UV-induced formation of activated Bi2O3 nanoflake: an enhanced visible light driven photocatalyst by platinum loading†
Zhanglian
Xu
*a,
Isao
Tabata
a,
Kazumasa
Hirogaki
a,
Kenji
Hisada
a,
Tao
Wang
b,
Sheng
Wang
b and
Teruo
Hori
a
aGraduate School of Engineering, University of Fukui, Fukui-shi, 910-8507, Japan. E-mail: xuzhanglian85@hotmail.com; Fax: +81 0776278641; Tel: +81 0776278641
bKey Laboratory of Advanced Textile Materials and Manufacturing Technology, Zhejiang Sci-Tech University, Hangzhou, 310018, P. R. China
First published on 1st November 2011
Abstract
Pt/Bi2O3 nanoflakes were designed as efficient visible light-driven photocatalyst which could be attributed to the synergy of its UV-induced nanostructure, surface phase and Pt-induced multielectron O2reduction.
Tailoring the architecture of nano/microcrystals has been of great research interest due to their unique shape and size-dependent properties. In this respect, bismuth oxide (Bi2O3) is an attractive material due to its good optical and electrical properties and semiconductor characteristics. It has been widely used in various applications such as microelectronics, gas sensor, and optical coatings.1,2 Furthermore, Bi2O3 has also proved to be an efficient photocatalyst for water splitting and pollutant decomposing under visible light irradiation. Recently, Bi2O3 nanocrystals with various morphologies have been achieved. For example, Liet al.3 reported the fabrication of Bi2O3 nanotubes from fast oxidizing the bismuth nanowires in air by a template-based heat-treatment method. Ultrathin Bi2O3 nanowires were prepared by thermal evaporation technique by Yang et al.4Bi2O3 microrods have been produced using BiI3 and O2 as a starting material.5 Chen et al. reported mesh-like Bi2O3 single crystalline nanoflakes via a bismuth oxalate precursor way.6 Our group also reported a free-standing 3D microjagged Bi2O3 prepared through a nontraditional template method.7 Accordingly, a direct correlation between the size, nanostructures and surface phase of the photocatalyst and its photocatalytic performance is of great significance, the availability of efficient Bi2O3 photocatalysts with combining the above factors is the main motive of this paper.
In this communication, we report the first example of porous bismuth carbonate microflowers prepared by urea-assisted alcoholysis of bismuth nitrate as shown in ESI.† Thermal treatment is generally considered to be the most classic way to obtain oxides. However, thermal treatment adversely affected their physical performances. Undesirable crystal aggregation was involved in the transformation from bismuth carbonate to bismuth oxide (Bi2O3). The porous structure of bismuth carbonate microflowers was totally destroyed forming Bi2O3 micronuts with compact surface and accompanying rapid decrease of specific surface area. In general, thermal-induced tubular morphology collapse of nanotubes and crystal aggregation of nanoparticles8,9 are regarded as unavoidable thermodynamic behaviors of nanomaterials. However, our recent research demonstrated that these thermodynamic behaviors can access to be solved by special methods.10 In this paper, UV light irradiation was available to ‘make Bi2O3 micronuts rebloom’. The surface mesopores in Bi2O3 micronuts were successfully reopened forming Bi2O3 nanoflakes which were simultaneously introduced active species on the surface. Further platinum deposition on the surface of Bi2O3 nanoflakes structure that results from combining the ideas of nanostructural control with a Pt/Bi2O3 multicomponent composite as a heterocatalyst should therefore be more promising for visible-light-driven photocatalysis. Pt-loaded Bi2O3 nanoflake was proven to exhibit remarkably high photocatalytic activity for aqueous acetaldehyde (AcH) decomposition because of the synergy of its UV-induced nanostructure, surface phase and Pt-induced multielectron O2reduction.
The X-ray diffraction (XRD) profiles shown in Fig. 1 are for products obtained from the hydrothermal reaction (Fig. 1a), after calcination and UV irradiation (Fig. 1b and c). The sample from the hydrothermal reaction was identified as crystalline bismuth carbonate (JCPDS No.41-1488). After calcination, the bismuth carbonate decomposed into Bi2O3, as confirmed by the presence of characteristic peaks of monoclinic Bi2O3 (α-Bi2O3, JCPDS No.41-1449) in the XRD pattern. No other phases were detected in the spectrum, indicating that the impurities in the sample after calcination were out of the detection range of XRD. The XRD profile of the irradiated Bi2O3 shows the main phase, α-Bi2O3, with two new minor peaks at 2θ = 27.9° and 32.3° (Fig. 1c). The two characteristic reflections can be clearly indexed with the formation of the oxidized Bi2O4-x species7 on the surface of α-Bi2O3.
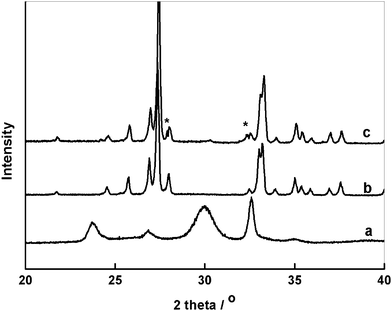 | ||
| Fig. 1 XRD patterns of a) bismuth carbonate microflowers before calcination and b) calcined Bi2O3 micronuts and c) UV-irradiated Bi2O3 nanoflakes. | ||
The morphology of the collected bismuth-based compounds is observed using the field-emission scanning electron microscope (FESEM). Fig. 2a shows SEM image of bismuth carbonate, which overall reveals many uniform flowerlike textures. These flowerlike bismuth carbonates are composed of many nanosheets as the petals with an average thickness of 20–30 nm, and these nanosheets interweave together forming an open porous structure. Fig. 2b and c depict a group of fully calcined Bi2O3 (500 °C, 1 h). Clear observation shows that the bismuth carbonate microflowers present obvious signs of aggregation after calcination, converting into Bi2O3 with nutlike shape and each micronut is constructed with two or three microflowers. The top-view SEM image (Fig. 2c) shows the micronut is actually reassembled by ordered nanosheets, rather than unorderly aggregation. This inspires us the possibility to separate these nanosheets again. Very interestingly, the ordered layers peel off leading to the formation of mesporous structure again when we employ the UV radiation, as shown in Fig. 2d. Energy dispersive X-ray analysis (Fig. S1a and b in ESI) indicates that bismuth carbonate converts into Bi2O3 in absence of carbon peak after calcination.
 | ||
| Fig. 2 SEM images of bismuth carbonate microflowers (a) and Bi2O3 micronuts after annealing (b, c) and UV-irradiated Bi2O3 nanoflakes (d). | ||
It has been reported that large surface areas are desirable for numerous applications of nanostructures to fulfil the demand of high efficiency and activity.11 The porosity and specific surface area changes in the morphology evolution of the products were investigated by nitrogen cryosorption studies. Bismuth carbonate microflowers were found to hold remarkable BET surface area of 120 m2 g−1. However, the specific surface area of the product rapidly decreased to 25 m2 g−1 due to the thermal aggregation when bismuth carbonate transformed into compact Bi2O3 micronuts. Excitingly, a significant increase in the surface area (95 m2 g−1) was observed after UV irradiation, in good agreement with SEM observations. The nitrogen absorption of the products exhibits the type IV characteristics (Fig. S2a in ESI). Fig. S2b in ESI shows the pore-size distribution. The pore-size distribution of bismuth carbonate microflowers predominantly locates in the range from 10 to 50 nm (mesopores), which can be attributed to the porous structure of microflowers. The pores from Bi2O3 micronuts reveal the absence of mesopores with the fusion of nanosheets, whereas the pores (range in 40–100 nm) are found again as the ordered nanosheets separated after UV irradiation. These mesopores are quite suitable to accommodate nanoscale Pt particles and allow for more efficient absorption of incident photons as well as decreased bulk recombination.
In this regard, two mainly striking features were produced by the employment of UV light irradiation including the formation of Bi2O3 nanoflakes with laminar porous nanostructure and the introduction of surface oxidized species on the surface of Bi2O3 nanomaterials. The band gap value of 2.4 eV is calculated for the irradiated Bi2O3 (Fig. S3 in ESI). This value is much lower than that of the calcined Bi2O3 (2.7 eV). The high band gap value of the irradiated material is likely attributed to originating from bulk O(2p)–Bi3+(6p°) excitations and surface O(2p)–Bi5+(6s°) transitions made available by UV irradiation treatment. Because highly oxidized species are formed by reacting with oxygen in solution in the case of calcined Bi2O3 under UV irradiation. Among these reactive species, the photogenerated superoxide radicals with the strongest oxidation ability and enough long lifetime12 can in return oxidize Bi3+ to Bi5+ with the formation of reactive Bi2O4-x at the surface. We have also proved that the surface oxidation won't occur without O2 in the solution. On the other hand, It is worthy to note that the UV light induced valence change from Bi3+ to Bi5+ would lead to the accumulation of electrons between the Bi2O3 nanosheets. With prolongation of UV light irradiation time, accommodating enough accumulated electrons between the nanosheets tended to repel each other and finally formed the laminar porous nanostructures. To confirm the above hypothesis, the intermediate product by irradiation for shorter time (10 min) was checked. From the SEM images (Fig. S4 in ESI), the compact surface of Bi2O3 micronuts became partially wavy and appeared a small part of clear furrows, verifying our proposed formation process very well. It thus reveals that the adopted UV irradiation is an ideal structural modification approach for Bi2O3, which should be minor impact for TiO2–based material. So far, only deliberate designed thermal treatment helps to obtain interesting anatanse/rutile nanocompositions with enhanced photocatalytic activity in TiO2–based material.13
Actually, Bi2O3 as a promising visible light driven photocatalyst has been more and more reported.14 Though its surface state can be easily modified by some high oxidative reagent such as Na2S2O815 and H2O27 or under some high-energy radiation like UV light,16 it is stable under visible light driven photocatalytic reaction. Thus we can say that the surface treatment of Bi2O3 under UV light radiation is a photooxidation process while the decomposition of organic pollution under visible light radiation is a photocatalytic reaction.
The photocatalytic activity is evaluated by the amount of CO2 that evolved from the oxidative decomposition of acetaldehyde solution (AcH aq) upon visible light irradiation, as described in the Experimental Section in the ESI. The photocatalytic activity of UV-irradiated Bi2O3 nanoflakes is better than that of calcined Bi2O3 micronut and nitrogen-doped TiO2 (N-TiO2) at each Pt-loading concentration, as shown in Fig. S5 in ESI. 1 wt% Pt loading produces the highest activity for all the photocatalysts and an excessive Pt loading decreases the photocatalytic activity due to the competitive photon absorption of the photocatalytically inactive Pt nanoparticles with the photocatalysts. Fig. 3a shows that the rate of CO2 generation over both UV-irradiated Bi2O3 nanoflakes and calcined Bi2O3 micronuts from aqueous AcH increased rapidly after Pt loading, achieving the maximum rate (131 μmol h−1) at 1 wt% Pt-loaded Bi2O3 nanoflake. This rate of CO2 generation is ca. 6.3 and 30 times higher than that over pure Bi2O3 nanoflake (21 μmol h−1) and Bi2O3 micronut (4.2 μmol h−1), respectively. It is necessary to note that the enhanced activity in UV-irradiated Bi2O3 nanoflake compared to calcined Bi2O3 micronut could be associated with the formation of surface phase junctions between the bulk Bi2O3 and surface Bi2O4-x when calcined Bi2O3 micronut was irradiated under UV light. We have proved that such surface phase junctions could promote the spatial charge separation in the surface region leading to high photocatalytic efficiency, and the high efficient photocatalytic activity can only be available under a certain ratio between bulk Bi2O3 and surface Bi2O4-x. Excess surface Bi2O4-x would instead decrease the photocatalytic activity.6 Furthermore, the UV-induce opening of laminar pores on the surface would be the other reason. These laminar pores could not only benefit photons' effective absorbance comparing to a flat surface due to light scattering,17,18 but also allow the rapid diffusion of the reactants and products during the reaction. Herein, N–TiO2 as a commercial visible light-responsive photocatalyst is also utilized in the control experiment. Although detectable amounts of CO2 gas were liberated over N–TiO2, the rate (ca. 25 μmol h−1 with optimal Pt loading) had just little increase compared to bare N–TiO2 and was much lower than that over irradiated Bi2O3 after Pt loading. The final molar amount of CO2 generation over Pt–Bi2O3 nanoflake is almost twice that of AcH used, indicating that the complete decomposition of AcH solution underwent in this reaction. We also investigate the stability of the UV-irradiated Pt/Bi2O3 catalyst during the decomposition of AcH. After ten reaction cycles, Pt/Bi2O3 nanoflakes showed a negligible decrease of the activity (Fig. S6 in ESI), and the XRD and SEM images of the photocatalysts after photocatalytic reaction showed no difference compared to the original samples (data no shown), suggesting that the photocatalyst is durable under aerobic, visible-light illumination conditions.
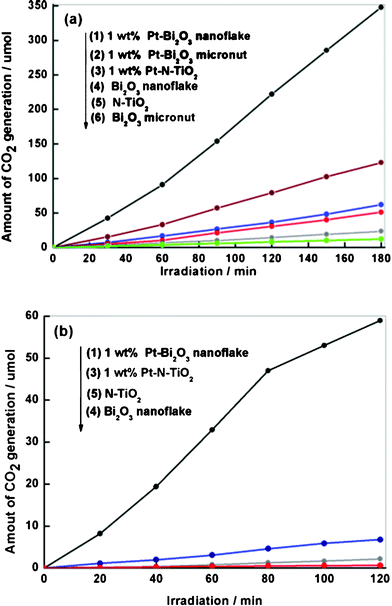 | ||
| Fig. 3 Time course of CO2 generation from the decomposition of (a) acetaldehyde (AcH) and (b) isopropyl alcohol (IPA) over 1 wt% Pt-Bi2O3 nanoflake (1), 1 wt% Pt-Bi2O3 micronut (2), 1 wt% Pt-N-TiO2 (3), UV-irradiated Bi2O3 nanoflake (4), N-TiO2 (5) and calcined Bi2O3 micronut (6) under visible light. | ||
In the end of this paper, we try to explore a plausible mechanism for the enhanced photocatalytic ability of Pt-loaded Bi2O3 nanoflakes. From the morphological view, UV irradiation induced reopening the bulk Bi2O3 micronuts forming Bi2O3 nanoflakes with large specific area. The shape and structure change is decisive to photocatalytic activity of materials. The calcined and UV-irradiated Bi2O3 after Pt loading were observed by TEM (Fig. S7 in ESI). For UV-irradiated Bi2O3 nanoflake, the laminar mesopores are though a little hard to observe clearly in TEM, as shown in Fig. S7b in ESI, some laminar mesopores are still visible and they do exist from SEM and pore size measurement. These laminar mesopores naturally act as Pt-buffering sites. Most Pt particles with small size (3–5 nm) preferentially entered these laminar pores leading to uniform loading on the surface of Bi2O3 nanoflakes or in the mesopores, while Pt nanoparticles can only be photodeposited on the smooth and compact surface of calcined Bi2O3 micronuts in Fig. S6a in ESI. The UV-induced formation of Bi2O3 nanoflakes results in a larger effective surface area and guarantees more active sites in close to the Pt nanoparticles, thus enabling diffusive transport of photogenerated electrons to these particles. Further from energetic view, the CB levels of Bi2O3 (+0.33 V versusNHE)19 with visible light absorption are more positive than the reduction potentials of O2 leading to the inefficient oxidation decomposition of isopropyl alcohol (IPA) in air (Fig. 3b). IPA is known as the counterpart of holes consumption. Photogenerated electron reduction of O2 to produce superoxide or active radicals predominately proceeds in the decomposition of IPA. Notably, the oxidation decomposition of IPA is accelerated by Pt loading. This result strongly indicates that the accumulated photogenerated electrons on the surface of Pt particles working as an electron magnet provide sufficiently negative potential to catalyze O2reduction. This is called multielectron-induced reduction, which has been reported in the case of Pt–WO3 photocatalysts.20 This is the first time to find Pt-induced multielectron reduction occurred in Bi2O3 samples. While single-electron reduction has been reported to mainly proceed over TiO2 and N–TiO2 photocatalysts,21–23 and this is reasonably supported by the Pt-induced improvement of the photoactivity of N-TiO2 was remarkably less than that for Bi2O3 (2.7-fold versus 100-fold). The cooperation of multi-electron assembled by Pt particles for O2reduction is superior to single-electron reduction and that's the reason why Pt loading contributes to high photocatalytic activity under visible light.
Conclusions
We have developed a simple and low cost approach to obtain Bi2O3 nanoflake from the surface compact Bi2O3 micronut through employing UV-visible radiation. The UV-visible radiation induced the formation of the nanostructure of Bi2O3 nanoflake with large specific area and the formation of surface active species (Bi2O4-x) on surface Bi2O3 simultaneously. After Pt loading, the Pt/Bi2O3 nanoflake showed higher photocatalytic efficiency for the degradation of aqueous acetaldehyde and isopropyl alcohol than commercial N–TiO2 and optimal Pt-loaded N–TiO2 under visible light. Such a large activity enhancement probably arises from the synergy of its UV-induced nanostructure, surface phase and Pt-induced multielectron O2reduction. This work provides a new route for the design of multicomponent photocatalysts with enhanced photoactivity as well as a new material which can be used in solar cells, nanodevices and other applications.References
- T. Hyodo, E. Kanazawa, Y. Takao, Y. Shimizu and M. Egashira, Electrochemistry, 2000, 68, 24 CAS.
- G. Bandoli, D. Barreca, E. Brescacin, G. A. Rizzi and E. Tondello, Chem. Vap. Deposition, 1996, 2, 238 CrossRef CAS.
- L. Li, Y. W. Yang, G. H. Li and L. D. Zhang, Small, 2006, 2, 548 CrossRef CAS.
- Y. F. Qiu, D. F. Liu, J. H. Yang and S. H. Yang, Adv. Mater., 2006, 18, 2604 CrossRef CAS.
- R. Chen, Z. R. Shen, H. Wang, H. J. Zhou, Y. P. Liu, D. T. Ding and T. H. Chen, J. Alloys Compd., 2011, 509, 2588 CrossRef CAS.
- T. Takeyama, N. Takahashi, T. Nakamura and S. Itoh, Solid State Commun., 2005, 133, 771 CrossRef CAS.
- Z. L. Xu, I. Tabata, K. Hirogaki, K. Hisada, T. Wang, S. Wang and T. Hori, Catal. Sci. Technol., 2011, 1, 397 CAS.
- X. M. Sun and Y. D. Li, Chem.–Eur. J., 2003, 9, 2229 CrossRef CAS.
- J. N. Nian and H. S. Teng, J. Phys. Chem. B, 2006, 110, 4193 CrossRef CAS.
- T. Wang, S. Wang, W. X. Chen, W. Wang, Z. L. Xu and T. Hori, J. Mater. Chem., 2009, 19, 4692 RSC.
- J. Tang, Z. Zou and J. Ye, Chem. Mater., 2004, 16, 1644 CrossRef CAS.
- T. Hirakawa, K. Yawata and Y. Nosaka, Appl. Catal., A, 2007, 325, 105 CrossRef CAS.
- J. Zhang, Q. Xu, Z. C. Feng, M. J. Li and C. Li, Angew. Chem., Int. Ed., 2008, 47, 1766 CrossRef CAS.
- (a) S. Y. Chai, Y. J. Kim, M. H. Jung, A. K. Chakraborty, D. Jung and W. I. Lee, J. Catal., 2009, 262, 144 CrossRef CAS; (b) L. Zhou, W. Z. Wang, H. L. Xu, S. M. Sun and M. Shang, Chem.–Eur. J., 2009, 15, 1776 CrossRef CAS; (c) X. H. Wu, W. Qin and W. D. He, J. Mol. Catal. A: Chem., 2007, 261, 167 CrossRef CAS; (d) M. Ge, Y. F. Li, L. Liu, Z. Zhou and W. Chen, J. Phys. Chem. C, 2011, 115, 5220 CrossRef CAS.
- A. S. Prakash, C. Shivakumara, M. S. Hegde, L. Dupont and J. M. Tarascon, Mater. Res. Bull., 2007, 42, 707 CrossRef CAS.
- A. Hameed, T. Montini, V. Gombac and P. Fornasiero, J. Am. Chem. Soc., 2008, 130, 9658 CrossRef CAS.
- G. K. Mor, K. Shankar, M. Paulose, O. K. Varghese and C. A. Grimes, Nano Lett., 2005, 5, 191 CrossRef CAS.
- H. X. Li, Z. F. Bian, J. Zhu, D. Q. Zhang, G. S. Li, Y. N. Huo, H. Li and Y. F. Lu, J. Am. Chem. Soc., 2007, 129, 8406 CrossRef CAS.
- Y. Xu and M. A. A. Schoonen, Am. Mineral., 2000, 85, 543 CAS.
- R. Abe, H. Takami, N. Murakami and B. Ohtan, J. Am. Chem. Soc., 2008, 130, 7780 CrossRef CAS.
- K. Ishibashi, A. Fujishima, T. Watanabe and K. Hashimoto, J. Phys. Chem. B, 2000, 104, 4933 CrossRef.
- T. Hirakawa and Y. Nosaka, Langmuir, 2002, 18, 3247 CrossRef CAS.
- A. J. Colussi and M. R. Hoffmann, J. Phys. Chem. B, 2004, 108, 17269 CrossRef.
Footnote |
| † Electronic Supplementary Information (ESI) available: Experimental section, EDXA data, BET and pore-size distribution data, UV-vis spectra data, SEM image and TEM image of as-prepared samples. See DOI: 10.1039/c1ra00638j/ |
| This journal is © The Royal Society of Chemistry 2012 |