Mixed pentafluorophenyl and o-fluorophenyl esters of aliphatic dicarboxylic acids: efficient tools for peptide and protein conjugation†
Adam T.
Ślósarczyk
a,
Ramesh
Ramapanicker
*ab,
Thomas
Norberg
a and
Lars
Baltzer
*a
aDepartment of Biochemistry and Organic Chemistry, Uppsala University, PO Box 576, SE-751 23, Uppsala, Sweden
bPresent adress: Department of Chemistry, Indian Institute of Technology, 208016, Kanpur, India
First published on 29th November 2011
Abstract
An efficient methodology for the heteroconjugation of biomolecules with exposed free amino groups has been developed. Mixed pentafluorophenyl and o-fluorophenyl esters of aliphatic dicarboxylic acids with aliphatic chains of varying sizes have been prepared and used to conjugate a 42-residue polypeptides with short model peptides as well as a model dodecapeptide with the antigenic determinant of type B blood, a carbohydrate derivative, to form a glycopeptide. The concept is based on the difference in reactivity towards primary amino groups between phenyl esters with leaving groups of unlike pKa. The reactivities of several pentafluorophenyl and o-fluorophenyl esters towards amino groups were carefully determined under reaction conditions to identify leaving group combinations that would provide optimal differences in reactivity for maximum yields of heteroconjugate formation while keeping the reasonable reaction times. Pentafluorophenyl esters react faster with an amino group and require a weaker base, while an o-fluorophenyl ester requires a stronger base and longer reaction time. The method described is economic, quick and gives complete control over the conjugation reaction. The size of the spacer is conveniently varied by selection of the appropriate aliphatic dicarboxylic acid. While the presented examples describe conjugation reactions of polypeptides with a maximum of 42 residues it is envisioned that the bifunctional linkers reported here will find their most important applications in the heteroconjugation of proteins using lysine side chains, a reaction for which currently few alternatives exist, if access to spacers of variable size is required.
Introduction
The conjugation of biomolecules to e.g. molecular probes, solid supports and other biomolecules has become an indispensable methodology in virtually all areas of research in biotechnology. The incorporation of fluorophores and radiolabels is the key to molecular imaging and bioanalytical spectroscopy, immobilization on solid support is essential in diagnostic applications and the conjugation of biomolecules to other biomolecules, for example linking peptides to carbohydrates, provides a route to mimicking endogenous conjugates in biomedicine that are difficult to obtain from natural sources. In addition, heteroconjugation of biomolecules is increasingly important in probing protein complexation in vivo by targeting two or more recognition sites simultaneously in unravelling biological networks.The formation of heteroconjugates from starting materials that are expensive and most often only available in low amounts, is only justifiable with robust and efficient linkers that are easy to handle and give high yields of the desired conjugates. Many bifunctional reagents that are readily available from commercial sources combine two different functional groups that react with amino groups, mercapto groups or carboxylic acids, the highly abundant side chain functional groups of amino acids. Combining functional groups that react with two different side chains in an orthogonal fashion ensures that the correct heteroconjugates are formed under mild conditions and with a high degree of selectivity. However, it is often of interest to link two biomolecules that carry the same functional group and then a bifunctional linker has to be developed according to a different strategy.
The use of bifunctional reagents such as diethylsquarate1 or thiophosgene2 to link two molecules, both of which carry amino groups, is based on the principle that both groups of the linker react with the same functional group but at different rates. They react rapidly with one amino group but more slowly with a second one, allowing essentially quantitative reaction of the linker with one biomolecule and separation of monoconjugate from unreacted biomolecule before the second biomolecule is introduced to form the final heteroconjugate. While this chemistry is robust, efficient and widely used, these bifunctional linkers do not provide an opportunity to vary the size and nature of the spacer. In the formation of a heterodimer for biological applications the ability to vary the size of the spacer is crucial, as heteroconjugates may not form if the components are forced in too close proximity for reaction to occur, and affinity may be decreased if the conjugated entity adversely affects the interaction with the target biomolecule of the primary biomolecule.
Herein, we report a very simple but extremely effective strategy to conjugate biomolecules with free amino groups to form heteroconjugates. The concept is of general applicability, perhaps most interestingly in the case of protein functionalization and protein heterodimerization. The strategy is based on the difference in reactivity towards primary amines between esters formed from phenyl groups with unlike pKa. Reactive phenyl esters are used frequently as efficient and reliable sources for amidation reactions. The reactivity of amines with these active esters depends on the stability of the liberated phenolate ions and thus on the pKa values of the corresponding phenols. The pKa values of phenols are significantly dependent on the nature of substituents present on the aromatic ring. It was assumed that a mixed diester of aliphatic dicarboxylic acids with two different phenols, having substantial differences in their pKa values, would exhibit differential reactivity at either ester groups towards amines. Here we present results that support this hypothesis, and the conclusion that this class of reagents is robust, highly selective and useful for a large number of bioconjugation reactions.
Results and discussion
In order to identify a pair of phenyl groups that would provide the required difference in reaction rate the reactivity of four different phenyl esters of hexanoic acid (1a–d) towards a helical dodecapeptide (2) bearing a single lysine residue was investigated (Scheme 1). The reactions were performed under two different sets of conditions and the results as obtained by analytical HPLC are listed in Table 1. Pentafluorophenyl hexanoate (1a) and p-nitrophenyl hexanoate (1b) reacted quantitatively with 2 in DMSO containing 10% of pyridine. However, the reactions with o-fluorophenyl (1c) and p-chlorophenyl (1d) hexanoates did not go to completion even after 24 h under these conditions. All four esters reacted faster in a more basic reaction medium with 1% of DIPEA in DMSO containing 10% of pyridine. While the reaction with the o-fluorophenyl ester, 1c was completed in 5 h, only 60% of the desired product was observed in the reaction with the p-chlorophenyl ester, 1d after 24 h.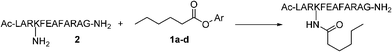 | ||
| Scheme 1 Reaction of the dodecapeptide (2) with phenolic esters (1a–d). | ||




The results suggested that it would be possible to react a pentafluorophenyl ester selectively with an amino group in the presence of an o-fluorophenyl or a p-chlorophenyl ester in a mildly basic medium. However, the p-chlorophenyl ester failed to react completely even when a more basic condition was employed. o-Fluorophenyl esters, on the other hand, reacted readily with the amino peptide 2 in the presence of 1% DIPEA and 10% pyridine. It was thus obvious that mixed pentafluorophenyl and o-fluorophenyl diesters derived from dicarboxylic acids could be used as reagents where amines react first with the pentafluorophenyl ester (10% pyridine in DMSO) and with the o-fluorophenyl ester only under more basic conditions (1% DIPEA and 10% pyridine in DMSO).
We synthesized mixed diesters of succinic acid, adipic acid and sebacic acid with pentafluorophenyl and o-fluorophenyl leaving groups. Adipoyl and sebacoyl chlorides were first treated with o-fluorophenol (1 equiv) to give the monoesters (2a–b) in good yields after aqueous workup (Scheme 2). We observed that addition of 1 equiv of pyridine improved the reaction yields significantly. There are versatile methods available in the literature that describe how to convert carboxilic acid into pentfluorophenyl ester,3,4 yet for the purpose of these studies, carbodiimide mediated method was selected.5 Therefore o-fluorophenyl esters, 2a–b with free carboxylic acid groups were then reacted with pentafluorophenol (DIC, pyridine, CH3CN) to give the diesters (3a–b) in very good yields (Scheme 2). The mixed diester 3c of succinic acid was synthesized from succinic anhydride (Scheme 3). Succinic anhydride was reacted with tert-butanol (DMAP, toluene, 110 °C) to give the mono-tert-butyl ester 4 in 60% yield. The monoester 4 was reacted with o-fluorophenol (DIC, pyridine, CH3CN, yield = 93%) to give the diester 5. The mixed diester, 5 was treated with TFA to remove the tert-butyl ester and the o-fluorophenyl monoester thus obtained was reacted with pentafluorophenol to give the required diester 3c in 93% (Scheme 3).
 | ||
| Scheme 2 Synthesis of mixed diesters of adipic acid and sebacic acid. | ||
 | ||
| Scheme 3 Synthesis of the mixed diester 3c of succinic acid. | ||
The applicability of these mixed esters 3a–c as linkers was examined by conjugating two oligopeptides. The active diesters 3a–c were treated with the dodecapeptide 2 (10% pyridine in DMSO) and found to react selectively with the pentafluorophenyl esters almost quantitatively to give the functionalized derivatives (6a–c) of 2 in very good yields (Scheme 4). There was no noticeable reaction of the amino group with the o-fluorophenyl esters under the reaction conditions employed. The active ester derivatives 6a–c were stable enough to be purified by reverse phase HPLC and could be stored for months after lyophilization.
 | ||
| Scheme 4 Reaction of mixed diesters (3a–c) with a dodecapeptide (2) containing a free amino group at the side chain of Lys 3. | ||
The o-fluorophenyl ester derivatives 6a–c were treated with a nonapeptide 7 containing a free N-terminus, under more basic conditions (1% DIPEA and 10% pyridine in DMSO). The active esters 6a and 6b reacted with 7 quantitatively to give the peptide conjugates 8a and 8b in very good yields (Scheme 5). However, the ester 6c did not undergo the conjugation reaction and the major product isolated was the carboxylic acid 9 resulting from the hydrolysis of 6c. The o-fluorophenyl ester group of 6c failed to react even with the amino group of phenylalanine methyl ester (H–Phe–OMe) and the acid 9 could be isolated as the major product (Scheme 6). The reduced reactivity of 6c could be due to steric factors resulting from the shorter alkyl chain separating the oligopeptide and the ester group. However, the conjugations of the two peptides 2 and 7 as amide derivatives of dicarboxylic acids, having six or eight methylene groups were very effective.
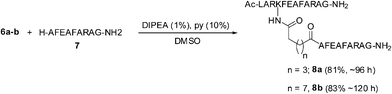 | ||
| Scheme 5 Conjugation reaction of a nonapeptide, 7 with o-fluorophenyl ester derivatives of a dodecapeptide, 2. | ||
 | ||
| Scheme 6 The o-flurophenyl ester derivative, 6c of 2 failed to react with amino groups and the major product resulted from the hydrolysis of the o-fluorophenyl ester. | ||
The effectiveness of the methodology was further proved by conjugating the peptide 2 with an amino derivative (10) of the blood group B antigenic determinant, a trisaccharide 3,4 (Scheme 7). The trisaccharide 10 was first treated with the diester, 3a to give the o-fluorophenyl ester derivative 11 in good yield under mildly basic conditions.
 | ||
| Scheme 7 Conjugation of the trisacharide derivative 10 with the dodecapeptide 2. | ||
This derivative (11) of the trisaccharide was then treated with the peptide 2 under more basic conditions (1% DIPEA and 10% pyridine in DMSO) to give the peptide-carbohydrate conjugate 12 (Scheme 7).
It was also possible to conjugate the dodecapeptide derivative 6a to two free lysine residues of a 42-residue helix loop helix6polypeptide, 13. The formation of the doubly conjugated product, 14 in good yields (Scheme 8) demonstrated further the usefulness of the methodology.
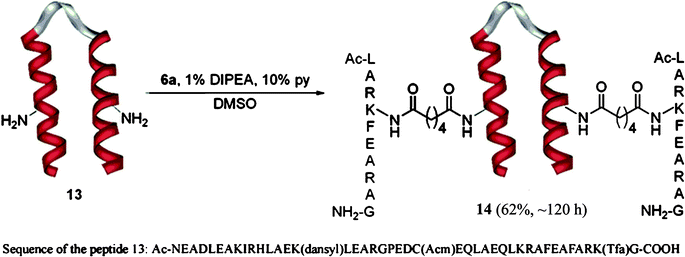 | ||
| Scheme 8 Double conjugation of the polypeptide 2 to the helix loop helix motif 13. The peptide contains 4 lysines residues two of which have free side chain amino groups. The side chain of lysine 15 is modified by incorporation of a fluorophore (dansyl = 5-(dimethylamino)naphthalene-1-sulfonyl) and the side chain of lysine 41 is protected with a Tfa (trifluoracetyl) group. The side chain of Cystein 24 is protected with an Acm (acetylaminomethyl) group. | ||
Conclusion
To conclude, we have developed a very efficient methodology for the conjugation of peptides with molecules carrying a free amino group. The use of differentially reactive esters of aliphatic dicarboxylic acids provides a cheap and efficient alternative to currently existing conjugation techniques for linking amines. The choice of esters was guided by a study of the reactivity of a set of phenyl esters with an amino group. The variation of the length of the linker is easily achieved by selecting a dicarboxylic acid of suitable length. We believe that linkers prepared according to this strategy has the potential to become important reagents in biotechnology research including not only peptides and carbohydrates but more importantly proteins.Experimental
Abbreviations used in the paper
DCM – Dichloromethane, DIC – N,N′-Diisopropylcarbodiimide, DIPEA – N,N'-Diisopropylethylamine, DMAP – 4-Dimethylaminopyridine, DMSO – Dimethyl sulfoxide, equiv – Equivalents, HCTU – (2-(6-Chloro-1H-benzotriazole-1-yl)-1,1,3,3-tetramethylaminium hexafluorophosphate), HOBt – Hydroxybenzotriazole, py – Pyridine, TEA – Triethylamine, TFA – Trifluoroacetic acid, TIS – Triisopropylsilane.General methods
All reagents and solvents were purchased from commercial sources and were used without further purification. Thin layer chromatography (TLC) was performed on 60 F254silica and 60 F254aluminum oxide plates (Merck) and spots were visualized with UV light (λ = 254 nm). Sep-Pak C–18 cartridges (0.7 g) were from Waters Corporation, Milford, MA, USA. NMR spectra were recorded on a Varian Unity INOVA (1H at 499.9 MHz) or a Varian Unity (13C at 100.6 MHz and 19F at 376.3 MHz) spectrometer. Chemical shifts are referenced directly via the internal standards: 1H NMR were referenced to the TMS signal (1H δ 0.0), 13C NMR were referenced to the middle deuterochloroform signal (13C δ 77.0), 19F NMR spectra were referenced to CCl3F indirectly via the lock signal. All the peptides and peptide conjugates were characterized by MALDI-TOF mass spectrometry, without an internal standard and calibration, on an Applied Biosystems Voyager-DE PRO instrument. Electrospray mass spectra (ESI-MS) for direct-infused dilute methanol solutions were recorded in both positive and negative ion modes using a Perkin-Elmer SCIEX API 150-EX mass spectrometer. HRMS characterization of the diesters was carried out on an Apex-Qe Ultra 7T instrument (Bruker Daltonics) using ESI in the positive ion mode. Melting points were measured on Stuart SMP10 Melting Point Apparatus and were uncorrected.Peptide synthesis
Peptides were synthesized using automated, solid-phase methodology on an Applied Biosystems 433A peptide synthesizer employing standard Fmoc/tBu strategy using the FastMoc synthesis program. The syntheses were performed on 0.1 mmol scale and H2N–RinkAmide-ChemMatrix (PCAS BioMatrix Inc) resin with a loading of 0.46 mmol g−1 was used as the solid support. All the coupling steps were conducted with a HCTU/6Cl-HOBt/DIPEA (Iris Biotech GmbH and Pepnet Inc.) activation cocktail. Fmoc deprotection was achieved by piperidine treatment. All reagents used in the peptide synthesis were prepared according to the manufacturer's protocols. The side chains of the amino acids (Iris Biotech GmbH and Pepnet Inc.) were protected by base-stable groups: tert-butyl ester (Asp, Glu), trityl (His, Asn, Gln) and 2,2,4,6,7-pentamethyldihydrobenzofuran-5-sulfonyl (Arg). Peptide cleavage from the solid support was achieved by treatment with the cleaving cocktail (TFA/TIS/H2O : 95/2.5/2.5 v/v) Purifications of the peptides were performed by HPLC (semi-preparative hypersil C–18 column 250 × 20 mm, 5 μm particle size) using two set of solvents: A (10% CH3CN/90% H2O/0.1% TFA), B (90% CH3CN/10% H2O/0.1% TFA). The synthesized peptides were characterized by MALDI-TOF MS using α-cyano-4-hydroxy-cinnamic acid as the matrix.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 10 mL), the phenol derivative (1 mmol) and DIC (126 mg, 1 mmol) were added at rt (25 °C). The reaction mixture was stirred for 2 h and the solvent was removed under reduced pressure. The crude esters (1a–d) were purified using column chromatography (silica gel) eluting with CH2Cl2.
:1, 10 mL) for 2 h at rt (25 °C). The reaction mixture was concentrated and the residue was subjected to column chromatography (silica gel) using CH2Cl2 as eluent to get the active diesters 3a and 3b.
:1, 10 mL) was stirred overnight (∼15 h) at rt (25 °C). The reaction mixture was concentrated under reduced pressure, diluted with CH2Cl2 (30 mL) and washed with 10% HCl (30 mL). The organic layer was concentrated and purified by column chromatography (silica gel) eluting with CH2Cl2 to get the diester 5 as colorless oil in 93% yield. 1H NMR (500 MHz, CDCl3) δ 7.17 (m, 4H), 2.88 (t, J = 6.9 Hz, 2H), 2.67 (t, J = 6.9 Hz, 2H), 1.46 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 197.9 , 164.5 (d, J = 49.7 Hz), 153.9 (d, J = 249.8 Hz), 127.3 (d, J = 7.0 Hz), 124.4 (d, J = 3.9 Hz), 123.5 , 116.7 (d, J = 18.4 Hz), 82.6 , 42.5 , 27.8; 19F NMR (376 MHz, CDCl3) δ –128.7 (m).
:3, 10 mL) and allowed to stand at rt (25 °C). The deprotection of the tert-butyl group was monitored by TLC (silica gel, CH2Cl2) and was found to be complete in 2 h. The reaction mixture was concentrated under reduced pressure and the crude product crystallized upon storage at −4 °C. Mono-o-flurophenyl succinate thus obtained was taken to the next step without further purification.
:1, 10 mL), the phenol derivative (1 mmol) and DIC (126 mg, 1 mmol) were added at rt (25 °C). The reaction mixture was stirred for 2 h and the solvent was removed under reduced pressure. The crude esters (1a–d) were purified using column chromatography (silica gel) eluting with CH2Cl2.
:1, 10 mL) for 2 h at rt (25 °C). The reaction mixture was concentrated and the residue was subjected to column chromatography (silica gel) using CH2Cl2 as eluent to get the active diesters 3a and 3b.
:1, 10 mL) was stirred overnight (∼15 h) at rt (25 °C). The reaction mixture was concentrated under reduced pressure, diluted with CH2Cl2 (30 mL) and washed with 10% HCl (30 mL). The organic layer was concentrated and purified by column chromatography (silica gel) eluting with CH2Cl2 to get the diester 5 as colorless oil in 93% yield. 1H NMR (500 MHz, CDCl3) δ 7.17 (m, 4H), 2.88 (t, J = 6.9 Hz, 2H), 2.67 (t, J = 6.9 Hz, 2H), 1.46 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 197.9 , 164.5 (d, J = 49.7 Hz), 153.9 (d, J = 249.8 Hz), 127.3 (d, J = 7.0 Hz), 124.4 (d, J = 3.9 Hz), 123.5 , 116.7 (d, J = 18.4 Hz), 82.6 , 42.5 , 27.8; 19F NMR (376 MHz, CDCl3) δ –128.7 (m).
:3, 10 mL) and allowed to stand at rt (25 °C). The deprotection of the tert-butyl group was monitored by TLC (silica gel, CH2Cl2) and was found to be complete in 2 h. The reaction mixture was concentrated under reduced pressure and the crude product crystallized upon storage at −4 °C. Mono-o-flurophenyl succinate thus obtained was taken to the next step without further purification.
A solution of mono-o-fluorophenyl succinate (700 mg, 3.3 mmol), pentafluorophenol (736 mg, 4 mmol) and DIC (505 mg, 0.8 mL, 4 mmol) in CH3CN/pyridine (9:1, 10 mL) was stirred for 2 h at rt (25 °C). The reaction mixture was concentrated under reduced pressure and purified by column chromatography (silica gel) eluting with CH2Cl2 to get the diester 3c as a white solid in 93% yield. m.p. 81 °C, 1H NMR (500 MHz, CDCl3) δ 7.19 (m, 4H), 3.14 (m, 2H), 3.09 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 178.1, 169.0, 168.0, 155.2, 152.7, 142.8, 127.4, 127.3, 124.5, 124.4, 123.6, 116.8, 116.7, 28.6, 28.4; 19F NMR (376 MHz, CDCl3) δ –129.1 (m), –152.9 (m), –158.1 (m), –162.6 (m). HRMS: calculated for C16H8F6NaO4 [M + Na]+: 401.0224, observed: 401.0222,.
Likewise, the peptide-linker conjugate 6b and 6c were synthesized from 2 and 3b or 3c in 84% and 78% yield respectively, MALDI-MS: Expected for 6b for [M + H]+: 1657.94, Observed: 1657.26; Expected for 6c for [M + H]+: 1572.79, Observed: 1572.34.
Likewise, the peptide-linker conjugate 8b was synthesized from 7 and 6b in 83% yield, MALDI-MS: Expected for [M + H]+: 2482.90, Observed: 2482.44
:3:3:2) showed complete conversion into a slower, ninhydrin-positive spot (1 h), then it was concentrated, co-concentrated with water several times, and lyophilized to give a colorless solid (59 mg). An aliquot of this material (25 mg) was dissolved in water (1 mL), and applied onto a 0.4 g C–18 Sep-Pak cartridge (packed in water). Elution with first water, then with increasing amounts of water-methanol (10% steps, 3 mL each, up to 20%) gave fractions containing pure product (compound 10), and these were pooled, evaporated shortly to remove methanol, then lyophilized to give colorless solid 10 (17 mg). The material showed high intensity m/z 630.2 [M + Na]+ and 606.2 [M − H]−ESI-MS positive and negative ions, respectively. An aliquot of this material (12 mg, 0.02 mmol) was taken up in dry DMSO (0.5 mL) containing pyridine (0.05 mL), and then 3a (20 mg, 0.05 mmol) was added. After 30 min, TLC (ethyl acetate-acetic acid-methanol-water, 6:3:3:2 detection with 5% sulfuric acid + charring) showed appearance of a fast rf 0.7 spot and disappearance of the starting sugar rf 0.3 spot. After 3 h, all starting material had disappeared and the mixture was diluted with water-acetic acid (98:2, 3 mL), washed with dichloromethane-ether 1:2, the aq phase was evaporated to approx. ½ volume, and applied to a 0.5 g C–18 Sep-Pak cartridge (packed in water). Elution with first water, then the same solvent with increasing amounts of CH3CN (10% steps, up to 60%) gave TLC-pure fractions containing 11 which were pooled, evaporated to half volume and then lyophilized. Yield: 11 mg, 69%. 1H NMR (D2O, δHOD = 4.68) δ 7.28 (d, J = 8.2 Hz, 2H), 7.13 (d, J = 8.2 Hz, 2H), 7.01–7.12 (m, 4H), 5.14 (d, J = 2.1 Hz, 1H), 5.11 (d, J = 4.0 Hz, 1H), 4.50 (d, J = 7.8 Hz, 1H), 3.54—4.20 (m, 16H), 3.29 (dd, J = 3.0, 10.0 Hz, 1H) , 3.09 (d, J = 2.7 Hz, 1H), 2.84 (m, 1H), 2.75 (m, 1H), 2.53 (m, 2H), 2.39 (m, 2H), 1.63 (m, 4H), 0.86 (d, J = 6.4 Hz, 3H), 13C NMR (100 MHz, D2O) δ 174.7, 173.9, 155.0, 152.6, 137.6, 137.4, 135.7, 135.6, 135.5, 129.0, 128.9, 128.9, 128.8, 128.0, 127.9, 125.2, 125.1, 125.1, 123.8, 123.7, 121.7, 121.6, 121.6, 121.5, 121.5, 116.9, 116.8, 116.7, 101.0, 98.6, 93.3, 76.8, 74.8, 72.3, 72.0, 71.4, 69.9, 69.7, 69.5, 68.8, 68.3, 67.9, 66.7, 63.79, 61.5, 61.1, 36.2, 34.0, 34.0, 33.2, 24.8, 24.0, 15.4; 19F NMR (376 MHz, CDCl3) δ –127.5; MALDI-MS: Expected for [M + Na]+: 852.30, Observed: 852.16
Acknowledgements
Dr hab. Piotr Stefanowicz, Faculty of Chemistry, University of Wrocław is gratefully acknowledged for providing HR-MS data.References
- R. Storer, C. Aciro and L. Jones, Chem. Soc. Rev., 2011, 40, 2330–2346 RSC.
- G. Hermanson, Bioconjugate techniques, 1996 Search PubMed.
- A. T. Gudkov and G. V. Shekbvatova, Zh. Obshch. Khim., 1978, 48, 2146 CAS.
- M. Green and J. Berman, Tetrahedron Lett., 1990, 31, 5851–5852 CrossRef CAS.
- L. Kisfaludy and I. Schon, Synthesis, 1983, 4, 325–327 CrossRef.
- L. Baltzer, Anal. Bioanal. Chem., 2011, 400, 1653–166 CrossRef CAS.
- H. Zahn and F. Schade, Chem. Ber., 1963, 96, 1747–1750 CrossRef CAS.
- S. Kreisky, Acta Chem. Scand., 1957, 11, 913–914 CrossRef CAS.
- A.-B. L. Fransson, Y. Xu, K. Leijondahl and J.-E. Bäckvall, J. Org. Chem., 2006, 71, 6309–6316 CrossRef CAS.
- T. Shimizu, R. Kobayashi, H. Ohmori and T. Nakata, Synlett, 1995, 650–652 CrossRef CAS.
- H. Backer and J. Homan, Receuil des Travaux Chimiques des Pays-Bas, 1939, 1939, 1048 Search PubMed.
- E. Kallin, H. Lönn and T. Norberg, Glycoconjugate J., 1988, 5, 3–8 CrossRef CAS.
- P.-M. Åberg, L. Blomberg, H. Lönn and T. Norberg, J. Carbohydr. Chem., 1994, 13, 141–161 CrossRef.
Footnote |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/c1ra00530h/ |
| This journal is © The Royal Society of Chemistry 2012 |