Octahedral Pt-Pd alloy catalysts with enhanced oxygen reduction activity and stability in proton exchange membrane fuel cells†
Young-Woo
Lee
,
A-Ra
Ko
,
Do-Young
Kim
,
Sang-Beom
Han
and
Kyung-Won
Park
*
Department of Chemical Engineering, Soongsil University, Seoul 156743, Republic of Korea. E-mail: kwpark@ssu.ac.kr; Fax: +82-2-812-5378; Tel: +82-2-820-0613
First published on 12th December 2011
Abstract
Structure-controlled Pt-based catalysts have been known to exhibit improved electrocatalytic activities due to particularly modulated surface structures favorable for hydrogen oxidation or oxygen reduction reactions. We prepare octahedral Pt-Pd alloy nanoparticles such as Pt3Pd1, Pt1Pd1, Pt1Pd3 by reducing metal ions with glycerol as a reducing agent in an aqueous solution. The octahedral Pt-Pd nanoparticles are well-defined alloy nanostructures with dominant {111} facets. Among them, the octahedral Pt3Pd1 shows excellent electrochemical properties i.e., much enhanced electrocatalytic activity and stability for oxygen reduction reactions in comparison with conventional Pt-based catalysts.
1. Introduction
Platinum is well known as the most effective catalyst for hydrogen oxidation and oxygen reduction reactions (ORR) in proton exchange membrane fuel cells and direct methanol fuel cells.1,2 In particular, however, the Pt cathode catalyst in ORR has critical problems such as kinetic limitation for oxygen diffusion rate and long-term stability of oxygen reduction at counter reaction.3 There have been many reports in the literature to solve such problems by controlling size and structure of Pt, bimetallic structures of Pt with other metal elements (Pt-M), and dual functional catalysts by supporting materials.4–9 It has been reported that bimetallic Pt-M electrocatalysts showed much improved ORR catalytic activity by alloying a 2nd metallic element such as Au, Fe, and Co with pure Pt because of the modified surface and electronic structures of Pt-M alloys which are favorable for ORR in comparison with pure Pt.10–12According to the typical ORR mechanism on Pt-based electrodes in acidic medium, Pt3M1 catalysts exhibit such an improved electrocatalytic activity in comparison with Pt-M alloy catalysts with different compositions between Pt and M.13 In particular, pure Pd as a 2nd metal with similar binding energies of O and OH exhibits comparable activity to that of Pt in an ORR. It has been reported that Pt-Pd alloy catalysts have exhibited excellent electrocatalytic activity due to the modified electronic structure of the electrodes in comparison with pure Pt. The modified electronic properties of the Pt-Pd catalysts might result from Pt containing a negative charge and vacant valence d-orbital and Pd containing a positive charge and fully occupied valence d-orbital. Recently, the Pt-Pd alloy catalysts with Pt-rich composition have showed a much higher oxygen reduction activity compared to a pure Pt catalyst.14,15
In order to enhance the ORR activity of metallic nanostructure catalysts, there have been many efforts to manipulate the structure and shape of the catalysts during the synthesis process of metallic nanoparticles.16–19 The structure or shape-controlled metallic catalysts have exhibited improved electrochemical activities due to a particular surface exposure, which is favorable for electrocatalytic reactions. In general, it has been reported that the order of the ORR activity in Pt-based catalysts with low index planes is as follows: (100) < (111) < (110) in HClO4 electrolyte.20,21 Lim et al. reported a bimetallic Pt-Pd nanostructure synthesized with L−ascorbic acid in an aqueous solution that could improve oxygen reduction activity in comparison with a polycrystalline Pt catalyst.22
Herein, we synthesized octahedral Pt-Pd alloy NPs with dominant {111} facets by reducing metal ions with glycerol as a reducing agent in aqueous solution. According to the metal reduction mechanism using glycerol, metal ions are reduced to metal atoms by glyceraldehydes containing an aldehyde (–CHO) functional group, which is transformed into glyceric acid containing a carboxyl (–COOH) group.23,24 The reducing agents containing many hydroxyl functional groups may result in a much faster reduction reaction than those containing fewer hydroxyl functional groups.25,26
The morphology, crystal structure, elemental composition, and chemical states of the NPs were investigated by transmission electron microscopy (TEM), energy dispersive X-ray spectroscopy (EDX), X-ray diffraction (XRD), and X-ray photoelectron spectroscopy (XPS). The electrochemical and electrocatalytic properties of the as-prepared catalysts were obtained in a typical electrochemical cell using a potentiostat.
2. Experimental
2.1. Synthesis of octahedral Pt-Pd alloy NPs
Octahedral Pt-Pd alloy NPs were prepared by simultaneously reducing Pd and Pt salts in aqueous solution. All chemicals used were of analytical grade. The 50 ml aqueous solution containing 269 mM glycerol (C3H5(OH)3, Fluka) as a reducing agent and 50 mg poly(vinyl pyrrolidone) (PVP, MW = 29![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, Aldrich) was prepared in a flask. To this mixture, the following metal salts for the Pt-Pd alloy NPs were dissolved with continuous stirring: 3.5 mM H2PtCl6 (Fluka) and 1.5 mM Na2PdCl4 (Aldrich) for Pt3Pd1 NPs, 2 mM H2PtCl6 and 2 mM Na2PdCl4, for Pt1Pd1 NPs, and 1.5 mM H2PtCl6 and 3.5 mM Na2PdCl4 for Pt1Pd3 NPs. The mixed solutions with a brown color were raised by 1 °C min−1 and kept for 3 h at 100 °C until Pt and Pd salts were completely reduced. The resulting colloid solutions with black color were cooled at 25 °C. The product was precipitated by centrifugation and washed several times with acetone, ethanol, and deionized water to remove PVP as a surfactant.
000, Aldrich) was prepared in a flask. To this mixture, the following metal salts for the Pt-Pd alloy NPs were dissolved with continuous stirring: 3.5 mM H2PtCl6 (Fluka) and 1.5 mM Na2PdCl4 (Aldrich) for Pt3Pd1 NPs, 2 mM H2PtCl6 and 2 mM Na2PdCl4, for Pt1Pd1 NPs, and 1.5 mM H2PtCl6 and 3.5 mM Na2PdCl4 for Pt1Pd3 NPs. The mixed solutions with a brown color were raised by 1 °C min−1 and kept for 3 h at 100 °C until Pt and Pd salts were completely reduced. The resulting colloid solutions with black color were cooled at 25 °C. The product was precipitated by centrifugation and washed several times with acetone, ethanol, and deionized water to remove PVP as a surfactant.
2.2. Preparation of octahedral Pt-Pd alloy NPs (20 wt%) on carbon supports
The carbon (Vulcan XC-72R) powder was stirred in 5 M HCl solution at 50 °C for 12 h and then washed with water and ethanol several times to remove impurity and HCl. An aqueous solution (10 ml) containing 1 ml of H2SO4 (0.5 M) and 1 ml of 1 M acetic acid was prepared in a flask. The as-synthesized Pt-Pd NPs (60.2 mg) and Vulcan XC-72R (240.8 mg) treated were added to the solution and mixed with continuous stirring at 25 °C for 24 h. The resulting powder was precipitated and washed with water and ethanol several times.2.3. Synthesis of spherical Pt3Pd1 alloy NPs (20 wt%) on carbon supports (S-Pt3Pd1/C)
The S-Pt-Pd/C catalyst was prepared by simultaneously reducing Pd and Pt salt by the polyol method. A solution of 3.5 mM H2PtCl6 (Fluka), 1.5 mM Na2PdCl4 (Aldrich), and 5 mM NaOH was dissolved in 50 ml of EG with Vulcan XC-72R treated in 5 M HCl solution at 50 °C for 12 h. The solution temperature was raised to 150 °C and was kept for 1 h until H2PtCl6 and Na2PdCl4 was completely reduced by EG. The resulting S-Pt-Pd/C was cooled at room temperature, washed with water and then with ethanol several times to remove EG.2.4. Structural analysis
For the structural analysis of the catalysts, XRD analysis was carried out using a Rigaku X-ray diffractometer with Cu Kα (λ = 0.15418 nm) source with a Ni filter. The source was operated at 40 kV and 100 mA. For all the XRD measurement, the resolution in the scans was kept at 0.02°. The morphology and size distribution of the catalysts were characterized by FE-TEM using a Tecnai G2 F30 system operating at 300 kV. TEM samples were prepared by placing drops of catalyst suspension dispersed in ethanol on a carbon-coated copper grid. Energy dispersive X-ray analysis of the catalysts was performed on a field emission transmission electron microscope (Tecnai G2 F30 system). XPS (Thermo Scientific, K-Alpha) analysis was carried out with the Al KαX-ray source of 1486.8 eV at the chamber pressure below 1 × 10−8 Torr and 200 W beam power. All high resolution spectra were collected using a pass energy of 46.95 eV. The step size and time per step were chosen to be 0.025 eV and 100 ms, respectively. Both ends of the baseline were set sufficiently far so as not to distort the shape of spectra, including tails. A small variation of the range of the base line did not affect the relative amount of fitted species (less than 1%). The C 1s electron binding energy was referenced at 284.6 eV and a nonlinear least-squares curve-fitting program was employed with a Gaussian–Lorentzian production function.2.5. Electrochemical analysis—oxygen reduction reaction
Electrochemical properties of the catalysts were measured in a three-electrode cell at 25 °C using a potentiostat (CH Instrument, CHI 700C). A Pt wire and Ag/AgCl (in saturated KCl) were used as a counter and reference electrode, respectively. The rotating ring disk electrode as a working electrode was polished with 1, 0.3, and 0.05 μm Al2O3 paste and then washed in deionized water. The catalyst ink was prepared by ultrasonically dispersing catalyst powders in an appropriate amount of Millipore water and 5 wt% Nafion® solution (Aldrich). The catalyst ink contained 1 mgmetal of all catalysts, 50 μL of Millipore water, 57.2 μL of 5 wt% Nafion® solution, and 43.8 μL of 2-propanol solution (C3H8O, Sigma). 0.9 μL of the catalyst ink was dropped onto a rotating ring disk electrode. After drying in a 50 °C oven, the total loading of all catalysts was ∼6.0 μgmetal on glassy carbon (area ∼0.126 cm2). To compare electrochemical properties and catalytic activity of the catalysts, CVs were obtained between −0.2 to +1.0 V in 0.1 M HClO4 solution. To clearly characterize electrochemical properties, electrochemical treatment was carried out between −0.2 and +1.0 V (50 CV cycles) with a scan rate of 50 mV s−1. The electrochemical active surface areas of these catalysts were determined by integrating hydrogen adsorption/desorption areas of the CVs. The oxygen reduction current–potential curves were obtained using linear sweep voltammetry at various rotation speeds in range from 100 to 2500 rpm in O2-saturated 0.1 M HClO4 solution by sweeping the potential from 0.8 to 0 V at a scan rate of 5 mV s−1. The accelerated durability test was carried out by applying linear potential sweeps for 2000 cycles between +0.4 and +0.9 V with a scan rate of 50 mV s−1 in O2-saturated 0.1 M HClO4 solution at 25 °C. To compare properties of the catalysts after the durability test, a CV curve was obtained between −0.2 to +1.0 V in 0.1 M HClO4 solution, and EASAs of the catalysts after the durability test were calculated based on the CV curve by integrating the area for hydrogen adsorption/desorption. The oxygen reduction current–potential curves after the durability test of the catalysts were obtained by sweeping the potential from 0.8 to 0 V at a scan rate of 5 mV s−1 and rotation disk speed of 1600 rpm.3. Results and discussion
3.1. Structure analysis of octahedral Pt-Pd alloy NPs
The octahedral Pt-Pd alloy NPs with various elemental compositions were prepared by simultaneously reducing H2PtCl6 and Na2PdCl4 in an aqueous solution containing glycerol as a reducing agent and PVP as a stabilizer at 100 °C for 3 h. The mole fractions of Pt to Pd metal salts for the Pt-Pd (X:Y) alloy NPs are as follows: 3.5 mM H2PtCl6 and 1.5 mM Na2PdCl4 for Pt-Pd (3:1) alloy NPs (denoted as Pt3Pd1), 2 mM H2PtCl6 and 2 mM Na2PdCl4 for Pt-Pd (1:1) alloy NPs (denoted as Pt1Pd1), and 1.5 mM H2PtCl6 and 3.5 mM Na2PdCl4 for Pt-Pd (1:3) alloy NPs (denoted as Pt1Pd3).
The structure analysis of the as-synthesized Pt-Pd NPs was examined by field-emission transmission electron microscopy (FE-TEM) in Fig. 1(a–c) (Fig. S1(a, d, and g), ESI†). The average size and yield of the octahedron in the Pt3Pd1, Pt1Pd1, and Pt1Pd3 NPs are ∼11.3 nm and ∼92.4%, ∼8.3 nm and ∼97.0%, and ∼8.4 nm and ∼84.7%, respectively (Fig. 1(a–c) and Fig. S2(a–c), ESI†). In the present synthesis of the octahedral Pt-Pd NPs, the glycerol can lead to faster reduction reaction of metal ions. Also, the fast reduction reaction might tend to thermodynamically minimize crystalline surface energy thus forming crystal facets with the lower surface energy in the structure. The surface energy of {111} facets in fcc metallic structures such as Pt and Pd is lower than that of other low-index planes.27 In the case of Pt, the surface energies of {111}, {100}, and {110} facets are 1.004, 1.378, and 2.009 eV atom−1, respectively. In the case of Pd, the surface energies of {111}, {100}, and {110} facets are 0.824, 1.152, and 1.559 eV atom−1, respectively. This shows that the octahedral Pt-Pd NPs can favorably expose {111} facets caused by the rapid reduction reaction in the synthetic atmosphere. Fig. 1(d–f) show line scanning profiles using energy dispersive X-ray spectroscopy (EDX) of the octahedral Pt-Pd NPs. As confirmed by EDX profiles (Fig. S1(c, f, and i), ESI†), the elemental compositions of Pt and Pd in the octahedral NPs are 74.74 and 25.26 at% for Pt3Pd1, 47.80 and 52.20 at% for Pt1Pd1, and 25.69 and 74.31 at% for Pt1Pd3, respectively. Fig. 1(g–i) show high-resolution TEM images of the octahedral Pt-Pd NPs. The as-synthesized Pt3Pd1, Pt1Pd1, and Pt1Pd3 NPs represent a d-spacing of the {111} plane corresponding to alloy structures between Pt and Pd metallic phases. The corresponding fast Fourier transform patterns (the insets of Fig. 1(g–i)) indicate that the octahedral Pt-Pd NPs are single nanocrystals enclosed by {111} facets. Furthermore, as shown in Fig. S1(b, e, and h), ESI,† the octahedral Pt3Pd1, Pt1Pd1, and Pt1Pd3 NPs can be homogeneously deposited on carbon (Vulcan XC-72R) as carbon supported catalysts for fuel cell applications.
![TEM images of Pt3Pd1 (a), Pt1Pd1 (b), and Pt1Pd3 (c). The inset shows HAADF-STEM (high-angle annular dark-field scanning transmission electron microscopy) images of Pt-Pd NPs for line-scanning profiles [white scale bar is 5 nm]. Line-scanning profiles across Pt-Pd NPs: Pt3Pd1 (d), Pt1Pd1 (e), and Pt1Pd3 (f) of the inset images of Fig. 1(a–c). HR-TEM Images of Pt-Pd NPs: Pt3Pd1 (g), Pt1Pd1 (h), and Pt1Pd3 (i). The insets indicate the FFT patterns of Pt-Pd NPs.](/image/article/2012/RA/c1ra00308a/c1ra00308a-f1.gif) | ||
| Fig. 1 TEM images of Pt3Pd1 (a), Pt1Pd1 (b), and Pt1Pd3 (c). The inset shows HAADF-STEM (high-angle annular dark-field scanning transmission electron microscopy) images of Pt-Pd NPs for line-scanning profiles [white scale bar is 5 nm]. Line-scanning profiles across Pt-Pd NPs: Pt3Pd1 (d), Pt1Pd1 (e), and Pt1Pd3 (f) of the inset images of Fig. 1(a–c). HR-TEM Images of Pt-Pd NPs: Pt3Pd1 (g), Pt1Pd1 (h), and Pt1Pd3 (i). The insets indicate the FFT patterns of Pt-Pd NPs. | ||
Fig. 2(a) shows X-ray diffraction (XRD) patterns of carbon supported octahedral Pt-Pd catalysts containing Pt3Pd1, Pt1Pd1, and Pt1Pd3 NPs. The XRD patterns of the octahedral Pt-Pd catalysts contain diffraction peaks corresponding to (111), (200) and (220) of a typical fcc crystal structure. Assuming a substitutional solid solution between metallic phases, the higher angle shift of the XRD peaks indicates alloy formation between Pt and Pd. As indicated in Fig. 2(b), the XRD peaks in the octahedral Pt-Pd NPs are shifted into higher 2θ values with an increasing amount of Pd, i.e., 67.67°, 67.75°, and 67.94° in (220) diffraction peaks corresponding to Pt3Pd1, Pt1Pd1, and Pt1Pd3 NPs, respectively. Based on the Vegard's law using the equation of dPtPd = X·dPt + (1-X)·dPd, the atomic composition of Pt and Pd in Pt3Pd1, Pt1Pd1, and Pt1Pd3 is 67.62 and 32.38 at%, 55.62 and 44.38 at%, and 27.14 and 72.86 at%, respectively. The lattice parameters of the octahedral Pt-Pd NPs reflecting the degree of alloying in the bimetallic structures are given in Fig. 2(c). Since the radius of the Pt atom (0.139 nm) is greater than the radius of a Pd atom (0.137 nm), the crystalline lattice parameters of the Pt-Pd NPs increase with increasing elemental composition of Pt. The octahedral Pt-Pd NPs appear on a straight fitted line based on the solid solution between Pt and Pd, which means a well-defined alloy formation between Pt and Pd. The surface chemical state and composition of individual components in the octahedral Pt-Pd/C catalysts were obtained by X-ray photoelectron spectroscopy (XPS) in comparison with Pt/C. For the octahedral Pt3Pd1, Pt1Pd1, and Pt1Pd3 alloy NPs, Pt 4f and Pd 3d spectra are shown in Fig. 3(a–h). The Pt 4f7/2 and 4f5/2 peaks typically appear at ∼71 and ∼74 eV, respectively, with a theoretical area ratio of 4:3. The Pd 3d5/2 and 3d3/2 peaks typically appear at ∼335 and ∼340 eV, respectively, with a theoretical area ratio of 3:2.28,29 The elemental compositions of Pt and Pd in the Pt-Pd NPs measured by XPS are as follows: 71.64 at% of Pt and 28.38 at% of Pd in the Pt3Pd1/C, 49.44 at% of Pt and 50.56 at% of Pd in the Pt1Pd1/C, and 34.07 at% of Pt and 65.93 at% of Pd in the Pt1Pd3/C. Furthermore, the Pt 4f peaks in the octahedral Pt-Pd/C and Pt/C consist of metallic and oxide states, i.e., the peaks for Pt0 and Pt2+ at 71.0 and 73.8 eV, respectively (Fig. 3(a–d)). The Pd 3d peaks in the octahedral Pt-Pd/C consist of metallic and oxide states, i.e., the peaks for Pd0, Pd2+, and Pd4+ at 335.2, 336.3 and 337.9 eV, respectively (Fig. 3(e–g)). By comparing the EDX, TEM, and XRD data of the NPs (Table 1), it can be concluded that the octahedral Pt-Pd NPs are well-defined alloy nanostructures. Furthermore, this represents that the octahedral Pt-Pd alloy NPs prepared by the present synthetic process show the homogeneous distribution of Pt and Pd atoms in surface and bulk of the alloy NPs.
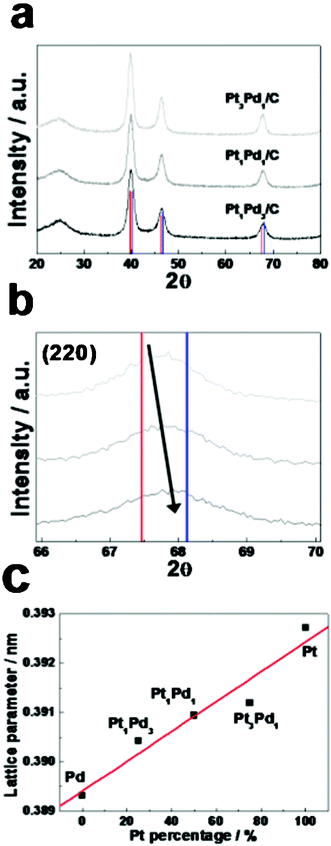 | ||
| Fig. 2 (a) Wide-range XRD patterns of Pt3Pd1/C, Pt1Pd1/C, and Pt1Pd3/C in comparison with XRD reference data of Pt (red) and Pd (blue). (b) The diffraction peaks of (220) planes in the catalysts are compared with XRD reference data of Pt (JCPDS No. 04-0802, red) and Pd (JCPDS No. 46-1043, blue). (c) The plot of lattice parameter versusPt atomic percentage of the Pt-Pd catalysts. | ||
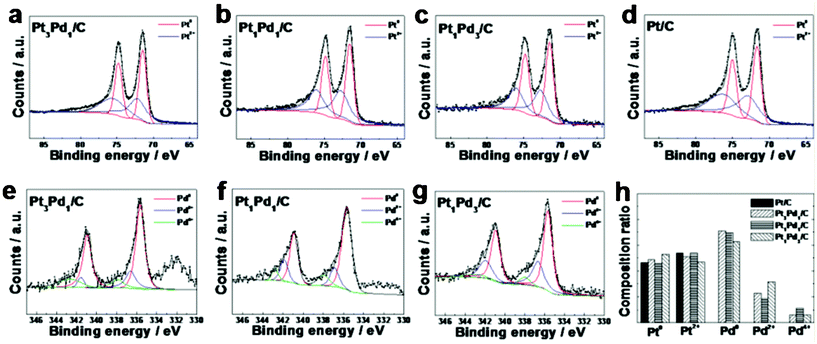 | ||
| Fig. 3 Surface and chemical analysis by XPS. Pt 4f spectra in Pt3Pd1/C (a), Pt1Pd1/C (b), Pt1Pd3/C (c), Pt/C (d). Pd 3d spectra in Pt3Pd1/C (e), Pt1Pd1/C (f), Pt1Pd3/C (g). Comparison of chemical states and ratios of Pt 4f and Pd 3d spectra of all catalysts measured and calculated from the XPS fitting data. | ||
3.2. Electrochemical analysis of octahedral Pt-Pd alloy NPs
To identify electrochemical properties of the as-prepared catalysts, cyclic voltammograms (CVs) were obtained in 0.1 M HClO4 between −0.2 and +1.0 V with a scan rate of 50 mV s−1 (Fig. S3, ESI†). The electrochemical active surface areas (EASAs) of the catalysts were measured by integrating the charges on the Hupd desorption region assuming a value of 210 μC cm−2 after double layer correction.10,11,30 The EASAs of Pt3Pd1/C, Pt1Pd1/C, Pt1Pd3/C, and Pt/C are 53.4 m2g−1Pt-Pd, 54.4 m2g−1Pt-Pd, 53.5 m2g−1Pt-Pd, and 95.3 m2g−1Pt, respectively. This represents that the octahedral Pt-Pd/C catalysts have high surface areas despite a relatively larger particle size. The ORR activity of the catalysts was measured using linear sweep voltammetry in O2-saturated HClO4 solution in comparison with the Pt/C (Fig. S4, ESI†). Based on the geometric area of the glassy carbon electrode, the ORR current densities of Pt3Pd1/C, Pt1Pd1/C, Pt1Pd3/C, and Pt/C at 0.55 V are 3.123, 0.718, 0.307, and 2.374 mA cm−2, respectively. The specific kinetic current densities (jk) of the catalysts can be measured from the loading mass or active surface area in the CVs. Fig. 4(a) shows the kinetic current densities per active surface area of as-prepared catalysts. The area-kinetic current densities (jk,Area) of Pt3Pd1/C, Pt1Pd1/C, Pt1Pd3/C, and Pt/C at 0.55 V in the ORR are 1.228, 0.277, 0.121, and 0.523 mA cm−2, respectively (Fig. 4(b)). Furthermore, the mass-kinetic current densities (jk,Mass) of Pt3Pd1/C, Pt1Pd1/C, Pt1Pd3/C, and Pt/C at 0.55 V are 0.937, 0. 302, 0.213, and 0.499 mA g−1Pt, respectively (Fig. 4(b)). In particular, the octahedral Pt3Pd1/C (Octa-Pt3Pd1/C) exhibits 2.35 times higher area-kinetic current density and 1.88 times higher mass-kinetic current density than those of the Pt/C. According to thermodynamic approach and density function theory of alloy formation, PtmXn (m > n, X = Pd, Ni, Co, Fe, Y) alloy catalysts have greater ORR activity and thermodynamically more stable state in comparison with different elemental compositions of PtmXn (m ≤ n) or pure Pt catalyst.31,32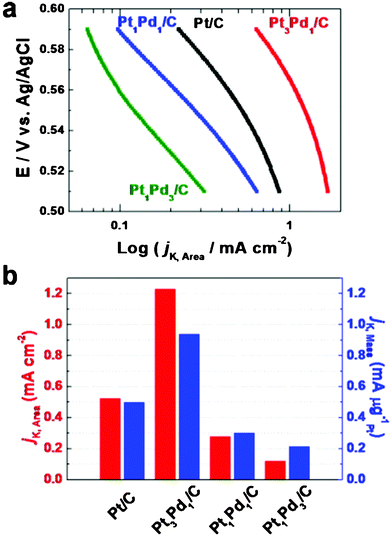 | ||
| Fig. 4 (a) Specific activity curves of as-prepared catalysts on oxygen reduction reaction at 0.55 V normalized to the EASAs calculated by integrating the hydrogen adsorption charge in the CVs. (b) Comparison of the ORR activities on as-prepared catalysts. Specific activity and mass activity were all measured at 0.55 V. | ||
The accelerated durability test of the ORR was performed by applying linear potential sweeps between 0.4 and 0.9 V in O2-saturated 0.1 M HClO4 solution at 25 °C. To confirm the effect of Pt3Pd1 alloy nanostructure on the ORR, we synthesized spherical Pt3Pd1 NPs deposited on carbon black (S–Pt3Pd1/C) by the polyol process using ethylene glycol with NaOH as additive agent (see Experimental section, ESI†). The average size of the S–Pt3Pd1 NPs is ∼3.86 nm. By EDX analysis, the elemental compositions of Pt and Pd in the as-synthesized S–Pt3Pd1 NPs are 71.64 at% and 28.36 at%, respectively (Fig. S5, ESI†). The ORR activity of the S–Pt3Pd1/C was measured using linear sweep voltammetry in O2-saturated HClO4 solution (Fig. S6, ESI†). The area-kinetic current densities (jk,Area) of Octa–Pt3Pd1/C, S–Pt3Pd1/C, and Pt/C at 0.55 V in the ORR are 1.228, 0.820, and 0.523 mA cm−2, respectively (Fig. 5(g)). Furthermore, the mass-kinetic current density (jk,Mass) of the Octa–Pt3Pd1/C (0.094 mA μg−1Pt) is 1.27 and 1.88 times higher than the S–Pt3Pd1/C (0.074 mA μg−1Pt) and Pt/C (0.050 mA μg−1Pt), respectively. Accordingly, the highly electrocatalytic activity for ORR may be due to the particular composition of the Pt3Pd1 alloy catalyst in comparison with pure Pt. Furthermore, it is confirmed that the octahedral Pt3Pd1 NPs with dominant {111} facets can provide much improved oxygen reduction reaction in comparison with spherical Pt3Pd1 NPs. Thus, we can demonstrate that the Octa–Pt3Pd1/C exhibits improved electrocatalytic properties due to octahedral shape with dominant {111} facets and particular elemental composition.
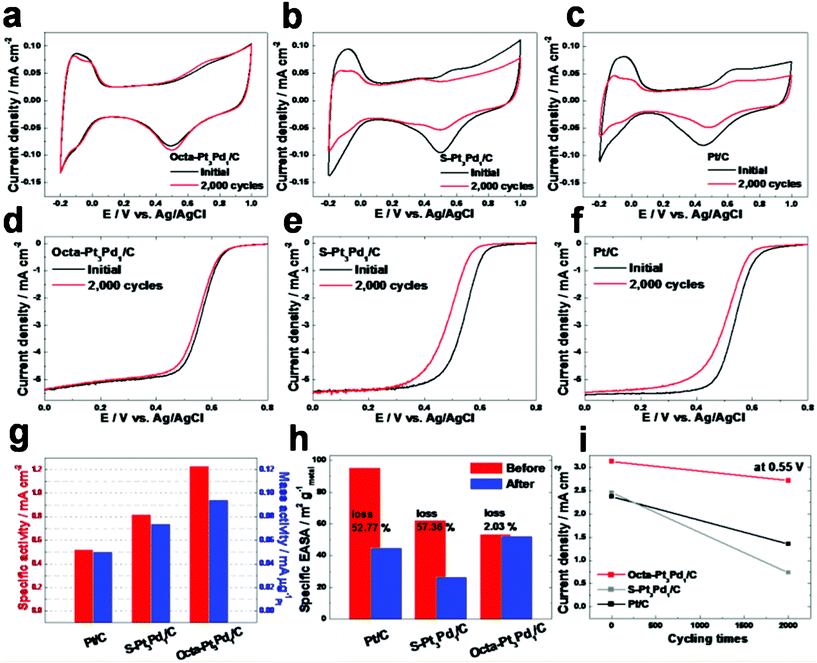 | ||
| Fig. 5 Cyclic voltammograms of Octa–Pt3Pd1/C (a), S–Pt3Pd1/C (b), and Pt/C (c) before and after durability test in Ar–saturated 0.1 M HClO4 with a scan rate of 50 mV s−1 at 25 °C. The accelerated durability test was carried out by applying linear potential sweeps between 0.4 and 0.9 V for 2000 cycles with a scan rate of 50 mV s−1 in O2–saturated 0.1 M HClO4 solution at 25 °C. ORR polarization curves of Octa–Pt3Pd1/C (d), S–Pt3Pd1/C (e), and Pt/C (f) before and after the durability test in O2–saturated 0.1 M HClO4 with a scan rate of 5 mV s−1. (g) Comparison of the ORR activities on as-prepared catalysts. Specific activity and mass activity were all measured at 0.55 V before the durability test. (h) Comparison of EASAs of Octa–Pt3Pd1/C, S–Pt3Pd1/C, and Pt/C before and after durability test. (i) Comparison of ORR current density at 0.55 V of Octa–Pt3Pd1/C, S–Pt3Pd1/C, and Pt/C before and after durability test in Fig. 5(d–f). | ||
The EASAs of Octa–Pt3Pd1/C, S–Pt3Pd1/C, and Pt/C before and after the ORR durability test were measured by integrating the charges on the Hupd desorption region of the CVs as shown in Fig. 5(a–c). In Fig. 5(h), the Octa–Pt3Pd1/C exhibits a negligible loss of 2.0% in EASA after 2000 cycles of the durability test whereas S–Pt3Pd1/C and Pt/C exhibit losses of 57.36% and 52.77% after the durability tests, respectively. Since the change of size and morphology for catalysts before and after durability test could affect the whole activity, their size distribution and shape should be observed and compared (Fig. S7–S9, ESI†). The Octa–Pt3Pd1/C shows a slight reduction of size and nearly the same morphology after the durability test, maintaining such an improved electrocatalytic activity (Fig. S7, ESI†). The improved stability of the Octa–Pt3Pd1/C might result from shape effect due to unique morphology.33,34 In contrast, after the durability test, S–Pt3Pd1/C and Pt/C exhibit non-uniform size distribution because of instability of metal catalysts resulting in deteriorated catalytic activity (Fig. S8 and S9, ESI†). Fig. 5(d–f) show the ORR measurements before and after the durability test between 0.0 and 0.8 V with a scan rate of 5 mV s−1 in O2-saturated 0.1 M HClO4 solution at 25 °C. In the case of half-wave potentials,35 the Octa–Pt3Pd1/C exhibits a slight decrease of ∼14 mV, whereas S–Pt3Pd1/C and Pt/C exhibit a rapid decrease of ∼50 and ∼34 mV, respectively. As indicated in Fig. 5(i), based on the geometric area of the glassy carbon electrode in the Fig. 5(d–f), the current density of the Octa–Pt3Pd1/C at 0.55 V after the durability test is slightly reduced from 3.123 mA cm−2 (before the durability test) to 2.712 mA cm−2. On the other hand, the current densities of S–Pt3Pd1/C and Pt/C after the durability test are rapidly reduced from 2.456 mA cm−2 and 2.374 mA cm−2 (before the durability test) to 0.735 mA cm−2 and 1.351 mA cm−2, respectively, at 0.55 V. Accordingly, we can expect that the highly improved electrocatalytic properties of the Octa–Pt3Pd1/C may result from thermodynamically stable surface exposure of {111} facets.
4. Conclusions
We have reported octahedral Pt-Pd alloy NPs synthesized using glycerol as a reducing agent. The Pt-Pd NPs such as Pt3Pd1, Pt1Pd1, and Pt1Pd3 for ORR have shown well-defined alloy formation and dominant {111} facets in the octahedral crystalline structure. In particular, the octahedral Pt3Pd1/C has exhibited remarkably excellent electrochemical properties i.e. much improved ORR electrocatalytic activity and stability. Thus, it is expected that the octahedral Pt3Pd1 alloy catalyst can be utilized as a promising cathode in fuel cell applications.Acknowledgements
This work was supported by New & Renewable Energy R&D Program (2008-N-FC08-P-01-3-030) and Manpower Development Program (20094020100010) for Energy & Resources of the Ministry of Knowledge and Economy, Korea.References
- S. Takaichi, H. Uchida and M. Watanabe, Electrochim. Acta, 2008, 53, 4699 Search PubMed
.
- Y.-W. Lee, A-R. Ko, S.-B. Han, H.-S. Kim, D.-Y. Kim, S.-J. Kim and K.-W. Park, Chem. Commun., 2010, 46, 9241 RSC
- N. M. Marković and P. N. Ross, Surf. Sci. Rep., 2002, 45, 117 CrossRef CAS
- Z. Lin, H. Chu, Y. Shen, L. Wei, H. Liu and Y. Li, Chem. Commun., 2009, 7167 RSC
- H. Liu and A. Manthiram, Energy Environ. Sci., 2009, 2, 124 RSC
- N. Maeda, T. Matsushima, H. Uchida, H. Yamashita and M. Watanabe, Appl. Catal., A, 2008, 341, 93 CrossRef CAS
- T. Saida, W. Sugimoto and Y. Takasu, Electrochim. Acta, 2010, 55, 857 Search PubMed
- S. Shen, T. S. Zhao, J. Xu and Y. Li, Energy Environ. Sci., 2011, 4, 1428 RSC
- S. An, J.-H. Park, C.-H. Shin, J. Joo, E. Ramasamy, J. Swang and J. Lee, Carbon, 2011, 49, 1108 Search PubMed
- Y. Gauthier, R. Baudoing-Savois, J. M. Bugnard, U. Bardi and A. Atrei, Surf. Sci., 1992, 276, 1 CrossRef
- Y. Xu, A. V. Ruban and M. Mavrikakis, J. Am. Chem. Soc., 2004, 126, 4717 CrossRef CAS
- Y. Zhou, K. Neyerlin, T. S. Olson, S. Pylypenko, J. Bult, H. N. Dinh, T. Gennett, Z. Shao and R. O'Hayre, Energy Environ. Sci., 2010, 3, 1437 RSC
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886 CrossRef CAS
- H. Li, G. Sun, N. Li, S. Sun, D. Su and Q. Xin, J. Phys. Chem. C, 2007, 111, 5605 CrossRef CAS
- Y. Wang and P. B. Balbuena, J. Phys. Chem. B, 2005, 109, 18902 CrossRef CAS
- S. E. Habas, H. Lee, V. Radmilovic, G. A. Somorjai and P. Yang, Nat. Mater., 2007, 6, 692 CrossRef CAS
- Y.-W. Lee, J.-K. Oh, H.-S. Kim, J.-K. Lee, S.-B. Han, W. Choi and K.-W. Park, J. Power Sources, 2010, 195, 5896 CrossRef CAS
- A. R. Tao, S. Habas and P. Yang, Small, 2008, 4, 310 CrossRef CAS
- Y.-W. Lee, S.-B. Han and K.-W. Park, Electrochem. Commun., 2009, 11, 1968 CrossRef CAS
- V. R.. Stamenkovic, B. Fowler, B. S. Mun, G. Wang, P. N. Ross, C. A. Lucas and N. M. Marković, Science, 2007, 315, 493 CrossRef CAS
- N. M. Marković, R. R. Adzic, B. D. Cahan and E. B. Yeager, J. Electroanal. Chem., 1994, 377, 249 CrossRef CAS
- B. Lim, M. Jiang, P. H. C. Camargo, E. C. Cho, J. Tao, X. Lu, Y. Zhu and Y. Xia, Science, 2009, 324, 1302 CrossRef CAS
- Y.-W. Lee, A-R. Ko, S.-B. Han, H.-S. Kim and K.-W. Park, Phys. Chem. Chem. Phys., 2011, 13, 5569 RSC
- C. Petibois, G. Cazorla and G. Déléris, Appl. Spectrosc., 2002, 56, 10 CrossRef CAS
- S.-W. Chou, C.-L. Zhu, S. Neeleshwar, C.-L. Chen, Y.-Y. Chen and C. C. Chen, Chem. Mater., 2009, 21, 4955 CrossRef CAS
- B. Lim, M. Jiang, J. Tao, P. H. C. Camargo, Y. Zhu and Y. Xia, Adv. Funct. Mater., 2009, 19, 189 CrossRef CAS
- L. Vitos, A. V. Ruban, H. L. Skriver and J. Kollár, Surf. Sci., 1998, 411, 186 CrossRef CAS
- K.-W. Park, J.-H. Choi, S.-A. Lee, C. Pak, H. Chang and Y.-E. Sung, J. Catal., 2004, 224, 236 CrossRef CAS
-
J. F. Moulder, W. F. Stickle, P. E. Sobol and K. D. BombenHandbook of X-ray photoelectron spectroscopy 1995Physical Electronics, Inc., Minnesota Search PubMed
- S. Sun, H. Yu, J. Hou, Z. Shao, B. Yi, P. Ming and Z. Hou, J. Power Sources, 2008, 177, 137 Search PubMed
- Z. Peng and H. Yang, J. Am. Chem. Soc., 2009, 131, 7542 CrossRef CAS
- J. Zhang, K. Sasaki, E. Sutter and R. R. Adzic, Science, 2007, 315, 220 CrossRef CAS
- B. Lim, X. Lu, M. Jiang, P. H. C. Camargo, E. C. Cho, E. P. Lee and Y. Xia, Nano Lett., 2008, 8, 4043 CrossRef CAS
- Z. Peng and H. Yang, J. Am. Chem. Soc., 2009, 131, 7542 CrossRef CAS
- D. Cao, L. Sun, G. Wang, Y. Lu and M. Zhang, J. Electroanal. Chem., 2008, 621, 31 Search PubMed
Footnote |
| † Electronic Supplementary Information (ESI) available: Fig. S1–S9 contains TEM, EDX, and electrochemical data. See DOI: 10.1039/c1ra00308a/ |
| This journal is © The Royal Society of Chemistry 2012 |