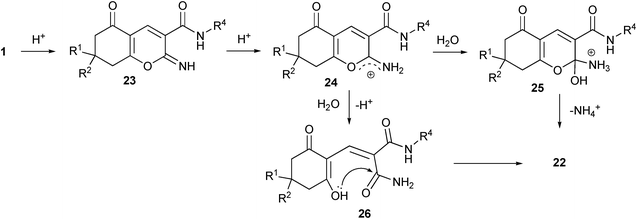 | ||
| Scheme 5 Two possible reaction mechanisms for hydrolysis of enolates 1. | ||
Experimental
General information
All solvents were used as acquired. Melting points were measured with a Buchi melting point apparatus and are uncorrected. 1H NMR spectra were recorded on a Varian Mercury VX 200 instrument using DMSO-d6 as solvent and TMS as an internal standard. 13C NMR experiments were performed using a Bruker AMX 500 spectrometer and Varian Mercury VX 200. IR spectra were recorded on a Perkin Elmer Spectrum One FTIR Spectrometer (pellets with potassium bromide KBr). The mass spectra were recorded on a Varian 1200 L GC-MS instrument with the use of direct exposure probe (DEP) method with EI at 70 eV. Elemental analysis was carried out on an EuroVector EA-3000 instrument.N1-Substituted 2-cyanoacetamides 20{1-36} were prepared by trans-cyanoacetylation of the corresponding amines with 1-cyanoacetyl-3,5-dimethylpyrazole in refluxing toluene following the known strategy.17,41 Dimedone 15{1} and 1,3-cyclohexadione 15{5} are commercially available compounds. Other substituted 1,3-cyclohexadiones 15{2-4, 6} were obtained using the general procedure described below.
General procedure for the preparation of substituted 1,3-cyclohexadiones 15{2-4, 6}42
Corresponding 4-arylbut-3-en-2-one (0.02 mol) was mixed with diethyl malonate 3.20 g (0.02 mol), potassium carbonate 1.8 g (0.013 mol) in 10 mL of ethanol. The mixture was refluxed for 15 h then cooled. The formed precipitate was filtered off and washed with water. Obtained crude ethyl 2,4-dioxo-6-arylcyclohexanecarboxylate was mixed with 20 mL of water and potassium carbonate 3 g (0.022 mol). The mixture was heated at 90 °C during 5 h. The obtained solution was cooled and hydrochloric acid was added to the solution till pH = 4–5 precipitating 2,4-dioxo-6-arylcyclohexanecarboxylic acid. The mixture was refluxed for 1 h then cooled. The precipitate of 2,4-dioxo-6-arylcyclohexane was filtered off, washed with water and dried. Overall yields calculated with respect to 4-arylbut-3-en-2-one: 25–30%.General procedure for the preparation of 2,5-dioxo-5,6,7,8-tetrahydro-2H-chromene-3-carboxamides 22{1-6,1-36}
Conclusions
In conclusion, we have developed a novel three-step one-pot protocol for the generation of 2,5-dioxo-5,6,7,8-tetrahydro-2H-chromene-3-carboxamide library 22 starting from 1,3-cyclohexanediones 15, DMFDMA 3 and N-substituted cyanacetamides 20via hydrolysis of enolate salts 21. This synthesis represents an additional instance of a controlled reaction pathways when the same reagents furnish different products dependently on the reaction conditions applied. Thus, the application of the N-substituted cyanoacetamide building blocks 20 in the reactions with enamines of type 19 allows the synthesis of several types of products: 3-quinolinecarbonitriles 7 (Scheme 1), 3-quinolinecarboamides 6 (Scheme 1), N1-substituted 3-quinolinecarboamides 9via reaction of the isolated intermediates 21 with N-nucleophiles (Scheme 1) and 3-chromenecarboxamides 22 (Scheme 4). The last reaction enlarges the diversity of synthetically available potentially bioactive 2-pyrone derivatives covering different parts of chemical space.References
- M. Colombo and I. Peretto, Drug Discovery Today, 2008, 13, 677–684 CrossRef CAS.
- C. Kalinski, M. Umkehrer, L. Weber, J. Kolb, C. Burdack and G. Ross, Mol. Diversity, 2010, 14, 513–522 CrossRef CAS.
- J. E. Biggs-Houck, A. Younai and J. T. Shaw, Curr. Opin. Chem. Biol., 2010, 14, 371–382 CrossRef CAS.
- C. Hulme and V. Gore, Curr. Med. Chem., 2003, 10, 51–80 CrossRef CAS.
- V. A. Chebanov, K. A. Gura and S. M. Desenko, Top. Heterocycl. Chem., 2010, 23, 41–84 CrossRef CAS.
- N. Elders, E. Ruijter, F. J. J. de Kanter, M. B. Groen and R. V. A. Orru, Chem.–Eur. J., 2008, 14, 4961–4973 CrossRef CAS.
- V. A. Chebanov, V. E. Saraev, S. M. Desenko, V. N. Chernenko, I. V. Knyazeva, U. Groth, T. N. Glasnov and C. O. Kappe, J. Org. Chem., 2008, 73, 5110–5118 CrossRef CAS.
- Y. Sakhno, S. Desenko, S. Shishkina, O. Shishkin, D. Sysoyev, U. Groth, C. Kappe and V. Chebanov, Tetrahedron, 2008, 64, 11041–11049 CrossRef CAS.
- Y. I. Sakhno, S. V. Shishkina, O. V. Shishkin, V. I. Musatov, E. V. Vashchenko, S. M. Desenko and V. A. Chebanov, Mol. Diversity, 2010, 14, 523–531 CrossRef CAS.
- E. Muravyova, S. Shishkina, V. Musatov, I. Knyazeva, O. Shishkin, S. Desenko and V. Chebanov, Synthesis, 2009, 1375–1385 CAS.
- J. Světlík and V. Kettmann, Tetrahedron Lett., 2011, 52, 1062–1066 CrossRef.
- Y. Suzuki, Y. Ohta, S. Oishi, N. Fujii and H. Ohno, J. Org. Chem., 2009, 74, 4246–4251 CrossRef CAS.
- G. W. Wang and J. Gao, Org. Lett., 2009, 11, 2385–2388 CrossRef CAS.
- H.-G. Cheng, C.-B. Chen, F. Tan, N.-J. Chang, J.-R. Chen and W.-J. Xiao, Eur. J. Org. Chem., 2010, 4976–4980 CrossRef CAS.
- S. Tu, B. Jiang, Y. Zhang, R. Jia, J. Zhang, C. Yao and F. Shi, Org. Biomol. Chem., 2007, 5, 355 CAS.
- S. Yermolayev, N. Gorobets, E. Lukinova, O. Shishkin, S. Shishkina and S. Desenko, Tetrahedron, 2008, 64, 4649–4655 CrossRef CAS.
- N. Y. Gorobets, B. H. Yousefi, F. Belaj and C. O. Kappe, Tetrahedron, 2004, 60, 8633–8644 CrossRef CAS.
- S. A. Yermolayev, N. Y. Gorobets and S. M. Desenko, J. Comb. Chem., 2009, 11, 44–46 CrossRef CAS.
- S. G. Dzhavakhishvili, N. Y. Gorobets, V. N. Chernenko, V. I. Musatov and S. M. Desenko, Russ. Chem. Bull., 2008, 57, 422–427 CrossRef CAS (translated from: Izvestiya Akademii Nauk. Seriya Khimicheskaya, No. 2, pp. –, February, 2008).
- S. A. Yermolayev, N. Y. Gorobets, O. V. Shishkin, S. V. Shishkina and N. E. Leadbeater, Tetrahedron, 2011, 67, 2934–2941 CrossRef CAS.
- F. Bondavalli, O. Bruno, E. Presti, G. Menozzi and L. Mosti, Synthesis, 1999, 1169–1174 CrossRef CAS.
- N. Y. Gorobets, Y. V. Sedash, S. V. Shishkina, O. V. Shishkin, S. A. Yermolayev and S. M. Desenko, ARKIVOC, 2009, 23–30 CAS.
- F. M. Abdelrazek and A. N. Elsayed, J. Heterocycl. Chem., 2009, 46, 949–953 CrossRef CAS.
- K. Khalil, H. Al-Matar and M. Elnagdi, Eur. J. Chem., 2010, 1, 252–258 CrossRef CAS.
- E. M. Arnett and J. A. Harrelson Jr, J. Am. Chem. Soc., 1987, 109, 809–812 CrossRef CAS.
- K. Byun, Y. Mo and J. Gao, J. Am. Chem. Soc., 2001, 123, 3974–3979 CrossRef CAS.
- I. Trummer, E. Ziegler and O. S. Wolfbeis, Synthesis, 1981, 225–227 CrossRef CAS.
- L. Mosti, P. Schenone and C. Menozzi, J. Heterocycl. Chem., 1985, 22, 1503–1509 CrossRef CAS.
- G. P. McGlacken and I. J. S. Fairlamb, Nat. Prod. Rep., 2005, 22, 369 RSC.
- C. D. Donner, M. Gill and L. M. Tewierik, Molecules, 2004, 9, 498–512 CrossRef CAS.
- A. Goel and V. J. Ram, Tetrahedron, 2009, 65, 7865–7913 CrossRef CAS.
- K. M. Chan, N. F. Rajab, D. Siegel, L. B. Din, D. Ross and S. H. Inayat-Hussain, Toxicol. Sci., 2010, 116, 533–548 CrossRef CAS.
- T. Grkovic, J. S. Blees, N. H. Colburn, T. Schmid, C. L. Thomas, C. J. Henrich, J. B. McMahon and K. R. Gustafson, J. Nat. Prod., 2011, 74, 1015–1020 CrossRef CAS.
- S. Gupta, L. Poeppelman, C. L. Hinman, J. Bretz, R. A. Hudson and L. M. V. Tillekeratne, Bioorg. Med. Chem., 2010, 18, 849–854 CrossRef CAS.
- Y. Shiono, M. Yokoi, T. Koseki, T. Murayama, N. Aburai and K.-i. Kimura, J. Antibiot., 2010, 63, 251–253 CrossRef CAS.
- J. Wang, B. Zhao, W. Zhang, X. Wu, R. Wang, Y. Huang, D. Chen, K. Park, B. C. Weimer and Y. Shen, Bioorg. Med. Chem. Lett., 2010, 20, 7054–7058 CrossRef CAS.
- D. Novikov, I. Yakovlev, V. Zakhs and A. Prep'yalov, Russ. J. Gen. Chem., 2002, 72, 1601–1615 CrossRef CAS (translated from: Zhurnal Obshchei Khimii, Vol. 72, No. 10, 2002, pp. 17013–1714).
- K. V. Sashidhara, J. N. Rosaiah, M. Kumar, R. K. Gara, L. V. Nayak, K. Srivastava, H. K. Bid and R. Konwar, Bioorg. Med. Chem. Lett., 2010, 20, 7127–7131 CrossRef CAS.
- R. Kuhn and D. Weiser, Angew. Chem., 1957, 69, 371–376 CrossRef CAS.
- J. A. Cabello, J. M. Campelo, A. Garcia, D. Luna and J. M. Marinas, J. Org. Chem., 1984, 49, 5195–5197 CrossRef CAS.
- W. Ried and B. Schleimer, Angew. Chem., 1958, 70, 164 Search PubMed.
- D. S. Khachatryan, N. M. Morlyan, R. G. Mirzoyan and S. O. Badanyan, Arm. Khim. Zh., 1980, 33, 850–855 CAS CA 1981, 94, 191844.
Footnote |
| † Electronic Supplementary Information (ESI) available: elemental analysis, 1H, 13C NMR, IR and mass spectral data including copies of spectra. See DOI: 10.1039/c1ra00723h/ |
| This journal is © The Royal Society of Chemistry 2012 |