Bimetallic Pd(II)/Fe(II)-mediated self-assembly of three-dimensional hybrid multilayers with a terpyridine-contained poly(vinylpyridine) derivative as a linker on substrate surface†
Jiang
Liu
,
An
Liu
,
Meng
Chen
and
Dong-Jin
Qian
*
Department of Chemistry, Fudan University, 220 Handan Road, Shanghai, 200433, P. R. China. E-mail: djqian@fudan.edu.cn; Fax: +86 21 65643666; Tel: +86 21 6564 3666
First published on 3rd November 2011
Abstract
Pd(II)/Fe(II)-mediated three-dimensional bimetal-organic multilayers have been constructed on the substrate surfaces with the use of a polymeric ligand of terpyridine-contained poly(vinylpyridine) (PVPTPy) derivative as a linker as well as inorganic salts of Na2PdCl4 and Fe(BF4)2 as connectors. The assembly process was monitored by measuring the absorption spectra, which revealed two main absorption bands at wavelengths of 250∼340 nm and about 580 nm corresponding to the electron transition of pyridyl or terpyridyl substituents as well as metal–ligand charge transfer, respectively. X-Ray photoelectron spectra revealed that the multilayer of (Pd)Fe-PVPTPy was composed of the elements of B(1s), Cl(2p), C(1s), Pd(3d), N(1s), O(1s), F(1s) and Fe(2p), which confirmed formation of the bimetal-organic multilayers. Morphologies of the multilayers were characterized by using scanning electron microscope and atomic force microscopy. Cyclic voltammograms revealed a couple of irreversible redox waves with cathodic peaks appearing in the potential range from −0.2 to −0.4 V (vs. Ag/AgCl), which related to the scan rates and layer numbers of the multilayers. This redox wave was designated as one electron transfer of Fe(II)-PVPTPy/Fe(III)-PVPTPy in the multilayers. The current density of the redox wave decreased with increasing layer numbers of the films due to an increase of the film resistance. Both the polymeric ligand and its bimetal-organic multilayers showed a broad emission at wavelengths of 390∼450 nm. A red shift for the maximum emission band was recorded in the (Pd)Fe-PVPTPy films. Because the transition metal ions can selectively coordinate with either pyridyl or terpyridyl coordinative site, the present method may be developed to design and construct the desired multi-functional metal–organic hybrid materials.
Introduction
Metal-mediated self-assembly of three-dimensional (3D) multilayers or frameworks has recently attracted growing attention because they can be designed as multifunctional materials for the development of opto-electronic devices and for gas separation and storage, ion exchange as well as catalysts.1,2 Different from the traditional open-framework inorganic materials like molecular sieves and zeolites,3 metal–organic multilayers are constructed based on coordination bond of transition metal ions and multidentate ligands (MDLs) with the chemical structure similar to coordination polymers (CPs).4 Thus, they are not only a kind of porous material but also supramolecular materials with functions of both metal ions and organic ligands. With the development of supramolecular assembly, many metal–organic multilayers, frameworks, or CPs with unique structure have been constructed in the past decades with the use of transition metal ions as connectors and multidentate ligands like carboxylate, amine or pyridine derivatives as linkers.5Besides the commercially available MDLs such as 4,4′-bipyridyl, many novel MDLs have been recently synthesized and used as linkers to construct 3D metal–organic multilayers at interfaces. For instance, Kurth and coworkers reported a “bidentate-like” bisterpyridine ligand, 2,2′:4′,4′′-terpyridinium,1′′,1′′′′-[1, 4- phenylenebis(methylene)] bis[6′-(2-pyridinyl)-, bis[hexafluorophosphate(1-)], which could coordinate with transition metal ions to produce viologen-like polyelectrolyte multilayers with interesting electronic, optical and magnetic properties.6 By using electrostatic layer-by-layer (LBL) self-assembly method, electrochromic multilayers of the viologen-like poly-electrolytes with anionic polymers have been constructed. Some ligand-like metal-complexes have similar structures to 4, 4′-bipyridyl, which have been largely developed and used as linkers by Constable and coworkers in the past decades. They have designed and synthesized many supramolecular 4,4′-bipyridyl-like metal-complexes,7 which could act as linkers to construct 1D, 2D and 3D coordination polymers or multilayers by the Langmuir–Blodgett (LB) and LBL methods.8 Furthermore, the tetrapyridylporphyrin and its metalated derivatives (metalloporphyrins) are tetradentate MDLs. Because of their optical, electrical and catalytic behaviors, many porphyrin-based organized multilayers have been synthesized in the solutions and at interfaces.9,10
We are interested in the design and construction of 3D organized metal–organic multilayers or frameworks at interfaces based on an interfacial coordination reaction between transition metal ions and various multidentate ligands. Because the MDLs were connected by metal ions via a coordination bond, the as-prepared 3D multilayers showed highly thermal, chemical, and structural stability as well as structural regularity and controllability,11 the features of which resulted in these multilayers as attractive supramolecular materials. For instance, we have found that multilayers of multiporphyrin arrays can stabilize in aqueous solutions with pHs from 1.4 to 14, and do not dissolve in commonly used organic solvents.12 Moreover, with the choice of MDLs, the porosity of the frameworks can be designed, which makes the immobilized multilayers act as attractive 3D porous networks to encapsulate small guest molecules on the solid surfaces.13 These immobilized porous multilayers could be used as heterogeneous catalysts for organic reactions or hydrogen evolution.14
In the present work, a terpyridine-contained poly(vinyl-pyridine) derivative (PVPTPy, Scheme 1) was synthesized and used as an MDL to bind with transition metal ions to form 3D multilayers directly on the substrate surface. This polymeric ligand contains two kinds of coordinative sites, pyridine (Py) and terpyridine (TPy), which can bind with different transition metal ions. Hence, based on the desired purposes, one can design and assemble 3D metal–organic multilayers by using different metal ions. The Py coordinative site has been widely used in the synthesis of inorganic complexes and construction of metal–organic frameworks.15 On the other hand, derivatives of TPy are considered as versatile building blocks for covalent or noncovalent architectures, which offer the possibility to build up a wide range of functionalized metallo-supramolecular materials and to construct polymer architectures by metalated complexation.16 Here, the Pd(II) ion was used as the connector to bind with Py coordinative site and Fe(II) ion was used as another connector to bind with the TPy site. As a result, bimetallic 3D hybrid multilayers could be constructed. The assembly process was monitored by UV-vis absorption spectra and the multilayer composition was detected using X-ray photoelectron spectra. Morphologies of the multilayers were characterized by using scanning electron microscopy and atomic force microscopy. Fluorescence emission properties of the terpyridine ligands, 3D LBL films and electrochemical behaviors of Fe(II) ions in the multilayers were investigated. Because various transition metal ions could be selectively used to bind with either Py or TPy coordinations site, the present method may be developed to design and assemble multi-functional supramolecular hybrid materials.
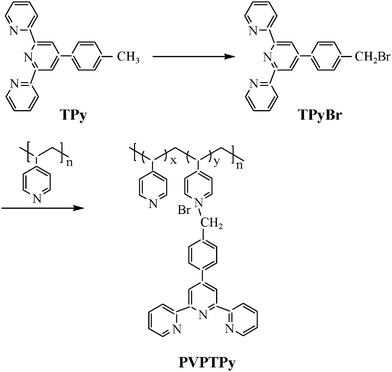 | ||
| Scheme 1 Schematic representation for the synthesis of the terpyridine-containing poly(vinylpyridine) derivative. | ||
Experimental section
Materials
4-(2,2′:6′2′′-Terpyridin-4′-yl)toluene, (p-chloromethyl-phenyl)trichlorosilane, 4,4′-bipyridyl, poly(4-vinylpyridine) (PVP), sodium tetrachloropalladate (II), iron(II) tetrafluoroborate hexahydrate and 4-pyridinethiol were purchased from Sigma-Aldrich Co. All chemicals were used as received without further purification. Ultrapure water (18.2 MΩ cm) was prepared with a Rephile filtration unit (China).Synthesis of terpyridine-containing poly(vinylpyridine) derivative
A schematic representation for the synthesis of the polymeric ligand of PVPTPy is shown in Scheme 1. Briefly, 4-(2,2′:6′2′′-terpyridin-4′-yl)benzyl bromide (TPyBr) was synthesized according to the literature method.171H NMR (CDCl3, δ, ppm, Fig. S1, ESI†), 8.76 (m, 4H), 8.69 (d, 2H), 7.91 (m, 4H), 7.55 (d, 2H), 7.38 (dd, 2H), 4.72 (s, 2H). Calc. For C22H16N3Br: C, 65.84; H, 3.99; N, 10.47%. Found: C, 65.29; H, 4.42; N, 10.20%. Selected IR (KBr, cm−1, Fig. S2†), 3052, 2927, 1590, 1567, 1515, 1471, 1386, 840, 790, 736, 504.The polymeric ligand PVPTPy was obtained by dissolving TPyBr in acetonitrile, which was then added to an acetonitrile solution of PVP in a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 (TPyBr : monomer of the PVP), and refluxed for 10 h under Ar atmosphere. After being evaporated on a rotary evaporator to reduce the solvent volume, the pale yellow product was filtered and washed using acetonitrile to remove unreacted TPyBr. Finally, the product was dried under vacuum at room temperature. 1H·NMR (CD3OD, δ, ppm, Fig. S1, ESI†), 8.66–8.76 (br, m, TPy), 7.38–8,00 (br, m, TPy), 8.04–8.67 (br, m, Py), 6.49–7.08 (br, s, Py), 1.21–2.18 (br, m, –CHCH2–). Selected IR (KBr, cm−1, Fig. S2, ESI†), 3022, 2927, 2860, 1592, 1561, 1466, 1415, 1383, 1221, 991, 825, 790, 738.
:10 (TPyBr : monomer of the PVP), and refluxed for 10 h under Ar atmosphere. After being evaporated on a rotary evaporator to reduce the solvent volume, the pale yellow product was filtered and washed using acetonitrile to remove unreacted TPyBr. Finally, the product was dried under vacuum at room temperature. 1H·NMR (CD3OD, δ, ppm, Fig. S1, ESI†), 8.66–8.76 (br, m, TPy), 7.38–8,00 (br, m, TPy), 8.04–8.67 (br, m, Py), 6.49–7.08 (br, s, Py), 1.21–2.18 (br, m, –CHCH2–). Selected IR (KBr, cm−1, Fig. S2, ESI†), 3022, 2927, 2860, 1592, 1561, 1466, 1415, 1383, 1221, 991, 825, 790, 738.
The molar fraction of the TPy substituents in the produced polymer of PVPTPy was estimated by absorption spectra in the dilute methanol solution.
Assembly of bimetal-organic multilayers at interfaces
Bimetal-mediated multilayers on the quartz substrate and indium tin oxide (ITO) surfaces were constructed according to the processes in Scheme 2, which could be divided into two parts. The first one was the assembly of the Pd-PVPTPy monolayer, which contained the following main steps: (1) formation of a coupling layer of O3SiC6H4CH2Cl (A) by immersing the hydrophilic substrate in the methanol solution of (p-chloromethylphenyl)trichlorosilane overnight at room temperature, (2) covalently attaching the 4,4′-bipyridyl (BPy) layer (B) by refluxing the substrate A in a 2 mg mL−1 BPy acetonitrile solution for 8 h, (3) Pd-mediated coordinative assembly of the BPy-Pd layer (C) by immersing the substrate B in an aqueous solution of 0.1 mmol L−1Na2PdCl4 for 20 min, (4) assembly of the first PVPTPy layer (D) by immersing the substrate C in the methanol solution of 2 mg mL−1PVPTPy for 20 min. As a consequence, the Pd-PVPTPy monolayer modified substrate was obtained, which was then used to coordinate with Fe(II) or Pd(II) to construct 3D multilayers as shown in part II (Scheme 2). All assembly experiments were performed at room temperature (about 20 °C).
The second part was the assembly of Pd/Fe-PVPTPy multilayers. As shown in Scheme 2 (Part II), two routes were used during experiments; one was by immersing the substrate D in a methanol solution of 1 mmol L−1Fe(BF4)2 and methanol solution of 2 mg mL−1PVPTPy alternatively to form (Pd)Fe-PVPTPy (E) bimetallic organic multilayers, another one was by immersing the substrate D in the aqueous solution of 0.1 mmol L−1Na2PdCl4 and mixture of Fe(BF4)2-PVPTPy (molar ratio of Fe(BF4)2 relative to TPy substituents in the polymer was 1:1) alternatively to form Pd/Fe-PVPTPy (F) bimetallic organic multilayers. The reaction time for each step was also 20 min. In both cases, the substrates were well washed and dried after the assembly of metal ions and the ligands. This process was repeated until the required layers of the films were obtained.
It is noted that the LBL multilayers could be assembled on both sides of the quartz or ITO substrate, which has been considered when the absorption intensity was calculated. For the ITO electrode, only one side of the substrate was covered with indium-tin oxide, so multilayers on the other side have no contribution to the redox behaviors. For the gold electrode, only one side was covered with the LBL multilayers.
Characterization of bimetal-organic multilayers
The UV-vis absorption spectra were measured by using a Shimadzu UV-1601 UV-vis spectrophotometer. Fluorescence spectra were recorded by using Shimadzu RF-5301PC spectrophotometer. Fourier transform infrared spectra (FITR) were measured by using a Nicolet NEXUS 470 spectrometer, operating at a resolution of 0.5 cm−1 at 25 °C.X-Ray photoelectron spectra (XPS) for the metal–organic multilayers on the quartz substrate surfaces were recorded using a VGESCALAB MKII multifunction spectrometer, with non-monochromatized Mg Kα X-rays as the excitation source. The system was carefully calibrated by Fermi-edge of nickel, Au 4f2/7 and Cu 2p2/3 binding energy. Pass energy of 70 eV and step size of 1 eV were chosen when taking spectra. In the analysis chamber pressures of 1∼2 × 10−7 Pa were routinely maintained. The binding energies obtained in the XPS analysis were corrected by referencing the C1s peak to 284.60 eV.
Scanning electron spectroscopic measurements were performed on a Shimadzu SSX-550 electron microscope. The samples were assembled on the gold substrate surface at different layers. This gold substrate surface was firstly covered by one layer of 4-pyridinethiol,18 which was then reacted with the 0.1 mmol L−1Na2PdCl4 aqueous solution to form a monolayer of pyridyl resulting in the surface layer similar to that of the substrate C in Scheme 2. Atomic force microscopy (AFM) images were also observed on the gold substrate surface by using an SPM-9500J3 scanning probe microscope (Shimadzu). Tapping mode was used with a tip fabricated from silicon (130 μm in length with ca. 40 kHz resonant frequency) in air.
Electrochemistry
The cyclic voltammogram (CV) for the multilayers of Pd/Fe-PVPTPy was measured by using an electrochemical analyzer (CHI 601b). A Pt wire and Ag/AgCl electrode were used as the auxiliary and reference electrodes, respectively, and the indium tin oxide (ITO) electrode coated with the bimetal-organic multilayers, was used as the working electrode with 10 mmol L−1KCl as the electrolyte. An initial potential of 0.8 V was applied for 2 s, and subsequently cyclic scans to a final potential of −0.6 V were done for 10 cycles. The CV curves and data reported in the present work were the 10th cycle. All electrochemical measurements were done under Ar atmosphere at room temperature.Results and discussion
Synthesis and characterization of the polymeric PVPTPy ligand
To construct 3D Pd/Fe-PVPTPy bimetal-organic hybrid multilayers, a polymeric multidentate ligand, shown in Scheme 1, was synthesized.17 Commercial PVP was used as the starting material and reacted with the synthesized 4-(2,2′:6′,2′′-terpyridin-4′-yl)benzyl bromide (TPyBr) to produce the polymeric ligand of PVPTPy, which contained two kinds of coordinative sites; one was a pyridyl (Py) and the other was a terpyridyl (TPy) substituent. These two kinds of coordinative sites could react with different transition metal ions, resulting in a possibility to design and construct 3D multi-functional building blocks.15,16
Fig. 1 shows the absorption spectra for compounds of PVP, TPyBr and PVPTPy in dilute methanol solutions, which revealed the following two features. The first one is the main absorption band for the reactants of PVP and TPyBr appearing at about 255 and 280 nm, corresponding to the π–π* transition of two compounds, respectively. The second one is that two absorption bands were recorded for the polymeric ligand of PVPTPy and appeared at about 254 and 280 nm, the data of which indicated that the produced PVPTPy was composed of PVP and TPyBr. Based on the absorption intensity of TPyBr and PVPTPy at 280 nm, the molar ratio of the TPy coordination site relative to that of Py in the polymer of PVPTPy was estimated to be about 1:9.
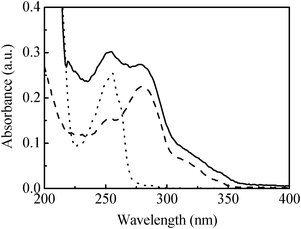 | ||
| Fig. 1 Absorption spectra for the compounds of PVP (······), TPyBr (-----) and PVPTPy (—) in the dilute methanol solutions. | ||
Assembly of three-dimensional Pd/Fe-PVPTPy bimetal-organic multilayers at interfaces
Two types of metal–organic multilayers have been designed and assembled. As shown in Scheme 2, the first one used Na2PdCl4 to connect the first layer of PVPTPy on the BPy-modified substrate surface (C, Scheme 2), and then Fe(BF4)2 used as the connector during the whole multilayer assembly process to form 3D (Pd)Fe-PVPTPy multilayers (E). The second one used Na2PdCl4 as the alternative layer (connector) and Fe-PVPTPy complex as the linker to form 3D Pd/Fe-PVPTPy multilayers (F). These assembly processes were monitored by absorption spectra, XPS and electrochemical analysis.We have previously reported that PdCl42− ions could coordinate with multidentate ligands containing pyridyl substituents, such as BPy, tetrapyridylporphyrin and PVP to form 2D or 3D coordination polymer multilayers or multiporphyrin arrays.10,11 Here, to confirm the possibility of the assembly of 3D multilayers with the linkers of PVPTPy, we firstly assembled Pd-PVPTPy multilayers with the use of Na2PdCl4 as the connector. Fig. 2 shows the absorption spectra of the Pd-PVPTPy multilayers after each assembly of the multidentate ligand of PVPTPy, which revealed a broad absorption band between 200 and 400 nm. This absorption was attributed to the π–π* transition of the Pd-Py coordination units in the Pd-PVPTPy multilayers and in agreement with the spectral feature of the Pd(II)-mediated tetrapyridylporphyrin (TPyP) arrays.12
 | ||
| Fig. 2 Absorption spectra for the Pd-PVPTPy multilayers on the quartz substrate surface. From bottom to top: BPy-modified quartz and then one to four layers of Pd-PVPTPy multilayers. Inset: a plot of absorption intensity at 250 nm to the layer numbers. | ||
A plot of the absorption intensity at 250 nm to the layer numbers assembled was inserted in Fig. 2, which revealed an almost linear increase during the multilayer assembly. This indicated that a similar amount of PVPTPy was coordinated for each assembly and confirmed the formation of the 3D Pd-PVPTPy multilayers.
Secondly, after the first layer of PVPTPy (Scheme 2, D) was assembled on the quartz substrate surface, construction of 3D (Pd)Fe-PVPTPy bimetal-organic multilayers was performed with the use of Fe(BF4)2 as the connector. The absorption spectra (Fig. S3, ESI†) for the (Pd)Fe-PVPTPy multilayers revealed two groups of absorption bands; one between 200 and 400 nm together with another one at about 580 nm. Based on the absorption spectra of Pd-PVPTPy multilayers in Fig. 2, and those in the previous work about the Fe-terpyridine complexes,7,8 we can conclude that the first absorption band corresponded to the π–π* transition of the Pd-Py coordination units, and that the second one corresponded to the metal–ligand change transfer (MLCT) of the Fe-TPy complex.
Research on the coordination chemistry of the 4′-(4′′′-pyridyl)-2,2′:6′,2′′-terpyridine (pyterpy; it contains two kinds of coordinative sites as PVPTPy) multidentate ligand has revealed that pyterpy could act as a tridentate ligand when reacted with Fe2+ ions in which the 4-pyridyl ring was not coordinated.7 On the other hand, when it reacted with PdCl42− ions, the pyridyl substituent acted as the coordination site.8 This means that with different transition metal ions, one could prepare bimetallic coordination polymers or frameworks with the use of pyterpy. Thus, besides using Na2PdCl4 or Fe(BF4)2 as the connector and PVPTPy as the linker to form Pd-PVPTPy or Fe-PVPTPy multilayers, we further synthesized a polymeric complex of Fe-PVPTPy, which was used as a linker to coordinate with the connector of Na2PdCl4. Such an assembly process resulted in the formation of a bimetallic coordination bond spontaneously as described below.
Fig. 3 shows absorption spectra of the 3D Pd/Fe-PVPTPy multilayers assembled by using Na2PdCl4 as the connector and Fe-PVPTPy complex as the linker. Similar to the spectral features in Fig. S3 (ESI†), two main absorption bands were recorded and fell into the range 250∼400 and ∼580 nm, corresponding to the π–π* transitions of PVPTPy and the MLCT of the Fe-TPy complex, respectively. A comparison of the absorption intensity in Fig. 2, S3 with that in Fig. 3 revealed a relatively stronger absorption intensity in the latter case. Because the absorption intensity was proportional to the amount of the ligand coordinated, we can conclude that more PVPTPy was assembled when the Fe-PVPTPy was used as the linker.
 | ||
| Fig. 3 Absorption spectra for the Pd/Fe-PVPTPy multilayers on the quartz substrate surface. From bottom to top: BPy-modified quartz, Pd-PVPTPy layer, and then one to four layers of Pd/Fe-PVPTPy multilayers (F, Scheme 2). | ||
X-Ray photoelectron spectra
To confirm the assembly processes of the metal–organic multilayers, elemental compositions for the main assembly steps were analyzed by the XPS spectra, which revealed several peaks in the binding energy from 180 to 800 eV.As an example, Fig. 4 shows the XPS spectra for the Pd/Fe-PVPTPy (F) multilayers on the quartz substrate surface, which revealed that eight elements were detected (besides the Si element from the substrate surface). The maximum peaks appeared at binding energies of 188.0, 197.8, 284.8, 338.0/343.4, 400.1, 532.3, 685.1 and 708.7 eV, which correspond to the elements of B(1s), Cl(2p), C(1s), Pd(3d), N(1s), O(1s), F(1s) and Fe(2p), respectively. The elements of C(1s) and N(1s) were from the polymeric ligand of PVPTPy; B(1s), F(1s) and Fe(2p) were from the connector of Fe(BF4)2, while Pd(3d) and Cl(2p) were from another connector of PdCl42−. Hence, these XPS data confirmed the formation of (Pd)Fe-PVPTPy bimetal-organic hybrid multilayers.
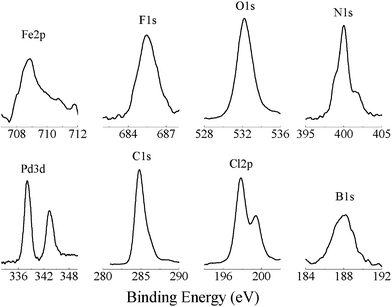 | ||
| Fig. 4 XPS spectra for the Pd/Fe-PVPTPy multilayer (F, in Scheme 2) on the quartz substrate surface. | ||
Table S1 (ESI†) summarizes the binding energy of the elements for other modified substrates of B, D (Pd-PVPTPy) and F (Pd/Fe-PVPTPy), the data of which reveal the following features: (i) four elements of C, N, O and Cl were detected for the BPy-modified substrate (B, Scheme 2), (ii) five elements were detected for the layer of Pd-PVPTPy (D), and (iii) eight elements (C, N, O, Cl, Pd, Fe, B and F) were detected for the Pd/Fe-PVPTPy metal–organic multilayers (F). These results confirm a successful assembly of the multilayers as shown in Scheme 2.
A close inspection of the data in Table S1 (ESI†) could find that the ratio of Pd(II) relative to Fe(II) was about 7.3:1 for the Pd/Fe-PVPTPy LBL multilayers, which was very close to the molar ratio of TPy to Py coordinative sites in the polymeric ligand of PVPTPy (9:1, as we estimated from the absorption spectra). Thus, based on the spectral features, XPS data and previous results on the Pd-PVP and Fe-TPy coordination polymers,7,8,11 we proposed a structural model for the Pd/Fe-PVPTPy bimetal-organic multilayers. As shown in Scheme 3, the Pd(II) ions coordinate with the N atoms of pyridyl coordinative site of the PVPTPy polymeric backbone, while Fe(II) ions coordinate with its terpyridyl coordinative site. Hence, after the LBL assembly, a bimetal-mediated 3D network structure was formed on the substrate surface.
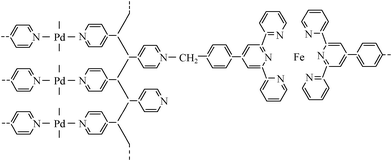 | ||
| Scheme 3 A proposed structural model for the (Pd)Fe-PVPTPy bimetal-organic multilayers. | ||
To confirm the proposed structural model in Scheme 3, we further did a control experiment by using Fe(BF)4 and PVP as connector and linker, respectively, for the formation of Fe-PVP LBL multilayers. The absorption spectra after each assembly of the ligand PVP are shown in Fig. S4 (ESI†), which reveal that the absorption intensity did not increase with the layer numbers assembled. The results indicated that, under the present experimental conditions, the Fe2+ ions were unable to coordinate with the pyridyl coordination site, though they could when additional heating or refluxing of the reaction system was performed. Thus, we suggest that the Pd(II) ions were coordinated with Py instead of Fe(II) ions in the present LBL multilayers as those having been previously reported.8,11
Another possibility in the LBL assembly is that Pd(II) ions could also coordinate with the terpyridine site of the PVPTPy ligand,7 which may result in mixed coordination with Fe(II)-TPy. To avoid such a possibility, we used the Fe-coordinated polymeric ligand of Fe-PVPTPy as the linker for the assembly of bimetal LBL multilayers of F in Scheme 2.
Morphologies of the bimetal-organic multilayers
To give more information about the as-prepared Pd/Fe-PVPTPy hybrid multilayers, we observed their morphologies using SEM and AFM. Here, the multilayers were prepared on the gold substrate surface, which was firstly modified by a monolayer of 4-pyridyl thiol (PySH). Assembly of the PySH monolayer on the gold surface was carried out according to literature methods,18 which was then reacted with Na2PdCl4 and Fe-PVPTPy alternatively.Fig. 5 shows several SEM images for the gold substrate covered by the monolayer of PySH (A) as well as one, three and five layers of the Pd/Fe-PVPTPy bimetal-organic hybrid films. These photos reveal the following features. Firstly, a smooth surface was observed for the gold surface covered by the initial layer of PySH (Fig. 5A), which indicated a uniform coverage for the self-assembled monolayer (SAM) of PySH on the gold surface. The Py-covered surface was used as a linker to coordinate with Pd(II) ions. Secondly, small domains were gradually formed after the Pd(II) ions were connected on the PySH modified gold surface (Fig. 5B). The size of these domains became larger when more layers of the Pd/Fe-PVPTPy were assembled. Finally, when more and more layers of Pd/Fe-PVPTPy were assembled, defects between these domains gradually disappeared to form a large rough surface (Fig. 5C and 5D).
 | ||
| Fig. 5 SEM images for the gold substrates covered by (A) the initial layer of PySH, denoted as AuSPy, as well as the LBL multilayers of (B) AuSPy(Pd/Fe-PVPTPy), (C) AuSPy(Pd/Fe-PVPTPy)3, and (D) AuSPy(Pd/Fe-PVPTPy)5. | ||
Three-dimensional images of Pd/Fe-PVPTPy were further characterized using AFM. As an example, Fig. 6A shows an AFM topographic image for the gold substrate surface covered by one layer of the multilayer (the same plate as observed in Fig. 5B), which indicated that the substrate was covered by a polymeric film with lots of small domains. A height file of the domains is shown in Fig. 6B, which suggests that the height of most domains is about 5 to 10 nm. Based on the structure of the film, these domains were composed of one layer of Pd(II) connector and one layer of Fe-PVPTPy.
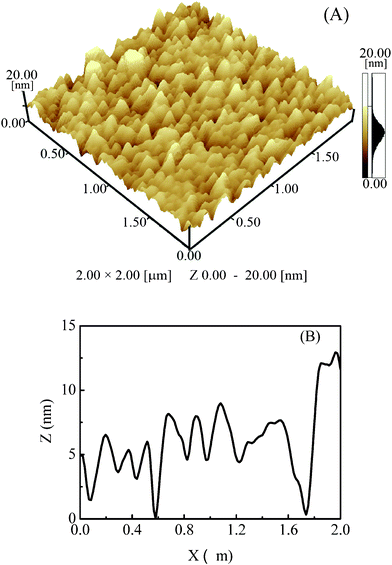 | ||
| Fig. 6 (A) AFM topographic image for the gold substrate surface covered by one layer of Pd/Fe-PVPTPy, (B) height profile corresponding to the AFM image. | ||
Images for the gold substrate covered by the monolayer of PySH (A) as well as one, three and five layers of the Pd/Fe-PVPTPy bimetal-organic hybrid films were shown in Fig. S5 (ESI†). These photos reveal similar rough surfaces but with a different height distribution. This difference was attributed to the fact that more and more Pd(II) and Fe-PVPTPy species were assembled.
Electrochemical studies
It has been known that the iron terpyridine complexes are a group of electroactive materials and recently often used as linkers to build up functional supramolecular hybrid materials.19 For instance, one couple of reversible Fe(II)/(III) oxidation-reduction process was recorded for the complex of Fe(pyterpy)2[BF4]2 in the MeCN solution.7 Derivatives of this iron complex have been widely used as complex-like ligands for the metal-directed assembly of 3D supramolecules with unique structure and opto-electrochemical properties.20Here, the redox behaviors for the ITO electrode modified by the LBL multilayers of (Pd)Fe-PVPTPy and Pd/Fe-PVPTPy were measured after each assembly of the ligands. As an example, Fig. 7A shows the CVs for the ITO electrode covered by one layer of Pd/Fe-PVPTPy in the 10 mM KCl electrolyte solution at the scan rates from 0.05 to 0.5 V s−1, which revealed a couple of irreversible redox waves. The cathodic peaks appeared in the potential range of −0.27∼−0.36 V vs.Ag/AgCl depending on the scan rates, while the anodic potentials appeared at about 0.35 V. These peaks correspond to the one electron transfer process of Fe(II)/Fe(III) in the modified Pd/Fe-PVPTPy films. The irreversible redox behaviors could be attributed to the film resistance caused by the polymeric ligand of PVPTPy and the positively charged alkylpyridinium moiety on the electrode surface, which has been often observed in the chemically modified electrodes of polymers and molecular assemblies.21
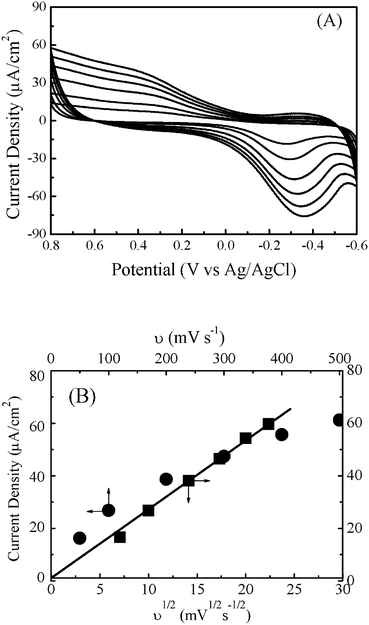 | ||
| Fig. 7 (A) Cyclic voltammograms for the ITO electrode modified by one layer of Pd/Fe-PVPTPy film in the 0.05 M KCl electrolyte solution at the potential scan rates from 50 to 500 mV s−1. (B) Plots of the cathodic current density to the scan rates (•) and root of the scan rates (■). | ||
With the increase of potential scan rates from 0.05 to 0.5 V s−1, it was found that the cathodic potentials shifted from −0.278 to −0.356 V. Based on these CV curves, the cathodic current density (ip/A, ip is current intensity, A is electrode surface area) was calculated. In the present work, we measured the CVs for the LBL film modified electrodes at scan rates from 50 to 500 mV s−1. As shown in Fig. 7A, the redox current density increased with increasing the scan rates.
For a film-modified electrode, a product of Dτ/d2, where D is the charge transport diffusion coefficient, τ is the experimental time scale (related to the time for the potential scan to traverse the wave) and d is the polymer film thickness, can be used to describe the charge transfer processes for a reversible reaction. Briefly, when Dτ/d2 ≫ 1, all electroactive sites in the film are in equilibrium with the electrode potential. That is, the film is so thin compared to the diffusion layer that the redox current density ip/A can be obtained by the equation22
| ip/A = n2ΓTF2ν/(4RT) | (1) |
| ip/A = (2.69 × 105) n3/2A D1/2Γν1/2d−1 | (2) |
Fig. 7B shows plots of cathodic current density to the scan rates and root of scan rates of the Pd/Fe-PVPTPy multilayer modified electrodes. A linear increase of the peak current density to the root of scan rates was obtained, but not to the scan rates. According to the eqn (1) and eqn (2), these observations suggest that, compared to the diffusion layer, the present Pd/Fe-PVPTPy multilayer was rather thicker. The redox reaction of Fe(II)/Fe(III) in the Pd/Fe-PVPTPy film was a diffusion-control process.
Increasing the layer numbers of PVPTPy resulted in a decrease of the current density. As shown in Fig. 8, the cathodic current density was about 26.8 μA cm−2 for the electrode modified by one layer of Pd/Fe-PVPTPy, which reduced to about 12.8 μA cm−2 when the electrode was modified by two layers of Pd/Fe-PVPTPy. Moreover, the peak separation ΔEp (ΔEp = Epa − Epc) was changed from 0.60 V to 0.70 V (peak position for the anodic current was estimated). Both features could be attributed to an increase of the film resistance after the multilayer formation.21 We have previously found that, because of the film resistance, electroactive species such as Fe(CN)63−/4− could not exchange an electron with the ITO electrode if its surface was covered by one layer of the Pd-PVP coordination polymer.13 Here, although the redox process was irreversible, electron transfer between the electrode surface and Fe(II)/Fe(III) ions in the multilayers was still possible, which may be attributed to the fact that the polymeric ligand of PVPTPy contained a relatively large terpyridine substituent resulting in a loosely packed arrangement of the molecules in the Pd/Fe-PVPTPy multilayers.
 | ||
| Fig. 8 Cyclic voltammograms for the ITO electrode modified by one (—) and two (----) layers of Pd/Fe-PVPTPy films in the 0.05 M KCl electrolyte solution at the potential scan rate of 100 mV s−1. | ||
Luminescent properties
Besides the strong coordination ability with the transition metal ions for the construction of building blocks, the terpyridine derivatives are also luminescent materials. These compounds can give off fluorescent emission or act as light-harvesting units to transfer the absorbed energy to central metal ions.23 For instance, our previous work has revealed that the terpyridine derivative of pyterpy and its metal complexes could give off yellow light emission in both solutions and LB films.24 It can also transfer the absorbed light energy to Eu(III) and Tb(III) ions and give off typical red and green emissions of the lanthanide complexes.Here, the luminescent behavior of the polymeric ligand of PVPTPy was investigated in methanol solutions and metal-mediated Pd/Fe-PVPTPy LBL multilayers. Fig. 9A shows the emission spectra for the compounds of TPyBr and PVPTPy in the dilute methanol solutions, excited at 342 and 350 nm, respectively. The excitation wavelength was determined based on the excitation spectrum of the ligand (not shown). A broad emission band was recorded in both cases, which was centered at wavelengths of 380 and 405 nm, respectively. That is, a slight red shift for the emission was observed when TPyBr was bound on the polymer of PVP. These emission spectra were similar to those having been observed for the terpyridine derivatives. For instance, the emission wavelength of pyterpy in solution was centered at about 364 nm,24 close to that observed in the present work.
 | ||
| Fig. 9 (A) Emission spectra for TPyBr (······) and PVPTPy (—) in the methanol solutions excited at 342 and 350 nm. (B) Emission spectrum for the three layers of Pd/Fe-PVPTPy hybrid multilayers on the quartz substrate surface excited at 376 nm. | ||
When the polymeric ligand of PVPTPy was assembled on the substrate surface to form Pd/Fe-PVPTPy multilayers, the emission wavelength was largely red shifted. As shown in Fig. 9B, the maximum emission band appeared at about 445 nm for three layers of Pd/Fe-PVPTPy LBL multilayers on a quartz surface, excited at 376 nm.
During experiments, we found that the Fe(II) ions could quench the emission from the ligands of TPyBr and PVPTPy. As shown in Fig. S6 (ESI†), the emission intensity for both ligands decreased with increasing concentrations of the Fe(BF4)2 salt in the solutions. It was almost completely quenched when the molar ratio of Fe/TPyBr was up to 2. However, such a fluorescent quench was not observed in the LBL multilayers, Probably because of the small molar fraction of Fe(II) ions in the LBL multilayers.
Conclusions
Bimetal-organic hybrid multilayers have been constructed with a terpyridine-contained polymer (PVPTPy) as a linker by the layer-by-layer method. The polymer contains two kinds of coordinative sites, that is pyridine and terpyridine, which can selectively coordinate with Pd(II) and Fe(II) transition metal ions resulting in the formation of Pd/Fe-PVPTPy bimetal-mediated metal–organic multilayers. An irreversible redox couple corresponding to one electron transfer process of the Fe(II)-/Fe(III)-PVPTPy redox couple was revealed. Increasing the layer numbers leaded to a decrease of the redox density due to the increase of the film resistance caused by the polymeric ligand of PVPTPy. Both the ligand in solutions and the Pd/Fe-PVPTPy LBL multilayers showed rather strong emissions in a range of wavelengths between 390 and 445 nm. Because various transition metal ions could be selectively used to coordinate with either pyridyl or terpyridyl coordinative site, we suggest that the present method may be developed to design and assemble bimetal-organic multi-functional supramolecular materials.Acknowledgements
The authors are grateful for the National Science Foundation of China (20873030, 91027042).References
- (a) C. M. Drain, A. Varotto and I. Radivojevic, Chem. Rev., 2009, 109, 1630–1658 CrossRef CAS; (b) I. Beletskaya, V. S. Tyurin, A. Y. Tsivadze, R. Guilard and C. Stern, Chem. Rev., 2009, 109, 1659–1713 CrossRef CAS; (c) B. H. Northrop, Y.-R. Zheng, K.-W. Chi and P. J. Stang, Acc. Chem. Res., 2009, 42, 1554–1563 CrossRef CAS.
- (a) J.-Z. Jiang and D. K. P. Ng, Acc. Chem. Res., 2009, 42, 79–88 CrossRef CAS; (b) B.-H. Ye, M.-L. Tong and X.-M. Chen, Coord. Chem. Rev., 2005, 249, 545–565 CrossRef CAS; (c) J. Sakamoto, J. van Heijst, O. Lukin and A. D. Schlüter, Angew. Chem., Int. Ed., 2009, 48, 1030–1069 CrossRef CAS.
- (a) R. J. White, R. Luque, V. L. Budarin, J. H. Clark and D. J. Macquarrie, Chem. Soc. Rev., 2009, 38, 481–494 RSC; (b) S. D. Ebbesen, B. L. Mojet and L. Lefferts, J. Phys. Chem. C, 2009, 113, 2503–2511 CrossRef CAS.
- (a) S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS; (b) Y. Lan, L. Xu, Y. Yan, J. Huang, A. de Keizer, N. A. M. Besseling and M. A. C. Stuart, Soft Matter, 2011, 7, 3565–3570 RSC.
- (a) J.-P. Zhang, Y.-Y. Lin, W.-X. Zhang and X.-M. Chen, J. Am. Chem. Soc., 2005, 127, 14162–14163 CrossRef CAS; (b) K. Ono, M. Yoshizawa, M. Akita, T. Kato, Y. Tsunobuchi, S. Ohkoshi and M. Fujita, J. Am. Chem. Soc., 2009, 131, 2782–2783 CrossRef CAS; (c) G. Bazzan, W. Smith, L. C. Francesconi and C. M. Drain, Langmuir, 2008, 24, 3244–3249 CrossRef CAS; (d) A. R. G. Smith, J. L. Ruggles, A. Yu and I. R. Gentle, Langmuir, 2009, 25, 9873–9878 CrossRef CAS; (e) J. L.Ruggles, K. M. Baldwin, S. A. Holt, G. J. Foran and I. R. Gentle, J. Phys. Chem. B, 2007, 111, 5651–5657 CrossRef CAS.
- (a) D. G. Kurth, J. P. Lopez and W.-F. Dong, Chem. Commun., 2005, 2119–2121 RSC; (b) F.-S. Han, M. Higuchi, T. Ikeda, Y. Negishi, T. Tsukuda and D. G. Kurth, J. Mater. Chem., 2008, 18, 4555–4560 RSC; (c) F.-S. Han, M. Higuchi and D. G. Kurth, Tetrahedron, 2008, 64, 9108–9116 CrossRef CAS; (d) F.-S. Han, M. Higuchi and D. G. Kurth, J. Am. Chem. Soc., 2008, 130, 2073–2081 CrossRef CAS; (e) D. G. Kuith and M. Higuchi, Soft Matter, 2006, 2, 915–927 RSC.
- (a) E. C. Constable, E. L. Dunphy, C. E. Housecroft, W. Kylberg, M. Neuburger, S. Schaffner, E. R. Schofield and C. B. Smith, Chem.–Eur. J., 2006, 12, 4600–4610 CrossRef CAS; (b) E. C. Constable and A. M. W. C. Thompson, J. Chem. Soc., Dalton Trans., 1992, 2947–2950 RSC.
- (a) C.-F. Zhang, A. Liu, M. Chen and D.-J. Qian, Chem. Lett., 2008, 37, 444–445 CrossRef CAS; (b) C.-F. Zhang, A. Liu, M. Chen, C. Nakamura, J. Miyake and D.-J. Qian, ACS Appl. Mater. Interfaces, 2009, 1, 1250–1258 CrossRef CAS.
- (a) X. Chen, L. Li and M. Liu, Langmuir, 2002, 18, 4449–4454 CrossRef CAS; (b) Y. Zhang, P. Chen, Y. Ma, S. He and M. Liu, ACS Appl. Mater. Interfaces, 2009, 1, 2036–2043 CrossRef CAS; (c) P. Ma, Y. Chen, Y. Bian and J. Jiang, Langmuir, 2010, 26, 3678–3684 CrossRef CAS.
- S. Takagi, M. Eguchi, D. A. Tryk and H. Inoue, J. Photochem. Photobiol., C, 2006, 7, 104–126 CrossRef CAS.
- (a) D.-J. Qian, T. Wakayama, C. Nakamura and J. Miyake, J. Phys. Chem. B, 2003, 107, 3333–3335 CrossRef CAS; (b) B. Liu, D.-J. Qian, H.-X. Huang, T. Wakayama, S. Hara, W. Huang, C. Nakamura and J. Miyake, Langmuir, 2005, 21, 5079–5084 CrossRef CAS; (c) B. Liu, D.-J. Qian, M. Chen, T. Wakayama, C. Nakamura and J. Miyake, Chem. Commun., 2006, 3175–3177 RSC; (d) B. Liu, M. Chen, C. Nakamura, J. Miyake and D.-J. Qian, New J. Chem., 2007, 31, 1007–1011 RSC; (e) C.-F. Zhang, M. Chen, C. Nakamura, J. Miyake and D.-J. Qian, Langmuir, 2008, 24, 13490–13495 CrossRef CAS.
- D.-J. Qian, C. Nakamura, T. Ishida, S. O. Wenk, T. Wakayama, S. Takeda and J. Miyake, Langmuir, 2002, 18, 10237–10242 CrossRef CAS.
- A. Liu, M. Chen and D.-J. Qian, Colloids Surf., A, 2010, 366, 183–190 CrossRef CAS.
- Z.-B. Ren, H.-L. Wang, Y.-Q. Cai, M. Chen and D.-J. Qian, Mater. Chem. Phys., 2011, 127, 310–315 CrossRef CAS.
- (a) S. Caramori, J. Husson, M. Beley, C. A. Bignozzi, R. Argazzi and P. C. Gros, Chem.–Eur. J., 2010, 16, 2611–2618 CrossRef CAS; (b) G. J. Halder and C. J. Kepert, J. Am. Chem. Soc., 2005, 127, 7891–7900 CrossRef CAS; (c) S. Pramanik, C. Zheng, X. Zhang, T. J. Emge and J. Li, J. Am. Chem. Soc., 2011, 133, 4153–4155 CrossRef CAS.
- (a) C. Haensch, M. Chiper, C. Ulbricht, A. Winter, S. Hoeppener and U. S. Schubert, Langmuir, 2008, 24, 12981–12985 CrossRef CAS; (b) S. K. Padhi, R. Sahu and V. Manivannan, Polyhedron, 2010, 29, 709–714 CrossRef CAS; (c) Y. T. Chan, C. N. Moorefield, M. Soler and G. R. Newkome, Chem.–Eur. J., 2010, 16, 1768–1771 CrossRef CAS.
- K. Hanabusa, A. Nakamura, T. Koyama and H. Shirai, Makromol. Chem., 1992, 193, 1309–1319 CrossRef CAS.
- (a) N. Nakamura, H.-X. Huang, D.-J. Qian and J. Miyake, Langmuir, 2002, 18, 5804–5809 CrossRef CAS; (b) T. Sawaguchi, F. Mizutani and I. Taniguchi, Langmuir, 1998, 14, 3565–3569 CrossRef CAS; (c) Z.-J. Zhang, S.-F. Hou, Z.-H. Zhu and Z.-F. Liu, Lanmguir, 2000, 16, 537–540 Search PubMed.
- Z.-L. Shi and N. Lin, J. Am. Chem. Soc., 2010, 132, 10756–10761 CrossRef CAS.
- (a) B. Liu, H.-X. Huang, C.-F. Zhang, M. Chen and D.-J. Qian, Thin Solid Films, 2008, 516, 2144–2150 CrossRef CAS; (b) E. C. Constable, C. E. Housecroft, M. Neuburger, D. Philips, P. R. Raithby, E. Schofield, E. Sparr, D. A. Tocher, M. Zehnder and Y. Zimmermann, J. Chem. Soc., Dalton Trans., 2000, 2219–2228 RSC.
- Z. Hu, J. Xu, Y. Tian, R. Peng, Y. Xian, Q. Ran and L. Jin, Carbon, 2010, 48, 3729–3736 CrossRef CAS.
- (a) S.-J. Dong, G.-L. Che and Y.-W. Xie, Chemically Modified Electrodes; Chinese Science Press: Beijing, 1995; pp 52–63 Search PubMed; (b) R. F. Murray, Electrochemical Chemistry; A. J. Bard ed, Mareel Dekker Press: New York, 1984; Vol. 13, pp 191 Search PubMed.
- P. Song, S.-G. Sun, P.-W. Zhou, J.-Y. Liu, Y.-Q. Xu and X.-J. Peng, Chin. J. Chem. Phys., 2010, 23, 558–564 CrossRef CAS.
- (a) C.-F. Zhang, H.-X. Huang, B. Liu, M. Chen and D.-J. Qian, J. Lumin., 2008, 128, 469–475 CrossRef CAS; (b) H.-F. Jiang, G. Wang, W.-Z. Zhang, X.-Y. Liu, Z.-Q. Ye, D.-Y. Jin, J.-L. Yuan and Z.-G. Liu, J. Fluoresc., 2010, 20, 321–328 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/c1ra00526j/ |
| This journal is © The Royal Society of Chemistry 2012 |