Two-step synthesis of Fe2O3 and Co3O4 nanoparticles: towards a general method for synthesizing nanocrystalline metal oxides with high surface area and thermal stability†
Junjiang
Zhu
*ab,
Xiaoying
Ouyang
c,
Ming-Yung
Lee
b,
Ryan C.
Davis
c,
Susannah L.
Scott
*bc,
Anna
Fischer
a and
Arne
Thomas
a
aDepartment of Chemistry, Technical University of Berlin, Englische Straße 20, 10587, Berlin, Germany. E-mail: ciaczjj@gmail.com (J. Zhu)
bDepartment of Chemical Engineering, University of California, Santa Barbara, 10 Mesa Road, Santa Barbara, CA 93106-5080, USA. E-mail: sscott@engineering.ucsb.edu (S. L. Scott)
cDepartment of Chemistry & Biochemistry, University of California, Santa Barbara, 10 Mesa Road, Santa Barbara, CA 93106-9510, USA
First published on 8th November 2011
Abstract
A simple, two-step method using activated carbon (AC) as a support/scaffold was developed to synthesize metal oxide nanocrystalline materials (NCMs). In the first step, metal nitrate precursors were deposited by wet impregnation onto the AC, then heated in argon at 350 °C to immobilize the metal oxides. In the second step, the AC was removed by calcination in air at 500 °C, to obtain the unsupported metal oxide NCMs. Characterization by N2-sorption isotherms, TGA, XPS and EXAFS reveals that the metal oxide particles are crystalline and nanometre-sized, with surface areas up to 148 m2 g−1. Moreover, the TEM images show particle sizes in the range 5–10 nm, even after calcination at 500 °C for 2 h. Their thermal stability and high surface areas, together with the nanometre-sized structures, make them promising materials for catalytic applications (e.g., CO oxidation).
Introduction
Nanocrystalline materials (NCMs) have attracted much attention in recent years because of a wide variety of potential applications, including catalysis, optics and chemical sensors.1–8 In catalysis, it is generally believed that, in addition to enhanced surface areas, NCMs often display interesting and unexpected properties that are qualitatively different from those of the corresponding bulk materials, or of the atomic or molecular species from which they are derived.9 One well-known example is the use of gold for low temperature CO oxidation: bulk gold shows no activity, while gold nanoparticles (NPs) show high reactivity even at 77 K.10–12Defining and understanding the origin of the novel properties of NCMs has stimulated much research, and consequently, methods to prepare them have been extensively reported, both for metal oxides13–18 and for noble metals.19–23 Compared to the preparation of noble metal-based materials, the synthesis of metal oxide NCMs can be more complex, because some metals are very reactive toward oxygen, and agglomeration of oxide NPs occurs readily at moderate temperatures. Straightforward, versatile routes for the synthesis of thermally stable metal oxide NCMs are therefore of considerable practical interest.
Activated carbon (AC) has long been recognized as a good support for both noble and base metal catalysts, due to its microporosity, multifunctional surface groups and high surface area.24–28 For a noble metal such as gold, Prati et al.29,30 found that when Au3+ is first reduced to Au0 in the presence of polyvinyl alcohol (PVA) and then immobilized on AC, small gold particles (∼6 nm) can be obtained after removing the PVA at 350 °C. We found that very small Co3O4·NPs (∼5 nm) are obtained when a metal nitrate-impregnated AC is heated in argon at 350 °C.31,32 AC has also been used to prepare unsupported metal oxides with high surface areas and designed structures.33–39 For example, Schüth et al.37 obtained metal oxides with surface areas ranging from 50 to 200 m2 g−1 by calcining metal nitrates deposited on AC in air at 450–800 °C. They reported better results for some metals (e.g., Cu) by heating in the presence of a limited amount of air, and suggested that slowing metal nitrate-catalyzed AC combustion reduces particle sintering. These observations inspired us to prepare metal oxide NPs using a prior immobilization step. By removing the AC scaffold in air in a subsequent step, we hoped to obtain unsupported, metal oxide NCMs with small particle sizes and high surface areas.
Results and discussion
For preparation of the metal oxide NCMs, a metal nitrate was first deposited by wet impregnation onto a microporous AC. The resulting material (denoted M/AC-1, where M is Co, Fe) was then dried and heated in a stream of argon at 350 °C, to generate and immobilize the metal oxide NPs on AC, denoted M/AC-2. Subsequently, the solid was allowed to cool to room temperature and heated again to 500 °C in air for 2 h, to remove the AC scaffold and recover the unsupported, nanocrystalline metal oxide.Fig. 1(A) and (B) show the thermal behavior of the materials at each stage during their synthesis, as monitored by thermogravimetric analysis (TGA). AC is thermally stable up to approx. 560 °C in oxygen (see Fig. S1†), and as expected no appreciable weight loss was observed for the AC at T < 500 °C in the inert atmosphere (curve “a”). Co/AC-1 (curve “b”) shows a stepwise weight loss during thermal treatment in the inert atmosphere. Stage (1), at T < 120 °C, is ascribed to loss of water and other adventitious adsorbates acquired during sample storage; stage (2), at 120 < T < 180 °C, is caused by transformation of [Co(OH2)6](NO3)2 introduced in the impregnation step to [Co3[NO3]2(OH)4]; stage (3), at 180 < T < 240 °C, corresponds to the decomposition of [Co3[NO3]2(OH)4] to a cobalt oxide (Co3O4).31 These transformations are similar to those observed previously in the synthesis of supported NiO nanoparticles.2 After formation of the supported metal oxide, the AC scaffold was removed in O2, as represented by curve “c”. It shows that (i) no appreciable weight loss occurs at 120 < T < 240 °C, confirming that most of the nitrate was removed during the first thermal treatment step; and (ii) AC is fully removed at T < 400 °C, suggesting that the final product calcined at 500 °C should not contain any residual AC scaffold. This is further evidenced by the TGA behavior of the product (curve “d”), which shows a weight loss of less than 5% at T = 510 °C in O2. Similar changes were observed for M = Fe, except that the temperature required for complete removal of the AC scaffold (curve “f”) was slightly higher than 500 °C. Nevertheless, the minor weight loss observed for Fe2O3 (<5%, curve “g”) suggests that AC had been completely removed from the final product.
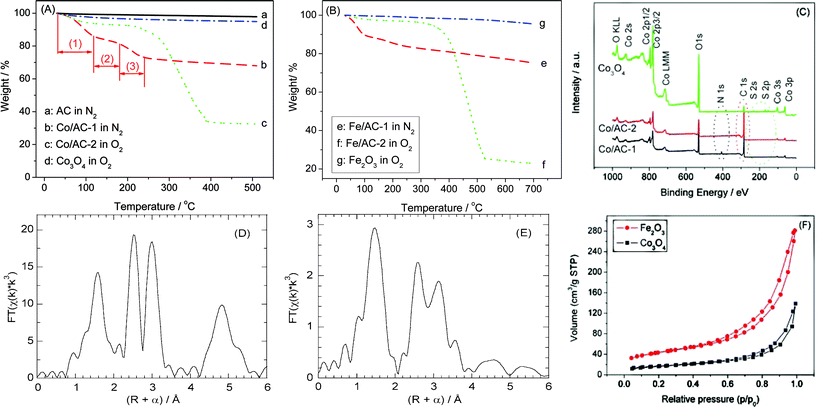 | ||
| Fig. 1 TGA curves for Co-containing (A) and Fe-containing samples (B), as well as XPS spectra for the Co-containing samples (C), before and after removal of the AC scaffold; R-space EXAFS at the Co K-edge for unsupported Co3O4_NSMs (D), and at the Fe K-edge for unsupported Fe2O3_NSMs (E), and N2-sorption isotherms for Co3O4_NSMs and Fe2O3_NSMs (F). “M/AC-1” and “M/AC-2” represent materials before and after treatment in the inert atmosphere, respectively. | ||
Thus the characterization and reactivity results below are considered to pertain exclusively to the unsupported metal oxides. By comparing the weight losses of samples Co/AC-2 and Fe/AC-2 with that of the unmodified AC, we estimate the loadings of Co3O4 and Fe2O3 prior to calcination to be 22 and 14 wt%, respectively.
Results obtained by X-ray photoelectron spectroscopy (XPS) confirm our conclusions based on TGA measurements. Fig. 1(C) shows that the N 1s signal is no longer present after the sample has been heated in Ar at 350 °C for 2 h, indicating that the metal nitrate decomposes completely to the metal oxide. Also, the intensity of the C 1s signal is strongly attenuated in the final product compared to the sample prior to calcination, indicating that AC is indeed removed in the second step. (The weak residual C 1s signal observed for the metal oxide arises due to the carbon tape used to hold the sample in place.) The small amount of S is presumably due to impurities in the AC scaffold: ca. 11 wt% non-combustible impurities are present according to the TGA of AC recorded in air, and may include S, Al, Si, etc.
In the powder X-ray diffraction (XRD) patterns of the metal oxide NCMs, the reflections are very weak and broad (see Fig. S3†), complicating their assignment and implying that the particle sizes are very small. This agrees with observations made previously for AC-supported cobalt oxide (Co3O4/AC), in which peak intensities were very low due to the small particle size (ca. 5 nm).31 Thus, we infer that the metal oxide NPs formed during the first preparation step do not grow appreciably when the AC support is removed. Low peak intensities could also indicate that the metal oxides are in an amorphous state, however, this possibility can be excluded since well-resolved lattice fringes are observed in the high resolution transmission electron microscopy (HR-TEM) images (Fig. 2(C)), typical of a crystalline structure. Consequently, phase assignment was carried out using extended X-ray absorption fine structure (EXAFS). The results shown in Fig. 1(D) and (E) are consistent with spinel Co3O440 and α-Fe2O341 for the cobalt and iron oxides, respectively. The ratio of peak heights for the M–O and M–M scattering paths is a reflection of the particle size.42 We estimate that the Fe2O3 particles are in the range 2–10 nm, by comparison to spectra reported by Kitagami et al.43 These observations confirm our idea that the two-step synthesis method leads to nanometre-sized, crystalline metal oxide particles.
 | ||
| Fig. 2 HR-TEM images for AC-supported Co3O4·NCMs, obtained in the first step (A), dark-field; and unsupported Co3O4·NCMs obtained in the second step (B), (C), bright-field. Note: for results involving Fe2O3, see the ESI.† | ||
N2-sorption isotherms were also recorded, in order to assess the porosity of the unsupported metal oxides (see Fig. 1(F)). Both show type II isotherms with a hysteresis loop, indicating a porous structure (presumably due to interparticle porosity). The surface areas calculated by the Brunauer–Emmett–Teller method are 60 and 148 m2 g−1 for Co3O4 and Fe2O3·NCMs, respectively. These high surface areas are consistent with the porous structures, as shown by Fig. 2(B)).
The morphology and average particle size of the metal oxide NCMs are directly accessible from the TEM images shown in Fig. 2. For the AC-supported metal oxides obtained in the first step, the particles are very small and difficult to see in bright-field mode, therefore the dark-field mode was also used. Fig. 2(A) shows that the metal oxide particles are highly dispersed, and that the particle size is generally ≤5 nm. A few large particles (<15 nm) are also present. They are attributed to concentration of the deposited metal ions on the support surface during the drying process, which is an unavoidable consequence of the impregnation method. After removal of the AC scaffold, agglomerated metal oxide nanoparticles are observed, however the small particle sizes from 5 to 10 nm show that they have not sintered to a larger extent. This is also seen by the porous morphology evident in Fig. 2B even after calcination at 500 °C for 2 h. This suggests that metal oxide NCMs prepared by this route have high thermal stability. The porous structure and small particle size are in agreement with those inferred by N2-sorption isotherms and EXAFS (Fig. 1).
These results indicate that AC-supported metal oxide NCMs formed by heating in an inert atmosphere during the first synthesis step retain their structure when the AC support is removed in the second step. In order to demonstrate the advantage of prior metal oxide formation and immobilization by the two-step method, Fe2O3·NPs were also prepared using the one-step method (direct calcination of the supported metal nitrate) according to Schüth et al.37 The product is denoted as “Fe2O3_NCM_I”, and its properties are compared with those of Fe2O3_NCM_II (prepared using the two-step method) in Table 1. Clearly, the latter has a higher surface area and pore volume, as well as a smaller particle size, indicating that immobilization in an inert atmosphere is indeed beneficial for the preparation of unsupported metal oxide NPs with high surface area and porosity. Presumably decomposition of the nitrate ions in the absence of air limits the exotherm in the subsequent calcination step, as proposed by Schüth et al.37 The resulting metal oxide NPs dispersed on AC are stable enough to resist agglomeration.
The high calcination temperature (500 °C) suggests that the NCMs may be robust enough for catalytic applications that require elevated temperatures, which represent a challenge for NCMs. Furthermore, the higher surface areas of metal oxides obtained using the two-step synthesis method described here should be beneficial for their catalytic activity. The catalytic performance of Fe2O3_NCM_II was studied in CO oxidation, a reaction that is often required to operate at elevated temperatures.44,45 For comparison, the catalytic performance of “Fe2O3_NCM_I” and an additional Fe2O3 sample prepared by an organic solution method without the AC scaffold (denoted as Fe2O3_Org, see SI†) were also studied. The results are shown in Fig. 3.
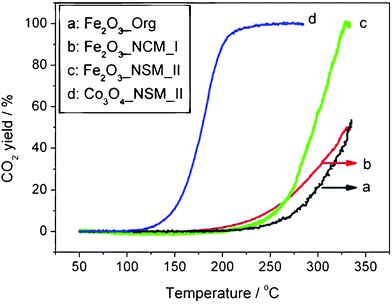 | ||
| Fig. 3 Temperature-programmed reaction profiles for CO oxidation over three unsupported Fe2O3 catalysts, as well as over Co3O4·NCMs. Conditions: 0.4 vol% CO + 10 vol% O2 in Ar; 50 mg catalyst; total flow rate 50 mL min−1. Temperature ramp 5 °C min−1. | ||
Although the temperatures for onset of reactivity are similar for all three iron oxide catalysts (ca. 200 °C), the conversion increases most rapidly with temperature for Fe2O3_NCM_II, and at 330 °C, the CO2 yield (100%) is far higher than for “Fe2O3_NCM_I” or Fe2O3_Org (both ca. 50%). Comparison of the TEM images for Fe2O3_NCM_II before and after its use in CO oxidation shows no appreciable change in the particle size (Fig. S6†), confirming its stability as a high-temperature catalyst. Even more interesting, the Co3O4·NCMs show much better catalytic activity than Fe2O3·NCMs, to be discussed in our forthcoming work.
Conclusions
In summary, we have synthesized crystalline, nanometre-sized iron and cobalt oxides by a two-step method using activated carbon (AC) as a support/scaffold, resulting in particle sizes ranging from 5 to 10 nm and surface areas up to 148 m2 g−1. The metal oxide NCMs persist at temperatures up to 500 °C without significant agglomeration, thus ensuring their stability under most reaction conditions. Furthermore, because the synthesis method described here is very simple, we expect it to be a general method for the preparation of metal oxide NCMs. The only requirement is that the metal precursor be soluble, so that it can be deposited on the AC support by wet impregnation.Acknowledgements
Financial support from the German Research Foundation (DFG, grant No. TH 1463/5-1), the Cluster of Excellence “Unifying Concepts in Catalysis” (EXL 31411), and the US Department of Energy (DE-FG02-03ER15467) are gratefully acknowledged. Portions of this work were performed at the Stanford Synchrotron Radiation Lightsource, a national user facility operated by Stanford University on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences.References
- M. M. Titirici, M. Antonietti and A. Thomas, Chem. Mater., 2006, 18, 3808 CrossRef CAS.
- J. R. A. Sietsma, J. D. Meeldijk, J. P. den Breejen, M. Versluijs-Helder, A. J. van Dillen, P. E. de Jongh and K. P. de Jong, Angew. Chem., Int. Ed., 2007, 46, 4547 CrossRef CAS.
- P. Poizot, S. Laruelle, S. Grugeon, L. Dupont and J. M. Tarascon, Nature, 2000, 407, 496 CrossRef CAS.
- L. J. Zhi, Y. S. Hu, B. El Hamaoui, X. Wang, I. Lieberwirth, U. Kolb, J. Maier and K. Mullen, Adv. Mater., 2008, 20, 1727 CrossRef CAS.
- A. T. Bell, Science, 2003, 299, 1688 CrossRef CAS.
- R. Schlogl and S. B. Abd Hamid, Angew. Chem., Int. Ed., 2004, 43, 1628 CrossRef.
- W. Y. Li, L. N. Xu and J. Chen, Adv. Funct. Mater., 2005, 15, 851 CrossRef CAS.
- H. Cao and S. L. Suib, J. Am. Chem. Soc., 1994, 116, 5334 CrossRef CAS.
- S. O'Brien, L. Brus and C. B. Murray, J. Am. Chem. Soc., 2001, 123, 12085 CrossRef CAS.
- M. Haruta, Catal. Today, 1997, 36, 153 CrossRef CAS.
- M. Haruta, CATTECH, 2002, 6, 102 CrossRef CAS.
- G. J. Hutchings and M. Haruta, Appl. Catal., A, 2005, 291, 2 CrossRef CAS.
- A. Murali, A. Barve, V. J. Leppert, S. H. Risbud, I. M. Kennedy and H. W. H. Lee, Nano Lett., 2001, 1, 287 CrossRef CAS.
- Y. D. Wang, C. L. Ma, X. D. Sun and H. D. Li, Inorg. Chem. Commun., 2002, 5, 751 CrossRef CAS.
- X. L. Xu, J. D. Guo and Y. Z. Wang, Mater. Sci. Eng., B, 2000, 77, 207 CrossRef.
- S. Castro-Garcia, S. Yanez-Vilar, M. Sanchez-Andujar, C. Gomez-Aguirre, J. Mira and M. A. Senaris-Rodriguez, J. Solid State Chem., 2009, 182, 2685 CrossRef.
- X. H. Liu, A. Yan, G. Qiu, N. Zhang, R. R. Shi, R. Yi, M. Tang and R. C. Che, Solid State Commun., 2007, 144, 315 CrossRef CAS.
- V. Polshettiwar, B. Baruwati and R. S. Varma, ACS Nano, 2009, 3, 728 CrossRef CAS.
- B. H. Wu, Y. J. Kuang, X. H. Zhang and J. H. Chen, Nano Today, 2011, 6, 75 CrossRef CAS.
- J. H. He, T. Kunitake and A. Nakao, Chem. Mater., 2003, 15, 4401 CrossRef CAS.
- Y. W. Tan, X. H. Dai, Y. F. Li and D. B. Zhu, J. Mater. Chem., 2003, 13, 1069 RSC.
- C. L. Lee, C. C. Wan and Y. Y. Wang, Adv. Funct. Mater., 2001, 11, 344 CrossRef CAS.
- J. J. Zhu, X. Xie, S. A. C. Carabineiro, P. B. Tavares, J. L. Figueiredo, R. Schomaecker and A. Thomas, Energy Environ. Sci., 2011, 4, 2020 RSC.
- F. Rodriguez-Reinoso, Carbon, 1998, 36, 159 CrossRef CAS.
- M. D. Hughes, Y. J. Xu, P. Jenkins, P. McMorn, P. Landon, D. I. Enache, A. F. Carley, G. A. Attard, G. J. Hutchings, F. King, E. H. Stitt, P. Johnston, K. Griffin and C. J. Kiely, Nature, 2005, 437, 1132 CrossRef CAS.
- D. S. Su, J. Zhang, B. Frank, A. Thomas, X. C. Wang, J. Paraknowitsch and R. Schlogl, ChemSusChem, 2010, 3, 169 CrossRef CAS.
- J. L. Figueiredo and M. F. R. Pereira, Catal. Today, 2010, 150, 2 CrossRef CAS.
- M. L. Toebes, F. F. Prinsloo, J. H. Bitter, A. J. van Dillen and K. P. de Jong, J. Catal., 2003, 214, 78 CrossRef CAS.
- L. Prati and G. Martra, Gold Bull., 1999, 32, 96 CAS.
- L. Prati and M. Rossi, J. Catal., 1998, 176, 552 CrossRef CAS.
- J. J. Zhu, K. Kailasam, A. Fischer and A. Thomas, ACS Catal., 2011, 1, 342 Search PubMed.
- J. J. Zhu, J. L. Faria, J. L. Figueiredo and A. Thomas, Chem.–Eur. J., 2011, 17, 7112 CrossRef CAS.
- H. J. Fan, Q. Xu, Y. Q. Guo, Q. Peng and Z. Z. Hou, Mater. Sci. Eng., A, 2006, 422, 272 CrossRef.
- H. J. Fan, Q. Xu, J. G. Li and Y. X. Cao, J. Am. Ceram. Soc., 2006, 89, 3065 CrossRef CAS.
- W. Rarog-Pilecka, E. Miskiewicz and Z. Kowalczyk, Catal. Commun., 2008, 9, 870 CrossRef CAS.
- H. N. Yang, L. F. Liu, F. L. Yang and J. C. Yu, J. Zhejiang Univ., Sci., A, 2008, 9, 981 CrossRef CAS.
- M. Schwickardi, T. Johann, W. Schmidt and F. Schüth, Chem. Mater., 2002, 14, 3913 CrossRef CAS.
- S. A. C. Carabineiro, S. S. T. Bastos, J. J. M. Orfao, M. F. R. Pereira, J. L. Delgado and J. L. Figueiredo, Appl. Catal., A, 2010, 381, 150 CrossRef CAS.
- S. A. C. Carabineiro, S. S. T. Bastos, J. J. M. Orfao, M. F. R. Pereira, J. J. Delgado and J. L. Figueiredo, Catal. Lett., 2010, 134, 217 CrossRef CAS.
- B. Ernst, A. Bensaddik, L. Hilaire, P. Chaumette and A. Kiennemann, Catal. Today, 1998, 39, 329 CrossRef CAS.
- F. Jiao, A. Harrison, J. C. Jumas, A. V. Chadwick, W. Kockelmann and P. G. Bruce, J. Am. Chem. Soc., 2006, 128, 5468 CrossRef CAS.
- T. Liu, L. Guo, Y. Tao, T. D. Hu, Y. N. Xie and J. Zhang, Nanostruct. Mater., 1999, 11, 1329 CrossRef CAS.
- M. Kitagami, T. Konishi, T. Kaneko, M. Sakamaki, L. Lin, J. G. Lin, D. Arvanitis and T. Fujikawa, Photon Factory Activity Report 2006 #24 Part B, 2007, 12C/2006G356 Search PubMed.
- Y. H. Zheng, Y. Cheng, Y. S. Wang, F. Bao, L. H. Zhou, X. F. Wei, Y. Y. Zhang and Q. Zheng, J. Phys. Chem. B, 2006, 110, 3093 CrossRef CAS.
- P. Li, D. E. Miser, S. Rabiei, R. T. Yadav and M. R. Hajaligol, Appl. Catal., B, 2003, 43, 151 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, additional N2-sorption isotherms, XRD, XPS, TGA, and TEM images of the metal oxides before and after their use as catalysts. See DOI: 10.1039/c1ra00552a |
| This journal is © The Royal Society of Chemistry 2012 |