Synthesis of fullerene-containing poly(ethylene oxide)-block-polystyrene as model shape amphiphiles with variable composition, diverse architecture, and high fullerene functionality†
Xue-Hui
Dong
,
Wen-Bin
Zhang
*,
Yiwen
Li
,
Mingjun
Huang
,
Shuo
Zhang
,
Roderic P.
Quirk
and
Stephen Z. D.
Cheng
*
Department of Polymer Science, College of Polymer Science and Polymer Engineering, The University of Akron, Akron, OH 44325-3909, USA. E-mail: scheng@uakron.edu; Fax: + 1 330 972 8626; Tel: +1 330 990 9801; wz8@uakron.edu; Fax: +1 330 972 8626; Tel: +1 330 972 6931
First published on 8th November 2011
Abstract
A series of [60]fullerene (C60)-containing poly(ethylene oxide)-block-polystyrene (PEO-b-PS) with various numbers and different locations of C60 along the polymer chains were designed and synthesized via a combination of “click” chemistry and living/controlled polymerization techniques such as anionic polymerization, atom transfer radical polymerization, and reversible addition–fragmentation chain transfer polymerization. One C60 was tethered either to the end of a PS block (PEO-b-PS-C60) or at the junction point between PS and PEO blocks [PEO-(C60)-PS]; while multiple C60s could be attached randomly along the PS block (PEO-b-PS/C60). The reaction conditions were carefully controlled to ensure a quantitative C60 functionality at precise locations in the case of PEO-b-PS-C60 and PEO-(C60)-PS and to avoid crosslinking in the synthesis of PEO-b-PS/C60. The results have implications in the precision synthesis of fullerenepolymers in general. These C60-containing diblock copolymers possess different composition, diverse architecture, and high fullerene functionality. They can serve as model “shape amphiphiles” for the construction of complex hierarchical structures via the interplay between C60–C60 aggregation and block copolymerself-assembly/micro-phase separation.
Introduction
Recognition of the importance of anisotropic shape and interactions has led to an exciting field of “shape amphiphiles” for the construction of novel functional hierarchical structures in the past few decades. Shape amphiphiles usually refer to the hybrids of molecular segments of distinct shapes and competing interactions1 and have been predicted by computer simulation to exhibit rich phase behaviours and various unusual structures due to packing constraints and amphiphilic interactions.2,3 Typical shape amphiphiles include polymer tethered nanoparticles,4–8 disc–rod mesogenic hybrids,1 sphere–cube or disc–cube conjugates,9 where [60]fullerene (C60) is a frequent spherical building block.Fullerene is an intriguing class of carbon-based molecular nanoparticles with wide-ranging applications due to its outstanding electronic properties and the possibility to perform precise chemical modification.10–12Self-assembly of C60-based shape amphiphiles is an important approach to control their electronic properties via manipulating their ordered structure formation of C60 in multi-dimensions across different length scales.1,3,4,13–16 To achieve tailor-made hierarchical structures, the precise control of important molecular parameters, such as surface chemistry, chain composition, and polymer architecture, is a prerequisite. Although living/controlled polymerization techniques developed in the past few decades have furnished the chemists with sufficient control over the composition, molecular weight, and polydispersity of polymers, the coupling between polymer chains and functional materials such as C60 (so-called “grafting-to” approach) is often plagued by the difficulty in purification as a result of incomplete functionalization and possible multi-addition and chain degradation.17–20 On the other hand, the “growing-from” approach in the synthesis of fullerenepolymers is usually problematic due to C60's reactive nature toward common reactive intermediates.17–21 Challenges in the synthesis of fullerenepolymers thus lie in the precise control of C60 locations on a polymer chain while ensuring a quantitative functionality and a well-defined stable structure. This is particularly true for C60-functionalizedblock copolymers, where different sites for functionalization are available.
Block copolymers have been extensively studied in the past three decades due to their versatile self-assembly behaviours and diverse supramolecular structures.22,23 For example, most of the amorphous–semicrystalline block copolymers can grow single crystals in dilute solutions using the self-seeding techniques,24 in which the amorphous blocks are tethered on both of the folded surfaces of the single crystal lamellae.24–26 Amphiphilic block copolymers are known to form various micellar morphologies in selective solvents27,28 or phase-separate into different nanostructures in the bulk or thin films.29,30 A typical block copolymer that has been studied in great detail is poly(ethylene oxide)-block-polystyrene (PEO-b-PS).24,26,28 The combination of this block copolymer and C60 hence gives shape amphiphiles of distinct structures (Fig. 1), including surfactant-like block copolymers with C60 at the chain end (PEO-b-PS-C60), lipid-like block copolymers with C60 at the junction point [PEO-(C60)-PS], and block copolymers with multiple C60s randomly tethered at the PS block (PEO-b-PS/C60). Driven by the interplay between the C60–C60 aggregation and the block copolymerself-assembly, they provide a versatile platform to achieve hierarchically ordered arrangement of C60s. Some pioneering works have already been carried out in this direction with certain successes. Amphiphilic block polymers with C60 at the junction point, such as PS-(C60)-poly(N-isopropylacrylamide) (PNIPAM) and PS-(C60)-PEO, were reported though a quantitative functionality was not shown.31,32 Later on, improved synthesis by “click” chemistry was reported in the synthesis of a series of C60-functionalized thermo-responsive diblock copolymers with controlled numbers of C60 at designated positions.33 Functionalizing polymers with multiple C60s was reported extensively with different fullerene loadings, especially in combination with conjugated polymers to form so-called “double-cable” polymers as promising organic photovoltaic materials.34–41 However, the lack of control over the primary structure of such polymers in terms of grafting position, grafting density, etc., and their propensity to undergo crosslinking lead to an unsuccessful finding of well-defined “double-cable” structures.41 It is, therefore, essential to control precise locations of C60s while ensuring a high degree of functionality during synthesizing polymers.
![Model shape amphiphiles based on the combination of C60 and block copolymers with different C60 locations [orange: polystyrene; green: poly(ethylene oxide)].](/image/article/2012/PY/c1py00435b/c1py00435b-f1.gif) | ||
| Fig. 1 Model shape amphiphiles based on the combination of C60 and block copolymers with different C60 locations [orange: polystyrene; green: poly(ethylene oxide)]. | ||
Recently, we have demonstrated a “click” chemistry approach as a general method to incorporate fullerenes into functional materials under mild conditions (Scheme 1) by the model synthesis of C60-endcapped PS and poly(ε-caprolactone) with a quantitative fullerene functionality.6,7 The highly reactive prototype alkyne-functionalized fullerene, Fulleryne01, can be synthesized in three steps in high yields, facilitating the modular and efficient synthesis of fullerenepolymers.7 Complicated purification procedures, such as fractionation, can be avoided. Similar methods have also been reported by other groups in the synthesis of fullerene-based hybrid materials, such as porphyrin–C60 conjugate,42fullerene sugar ball,43 C60–PNIPAM,33 C60–viral nanoparticle conjugates44 among others.45–47 It is thus quite advantageous for a variety of applications, particularly in heterogeneous systems, biological environments, and multiple functionalizations.
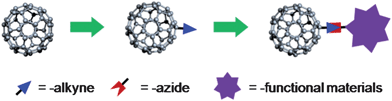 | ||
| Scheme 1 General functionalization methodology to Fullerene materials via “Click” chemistry. | ||
In this publication, we report our recent efforts in the synthesis of shape amphiphiles based on PEO-b-PS and C60 using combined techniques of living/controlled polymerization and “click” chemistry. Specifically, C60 is located at the end of the PS block (PEO-b-PS-C60), or at the junction point between PS and PEO blocks [PEO-(C60)-PS], or randomly tethered along the PS block (PEO-b-PS/C60) (Fig. 1). The “click” reaction ensures a quantitative C60 functionality at precise locations in the cases of PEO-b-PS-C60 and PEO-(C60)-PS and avoids cross-linking in the synthesis of PEO-b-PS/C60.
Experimental section
Chemicals and solvents
The following chemicals were used as received: [60]fullerene (MTR Ltd, >99.5%), sodium azide (NaN3, Aldrich, ReagentPlus®, >99.5%), epichlorohydrin (Acros, 99%), N,N-dimethylformamide (DMF, Aldrich, 99.9%), methanol (MeOH, Fisher Scientific, reagent grade), deuterated chloroform (CDCl3 Aldrich, 99.8 atom% D), N,N,N′,N′′,N′′-pentamethyldiethylene-triamine (PMDETA, Aldrich, 99%), triethylamine (TEA, Aldrich, 99%), 2-bromoisobutyryl bromide (Aldrich, 98%), oxalyl chloride (Aldrich, ReagentPlus®, >99%), ammonium chloride (NH4Cl, Aldrich, >99.5%), and sodium hydroxide (NaOH, Aldrich, >97.0%). 2,2′-Azobisisobutyronitrile (AIBN, Aldrich, 98%) was purified by recrystallization from ethanol. Cuprous bromide (CuBr, Aldrich, 98%) was freshly purified by stirring in acetic acid overnight, washed with acetone, and dried in vacuum. Dichloromethane (CH2Cl2, Fisher Scientific, reagent grade), styrene (Aldrich, 99%), and vinyl benzyl chloride (VBC, Aldrich, >90%) were purified by stirring over freshly ground calcium hydride for 12 hours and redistilled under vacuum before use. Fulleryne01 and S-1-dodecyl-S′-(r,r′-dimethyl-r′′-acetic acid)trithiocarbonate (TC) were prepared according to reported procedures.7,48 Preparation of poly(ethylene oxide) (PEO-OH) and C60-endcapped poly(ethylene oxide) (PEO-C60) is described in the ESI†.Instrumentation and characterizations
All 1H and 13C NMR spectra were acquired in CDCl3 using a Varian Mercury 300 or Varian 500 NMR spectrometer. The 1H NMR spectra were referenced to the residual proton impurities in CDCl3 at δ 7.27 ppm. The 13C NMR spectra were referenced to 13CDCl3 at δ 77.00 ppm. The molecular weight of the PEOhomopolymer was calculated based on the integration ratio between the peaks at 1.20 ppm [–C(CH3)3] and 3.64 ppm (–OCH2CH2–) in 1H NMR spectra (see ESI for details, eqn (S1)†). The molecular weight of the PS blocks in the block copolymer was calculated based on the integration ratio between the peak at 3.64 ppm (–OCH2CH2–, SA) and broad peaks between 7.40 and 6.30 ppm (the protons on the phenyl rings on PS blocks, SB) in 1H NMR spectra using eqn (1):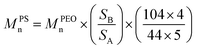 | (1) |
The degree of polymerization (N) and number average molecular weight (Mn) of poly(vinyl benzyl chloride) (PVBC) segments and PS segments of PEO-b-PS/Cl can be calculated based on eqn (2):
 | (2) |
Fourier transform infrared (FTIR) spectra were recorded on an Excalibur Series FT-IR spectrometer (DIGILAB, Randolph, MA) by casting polymer films onto KBr plates from polymer solutions or by making a KBr pellet from a mixture with the sample. The data were processed using Win-IR software.
Ultraviolet-visible light (UV-Vis) spectra were recorded on a HP 8453 UV-Vis Spectrometer System. Concentration dependent UV spectra were used to measure the degree of fullerene functionality based on the Beer–Lambert Law49 because the chromospheres in Fulleryne01 and the fullerenepolymers are identical. A calibration curve is then obtained using Fulleryne01 dilute solution in toluene (Fig. S1 in ESI†). The linear relationship is fitted to the following equation:
| A = kc + b | (3) |
| fC60 = cFulleryne01/cpolymer × 100% | (4) |
The C60 content in these polymers, C60 wt%, is estimated using the following equation:
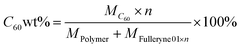 | (5) |
Size-exclusion chromatographic analyses (SEC) were performed using a Waters 150-C Plus instrument equipped with three HR-Styragel columns [100 Å, mixed bed (50/500/103/104 Å), mixed bed (103/104/106 Å)] and a double detector system with THF as eluent at a flow rate of 1.0 mL min−1 at 30 °C; the detector system consisted of a differential refractometer (Waters 410) and a laser light scatteringdetector (Wyatt Technology, DAWN EOS, λ = 670 nm). Regular SEC calibrations were conducted with PS standards (Polymer Laboratories).
General synthetic procedures
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)–), 3.64 (br, 770H, –CH2CH2O–), 1.93 (s, 6H, –(CO)C(CH3)2Br), 1.20 (s, 9H, (CH3)3C–O–). 13C NMR (CDCl3, 500 MHz, ppm, δ): 71.1 (–OCH2CH2–), 61.1 ((CH3)3C–O–), 55.6 (–(CO)C(CH3)2Br), 30.7 (–(CO)C(CH3)2Br), 27.5 ((CH3)3C–O–). FT-IR (cm−1): 2882, 1730 (CO), 1464, 1342, 1280, 1240, 1107, 958, 842. SEC (THF, RI detector): Mn = 11.1 g mol−1, Mw = 12.0 g mol−1, PDI = 1.07.
O)–), 3.64 (br, 770H, –CH2CH2O–), 1.93 (s, 6H, –(CO)C(CH3)2Br), 1.20 (s, 9H, (CH3)3C–O–). 13C NMR (CDCl3, 500 MHz, ppm, δ): 71.1 (–OCH2CH2–), 61.1 ((CH3)3C–O–), 55.6 (–(CO)C(CH3)2Br), 30.7 (–(CO)C(CH3)2Br), 27.5 ((CH3)3C–O–). FT-IR (cm−1): 2882, 1730 (CO), 1464, 1342, 1280, 1240, 1107, 958, 842. SEC (THF, RI detector): Mn = 11.1 g mol−1, Mw = 12.0 g mol−1, PDI = 1.07.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :v = 50:50), the PEO-b-PS block copolymer was precipitated and collected by vacuum filtration. The block copolymer (0.93 g) was obtained after drying in vacuo for 24 h. 1H NMR (CDCl3, 500 MHz, ppm, δ): 6.30–7.40 (br, 430H, phenyl rings), 3.64 (br, 770H, –CH2CH2O–), 1.67–2.15 (br, 87H, –CH2CH(–Ar)–), 1.20–1.67 (br, 174H, –CH2CH(–Ar)–), 1.20 (s, 9H, (CH3)3C–O–), 0.93 (m, 6H, –O(CO)C(CH3)2CH2–). 13C NMR (CDCl3, 500 MHz, ppm, δ): 144.8–146.6, 127.0–128.5, 125.4, 71.1, 64.6, 61.1, 41.0–47.8, 44.0, 27.5. FT-IR (cm−1): 3082, 3059, 3026, 2920, 2882, 1943, 1884, 1807, 1728, 1643, 1601, 1493, 1452, 1350, 1298, 1250, 1107, 1029, 951, 844, 758, 699, 542. SEC (THF, RI detector): Mn = 21.8 kg mol−1, Mw = 23.6 kg mol−1, PDI = 1.08.
:v = 50:50), and a white solid was collected by filtration. The PEO-b-PS-N3 was obtained in 87% yield (0.69 g) after drying under vacuum for 24 hours. 1H NMR (CDCl3, 500 MHz, ppm, δ): 6.30–7.40 (br, 430H, phenyl rings), 3.64 (br, 770H, –CH2CH2O–), 1.67–2.15 (br, 87H, –CH2CH(–Ar)–), 1.20–1.67 (br, 174H, –CH2CH(–Ar)–), 1.20 (s, 9H, (CH3)3C–O–), 0.93 (m, 6H, –O(CO)C(CH3)2CH2–). 13C NMR (CDCl3, 500 MHz, ppm, δ): 144.9–146.4, 127.1–128.6, 125.5, 71.1, 64.6, 61.1, 41.1–47.9, 44.1, 27.5. FT-IR (given in Fig. S2†, cm−1): 3082, 3060, 3027, 2920, 2869, 2098 (N3), 1943, 1884, 1807, 1730, 1641, 1601, 1493, 1452, 1349, 1300, 1249, 1110, 1029, 950, 844, 758, 699, 542. SEC (THF, RI detector): Mn = 22.2 kg mol−1, Mw = 23.4 kg mol−1, PDI = 1.06.
:v = 90:10) to give a colored fraction, which was collected, concentrated, and recrystallized in amyl acetate to give PEO-b-PS-C60 as a brown powder (310 mg, 77%). 1H NMR (CDCl3, 500 MHz, ppm, δ): 7.80 (m, 1H), 6.30–7.40 (br, 430H, phenyl rings), 5.20 (br, 1H, –CH2CH(–Ar)–N–), 3.64 (br, 770H, –CH2CH2O–), 2.90 (br, 1H, –CH2CH2CH(–OCO–)CH2–), 1.67–2.15 (br, 87H, –CH2CH(–Ar)–), 1.20–1.67 (br, 174H, –CH2CH(–Ar)–), 1.20 (s, 9H, (CH3)3C–O–), 0.93 (m, 6H,–O(CO)C(CH3)2CH2–). 13C NMR (CDCl3, 500 MHz, ppm, δ): (given in Fig. S3†). FT-IR (given in Fig. S2†, cm−1): 3082, 3059, 3026, 2918, 2867, 1945, 1871, 1801, 1724 (CO), 1601, 1493, 1452, 1349, 1299, 1249, 1107, 1030, 949, 842, 758, 699, 540.
O)C(CH3)2Br), 1.20 (s, 9H, (CH3)3C–O–). 13C NMR (CDCl3, 500 MHz, ppm, δ): 171.5 (–O(CO)C(CH3)2Br), 71.1 (–OCH2CH2–), 61.1 ((CH3)3C–O–), 55.6 (–(CO)C(CH3)2Br), 50.9 (–CH2CH(–CH2N3)O–), 30.7 (–(CO)C(CH3)2Br), 27.5 ((CH3)3C–O–). FT-IR (cm−1): 2881, 2100 (N3), 1737 (CO), 1467, 1359, 1280, 1240, 1147, 1107, 1061, 946, 842. SEC (THF, RI detector): Mn = 9.5 kg mol−1, Mw = 10.4 kg mol−1, PDI = 1.10.
O)C(CH3)2CH2–). 13C NMR (CDCl3, 500 MHz, ppm, δ): 144.6–146.6, 127.0–128.5, 125.4, 71.1, 61.1, 41.0–47.7, 44.0, 27.5. FT-IR (given in Fig. S12†, cm−1): 3082, 3059, 3026, 2922, 2101(N3), 1945, 1873, 1804, 1729 (CO), 1601, 1493, 1452, 1349, 1298, 1110, 1029, 946, 844, 696, 540. SEC (THF, RI detector): Mn = 21.2 kg mol−1, Mw = 23.5 kg mol−1, PDI = 1.10.
O)CH2–), 1.67–2.15 (br, 88H, –CH2CH(–Ar)–), 1.20–1.67 (br, 176H, –CH2CH(–Ar)–), 1.20 (s, 9H, (CH3)3C–O–), 0.93 (m, 6H, –O(CO)C(CH3)2CH2–). 13C NMR (CDCl3, 500 MHz, ppm, δ): Fig. S13†. FT-IR (given in Fig. S12†, cm−1): 3082, 3059, 3026, 2882, 1724, 1601, 1491, 1449, 1352, 1273, 1109, 842, 696, 544.
O)–), 3.64 (br, 836H, –CH2CH2O–), 3.27 (t, 2H, –SCH2CH2–), 1.69 (s, 6H, –O(CO)C(CH3)2S–), 1.65 (m, 2H, –SCH2CH2–), 1.2–1.4(m, 18H, –SCH2CH2(CH2) 9CH3), 1.20 (s, 9H, (CH3)3C–O–), 0.9 (t, 3H, –SC11H22CH3). 13C NMR (CDCl3, 500 MHz, ppm, δ): 71.1 (–OCH2CH2–), 61.1 ((CH3)3C–O–), 36.8 (–SCH2CH2(CH2)9CH3), 31.8 (–SCH2CH2(CH2)9CH3), 28.8–29.5 (–SCH2CH2(CH2)8 CH2CH3), 27.5 ((CH3)3C–O–), 25.2 (–O(CO)C(CH3)2S–), 22.6 (–SCH2CH2(CH2)8CH2CH3), 14.0 (–SCH2CH2(CH2)8CH2CH3). FT-IR (cm−1): 2889, 1966, 1733, 1695, 1643, 1467, 1342, 1280, 1242, 1147, 1107, 1061, 963, 842. SEC (THF, RI detector): Mn = 13.4 kg mol−1, Mw = 14.4 kg mol−1, PDI = 1.06.
:v = 50:50), the PEO-b-PS/Cl was precipitated and collected by vacuum filtration. The block copolymer was obtained after drying in vacuo for 24 h as a white powder (0.69 g). 1H NMR (CDCl3, 500 MHz, ppm, δ): 6.30–7.40 (br, 340H, phenyl rings), 4.52 (br, 9H, –CH2Cl), 3.64 (br, 836H, –CH2CH2O–), 1.67–2.15 (br, 70H, –CH2CH(–Ar)–), 1.20–1.67 (br, 140H, –CH2CH(–Ar)–), 1.27 (m, 18H, –SCH2CH2(CH2)9CH3), 1.20 (s, 9H, (CH3)3C–O–), 0.93 (m, 9H,–O(CO)C(CH3)2CH2–, –SC11H22CH3). 13C NMR (CDCl3, 500 MHz, ppm, δ): 145.1–150.0, 127.3–128.0, 125.5, 71.1, 63.0, 61.1, 41.1–47.9, 40.4, 31.9, 29.5, 27.5, 22.6, 14.0. FT-IR (cm−1): 3082, 3059, 3026, 2922, 2875, 1945, 1872, 1803, 1724, 1643, 1601, 1493, 1452, 1351, 1298, 1250, 1107, 1029, 951, 909, 843, 758, 699, 540. SEC (THF, RI detector): Mn = 15.8 kg mol−1, Mw = 18.0 kg mol−1, PDI = 1.17.
:v = 50:50). PEO-b-PS/N3 was obtained as white powder after drying in vacuo for 24 h (440 mg, 88%). 1H NMR (CDCl3, 500 MHz, ppm, δ): 6.30–7.40 (br, 340H, phenyl rings), 4.23 (br, 9H, –Ar–CH2N3), 3.64 (br, 836H, –CH2CH2O–), 1.67–2.15 (br, 70H, –CH2CH(–Ar)–), 1.20–1.67 (br, 140H, –CH2CH(–Ar)–), 1.27 (m, 18H, –S–CH2CH2(CH2)9CH3), 1.20 (s, 9H, (CH3)3C–O–), 0.93 (m, 9H, –O(CO)C(CH3)2CH2–, –SC11H22CH3). 13C NMR (CDCl3, 500 MHz, ppm, δ): 145.0–150.1, 127.5–128.0, 125.4, 71.1, 63.0, 61.1, 41.1–48.0, 40.4, 31.9, 29.5, 27.5, 22.6, 14.0. FT-IR (given in Fig. S16†, cm−1): 3082, 3060, 3026, 3026, 2883, 2098 (–N3), 1945, 1872, 1803, 1724, 1643, 1601, 1493, 1467, 1453, 1360, 1344, 1280, 1148, 1109, 1061, 945, 842, 757, 696, 544. SEC (THF, RI detector): Mn = 16.0 kg mol−1, Mw = 18.1 kg mol−1, PDI = 1.13.
O)C(CH3)2CH2–, –SC11H22CH3). 13C NMR (CDCl3, 500 MHz, ppm, δ): Fig. S17†. FT-IR (given in Fig. S16†, cm−1): 3082, 3060, 3026, 3026, 2882, 1945, 1872, 1803, 1728 (CO), 1601, 1491, 1449, 1352, 1273, 1109, 842, 696, 544, 528.
:v = 50:50), the PEO-b-PS block copolymer was precipitated and collected by vacuum filtration. The block copolymer (0.93 g) was obtained after drying in vacuo for 24 h. 1H NMR (CDCl3, 500 MHz, ppm, δ): 6.30–7.40 (br, 430H, phenyl rings), 3.64 (br, 770H, –CH2CH2O–), 1.67–2.15 (br, 87H, –CH2CH(–Ar)–), 1.20–1.67 (br, 174H, –CH2CH(–Ar)–), 1.20 (s, 9H, (CH3)3C–O–), 0.93 (m, 6H, –O(CO)C(CH3)2CH2–). 13C NMR (CDCl3, 500 MHz, ppm, δ): 144.8–146.6, 127.0–128.5, 125.4, 71.1, 64.6, 61.1, 41.0–47.8, 44.0, 27.5. FT-IR (cm−1): 3082, 3059, 3026, 2920, 2882, 1943, 1884, 1807, 1728, 1643, 1601, 1493, 1452, 1350, 1298, 1250, 1107, 1029, 951, 844, 758, 699, 542. SEC (THF, RI detector): Mn = 21.8 kg mol−1, Mw = 23.6 kg mol−1, PDI = 1.08.
:v = 50:50), and a white solid was collected by filtration. The PEO-b-PS-N3 was obtained in 87% yield (0.69 g) after drying under vacuum for 24 hours. 1H NMR (CDCl3, 500 MHz, ppm, δ): 6.30–7.40 (br, 430H, phenyl rings), 3.64 (br, 770H, –CH2CH2O–), 1.67–2.15 (br, 87H, –CH2CH(–Ar)–), 1.20–1.67 (br, 174H, –CH2CH(–Ar)–), 1.20 (s, 9H, (CH3)3C–O–), 0.93 (m, 6H, –O(CO)C(CH3)2CH2–). 13C NMR (CDCl3, 500 MHz, ppm, δ): 144.9–146.4, 127.1–128.6, 125.5, 71.1, 64.6, 61.1, 41.1–47.9, 44.1, 27.5. FT-IR (given in Fig. S2†, cm−1): 3082, 3060, 3027, 2920, 2869, 2098 (N3), 1943, 1884, 1807, 1730, 1641, 1601, 1493, 1452, 1349, 1300, 1249, 1110, 1029, 950, 844, 758, 699, 542. SEC (THF, RI detector): Mn = 22.2 kg mol−1, Mw = 23.4 kg mol−1, PDI = 1.06.
:v = 90:10) to give a colored fraction, which was collected, concentrated, and recrystallized in amyl acetate to give PEO-b-PS-C60 as a brown powder (310 mg, 77%). 1H NMR (CDCl3, 500 MHz, ppm, δ): 7.80 (m, 1H), 6.30–7.40 (br, 430H, phenyl rings), 5.20 (br, 1H, –CH2CH(–Ar)–N–), 3.64 (br, 770H, –CH2CH2O–), 2.90 (br, 1H, –CH2CH2CH(–OCO–)CH2–), 1.67–2.15 (br, 87H, –CH2CH(–Ar)–), 1.20–1.67 (br, 174H, –CH2CH(–Ar)–), 1.20 (s, 9H, (CH3)3C–O–), 0.93 (m, 6H,–O(CO)C(CH3)2CH2–). 13C NMR (CDCl3, 500 MHz, ppm, δ): (given in Fig. S3†). FT-IR (given in Fig. S2†, cm−1): 3082, 3059, 3026, 2918, 2867, 1945, 1871, 1801, 1724 (CO), 1601, 1493, 1452, 1349, 1299, 1249, 1107, 1030, 949, 842, 758, 699, 540.
O)C(CH3)2Br), 1.20 (s, 9H, (CH3)3C–O–). 13C NMR (CDCl3, 500 MHz, ppm, δ): 171.5 (–O(CO)C(CH3)2Br), 71.1 (–OCH2CH2–), 61.1 ((CH3)3C–O–), 55.6 (–(CO)C(CH3)2Br), 50.9 (–CH2CH(–CH2N3)O–), 30.7 (–(CO)C(CH3)2Br), 27.5 ((CH3)3C–O–). FT-IR (cm−1): 2881, 2100 (N3), 1737 (CO), 1467, 1359, 1280, 1240, 1147, 1107, 1061, 946, 842. SEC (THF, RI detector): Mn = 9.5 kg mol−1, Mw = 10.4 kg mol−1, PDI = 1.10.
O)C(CH3)2CH2–). 13C NMR (CDCl3, 500 MHz, ppm, δ): 144.6–146.6, 127.0–128.5, 125.4, 71.1, 61.1, 41.0–47.7, 44.0, 27.5. FT-IR (given in Fig. S12†, cm−1): 3082, 3059, 3026, 2922, 2101(N3), 1945, 1873, 1804, 1729 (CO), 1601, 1493, 1452, 1349, 1298, 1110, 1029, 946, 844, 696, 540. SEC (THF, RI detector): Mn = 21.2 kg mol−1, Mw = 23.5 kg mol−1, PDI = 1.10.
O)CH2–), 1.67–2.15 (br, 88H, –CH2CH(–Ar)–), 1.20–1.67 (br, 176H, –CH2CH(–Ar)–), 1.20 (s, 9H, (CH3)3C–O–), 0.93 (m, 6H, –O(CO)C(CH3)2CH2–). 13C NMR (CDCl3, 500 MHz, ppm, δ): Fig. S13†. FT-IR (given in Fig. S12†, cm−1): 3082, 3059, 3026, 2882, 1724, 1601, 1491, 1449, 1352, 1273, 1109, 842, 696, 544.
O)–), 3.64 (br, 836H, –CH2CH2O–), 3.27 (t, 2H, –SCH2CH2–), 1.69 (s, 6H, –O(CO)C(CH3)2S–), 1.65 (m, 2H, –SCH2CH2–), 1.2–1.4(m, 18H, –SCH2CH2(CH2) 9CH3), 1.20 (s, 9H, (CH3)3C–O–), 0.9 (t, 3H, –SC11H22CH3). 13C NMR (CDCl3, 500 MHz, ppm, δ): 71.1 (–OCH2CH2–), 61.1 ((CH3)3C–O–), 36.8 (–SCH2CH2(CH2)9CH3), 31.8 (–SCH2CH2(CH2)9CH3), 28.8–29.5 (–SCH2CH2(CH2)8 CH2CH3), 27.5 ((CH3)3C–O–), 25.2 (–O(CO)C(CH3)2S–), 22.6 (–SCH2CH2(CH2)8CH2CH3), 14.0 (–SCH2CH2(CH2)8CH2CH3). FT-IR (cm−1): 2889, 1966, 1733, 1695, 1643, 1467, 1342, 1280, 1242, 1147, 1107, 1061, 963, 842. SEC (THF, RI detector): Mn = 13.4 kg mol−1, Mw = 14.4 kg mol−1, PDI = 1.06.
:v = 50:50), the PEO-b-PS/Cl was precipitated and collected by vacuum filtration. The block copolymer was obtained after drying in vacuo for 24 h as a white powder (0.69 g). 1H NMR (CDCl3, 500 MHz, ppm, δ): 6.30–7.40 (br, 340H, phenyl rings), 4.52 (br, 9H, –CH2Cl), 3.64 (br, 836H, –CH2CH2O–), 1.67–2.15 (br, 70H, –CH2CH(–Ar)–), 1.20–1.67 (br, 140H, –CH2CH(–Ar)–), 1.27 (m, 18H, –SCH2CH2(CH2)9CH3), 1.20 (s, 9H, (CH3)3C–O–), 0.93 (m, 9H,–O(CO)C(CH3)2CH2–, –SC11H22CH3). 13C NMR (CDCl3, 500 MHz, ppm, δ): 145.1–150.0, 127.3–128.0, 125.5, 71.1, 63.0, 61.1, 41.1–47.9, 40.4, 31.9, 29.5, 27.5, 22.6, 14.0. FT-IR (cm−1): 3082, 3059, 3026, 2922, 2875, 1945, 1872, 1803, 1724, 1643, 1601, 1493, 1452, 1351, 1298, 1250, 1107, 1029, 951, 909, 843, 758, 699, 540. SEC (THF, RI detector): Mn = 15.8 kg mol−1, Mw = 18.0 kg mol−1, PDI = 1.17.
:v = 50:50). PEO-b-PS/N3 was obtained as white powder after drying in vacuo for 24 h (440 mg, 88%). 1H NMR (CDCl3, 500 MHz, ppm, δ): 6.30–7.40 (br, 340H, phenyl rings), 4.23 (br, 9H, –Ar–CH2N3), 3.64 (br, 836H, –CH2CH2O–), 1.67–2.15 (br, 70H, –CH2CH(–Ar)–), 1.20–1.67 (br, 140H, –CH2CH(–Ar)–), 1.27 (m, 18H, –S–CH2CH2(CH2)9CH3), 1.20 (s, 9H, (CH3)3C–O–), 0.93 (m, 9H, –O(CO)C(CH3)2CH2–, –SC11H22CH3). 13C NMR (CDCl3, 500 MHz, ppm, δ): 145.0–150.1, 127.5–128.0, 125.4, 71.1, 63.0, 61.1, 41.1–48.0, 40.4, 31.9, 29.5, 27.5, 22.6, 14.0. FT-IR (given in Fig. S16†, cm−1): 3082, 3060, 3026, 3026, 2883, 2098 (–N3), 1945, 1872, 1803, 1724, 1643, 1601, 1493, 1467, 1453, 1360, 1344, 1280, 1148, 1109, 1061, 945, 842, 757, 696, 544. SEC (THF, RI detector): Mn = 16.0 kg mol−1, Mw = 18.1 kg mol−1, PDI = 1.13.
O)C(CH3)2CH2–, –SC11H22CH3). 13C NMR (CDCl3, 500 MHz, ppm, δ): Fig. S17†. FT-IR (given in Fig. S16†, cm−1): 3082, 3060, 3026, 3026, 2882, 1945, 1872, 1803, 1728 (CO), 1601, 1491, 1449, 1352, 1273, 1109, 842, 696, 544, 528.
Results and discussions
Giant surfactant with a diblock copolymer tail (PEO-b-PS-C60)
A nanoparticle tethered with a polymer chain resembles the structure of a small-molecule surfactant in that they both possess a compact head and a long tail, though their sizes are very different. Hence, it is a type of “giant surfactant”.8 An important molecular parameter in their self-assembly is the composition of the single polymer tail (e.g.homopolymervs.block copolymer). The synthesis of a giant surfactant with a single block copolymer tail is thus the first target. The synthesis began with hydroxyl-functionalized PEOs (PEO-OH), which could be synthesized by anionic polymerization with controlled molecular weights and low polydispersity (see ESI† for details). Combined with ATRP and RAFT polymerization techniques, the PS block can be synthesized in a controlled fashion with exact locations of reactive azidegroups. The synthetic scheme for PEO-b-PS-C60 is outlined in Scheme 2, which is similar to the synthesis of PS-C60 as reported previously,7 except that a macroinitiator (PEO-Br) is used in place of the ethyl α-bromoisobutyrate. The macroinitiator was obtained by esterification of PEO-OH with α-bromoisobutyryl bromide in 88% yield. Following ATRP at 90 °C and subsequent nucleophilic substitution with NaN3, the PEO-b-PS-N3 is “clicked” with Fulleryne01 to give the target C60-endcapped block copolymer (PEO-b-PS-C60) in a straightforward way.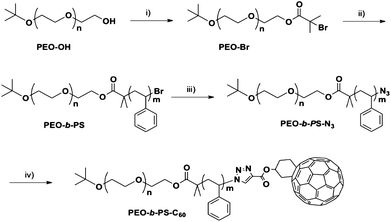 | ||
| Scheme 2 Synthesis of chain-end-functionalized PEO-b-PS (PEO-b-PS-C60): (i) 2-bromoisobutyryl bromide, TEA, CH2Cl2, rt, 88%; (ii) styrene, CuBr, PMDETA, toluene, 90 °C, 38%; (iii) NaN3, DMF, rt, 87%; and (iv) Fulleryne01, CuBr, PMDETA, toluene, rt, 77%. | ||
The chemical structures, molecular weights, and polydispersities were characterized by 1H NMR, 13C NMR, FT-IRspectrometry, and SECchromatography. After esterification, new chemical shifts appear in the 1H NMR spectrum at 1.92 and 4.30 ppm, which can be assigned to protons on –C(CH3)2Br and adjacent to the ester linkage [–(CO)OCH2–], respectively (Fig. 2a). The degree of functionalization can be determined by the integration ratio among the chemical shifts at 4.30, 1.92, and 1.20 ppm. The ratio of 1.78:6.34:9.00 is close to the expected 2:6:9 and suggests a high degree of α-bromoisobutyrate functionality. The block copolymer formation was evidenced by new chemical shifts appearing in the 1H NMR spectrum at the aromatic region (Fig. 2b). An apparently complete shift of the SEC trace to a lower elution volume further confirms the success of polymerization (Fig. 3). The block copolymer was obtained as a white powder. After the “click” reaction, it turned into a brown powder. The typical absorption band of azide (2100 cm−1) on the FT-IR spectrum disappears completely (Fig. S2†). In the 1H NMR spectrum of PEO-b-PS-C60, the signals, though weak, can be identified and assigned to the Fulleryne01 component at δ 2.9 ppm (protoni), to the one near triazole at δ 5.2 ppm (protong), and to the one on the triazole at δ 7.8 ppm (protonh), respectively as indicated in Fig. 2c. The existence of C60 was confirmed by 13C NMR, in which the sp3 carbons as well as sp2 carbons of the C60 moiety (60.8, 63.6, and 135–155 ppm overlapping with aromatic carbons) can be clearly observed (Fig. S3 in the ESI†). In the SEC chromatogram, after C60 functionalization, the retention volume essentially remained the same, yet a high molecular weight shoulder peak appears (Fig. 3). The question then arises concerning the explanation of this high molecular weight shoulder and the degree of fullerene functionality. However, due to the high molecular weight, it was difficult to measure the degree of C60 functionality either by MALDI-TOF mass spectra or by comparing the integration ratio in 1H NMR spectra accurately.
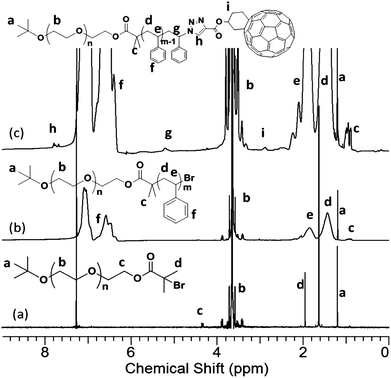 | ||
| Fig. 2 1H NMR spectra of (a) PEO-Br; (b) PEO-b-PS; and (c) PEO-b-PS-C60. The results were based on the samples with MnPEO = 8.4 kg mol−1 and MnPS = 9.0 kg mol−1. | ||
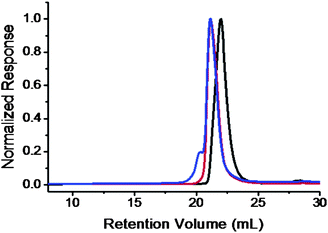 | ||
| Fig. 3 SEC overlay of PEO-Br (black); PEO-b-PS (red); and PEO-b-PS-C60 (blue) chromatograms. The results were based on the samples with MnPEO = 8.4 kg mol−1 and MnPS = 9.0 kg mol−1. | ||
Previously, the “click” chemistry approach to fullerenepolymers was demonstrated by the synthesis of PS-C60 in which such a high molecular weight shoulder was not observed in SEC and the possibility of high addition was thus excluded.7 In the case of PCL-C60, a shoulder peak was indeed observed, but MALDI-TOF mass spectrometry suggested that the formation of multi-addition products is very unlikely.6 The C60-containing block copolymers in this paper are very difficult to characterize by MALDI-TOF mass spectrometry due to their high molecular weights and the presence of two repeating units of different ionization efficiencies. As a reference for comparison, PEO-C60 with a low PEO molecular weight (MnPEO = 2.5 kg mol−1) was synthesized as a model compound. The detailed procedure and characterization are described in the ESI (Scheme S1, and Fig. S4–S9†). Fig. S4† shows the MALDI-TOF mass spectrum of the product. A narrow distribution with the molecular weights in accordance with the proposed structure is observed, confirming the cleanness of the reaction and the stability of the resulting fullerenepolymer. The reason that a perfect Gaussian distribution was not observed but slightly shifts towards the smaller molecular weight fraction side is due probably to the fact that small molecules are easier to be ionized and thus give rise to stronger signal. Nevertheless, no higher addition product signal was observed. Considering the low molecular weight and high ionization efficiency of PEO, any higher addition product, if present, shall be observed by MALDI-TOF mass spectroscopy that are known to be sensitive to impurities of 1% or less. The result indicates that PEO-C60 is free of homo-polymer and multi-addition impurities, which is consistent with the previous finding. This is further confirmed by the quantitative fullerene functionality (100.6%) for PEO-C60 as determined by the Beer–Lambert law within the range of error (Fig. S5 in the ESI†). Despite this evidence, the SEC trace of PEO-C60 (Fig. S6†) still shows a shoulder peak, which can only be invoked by the aggregation of PEO-C60 in THF caused by the poor solubility of C60 in THF and the amphiphilic feature of the molecule.
Similar results were also obtained in the case of PEO-b-PS-C60. The degree of fullerene functionality was measured to be 100.4% (Fig. S10†), again a quantitative functionalization though a shoulder peak is still observed in the SEC trace (Fig. 3). It is consistent with the above explanation for PEO-C60 since multi-addition would significantly decrease the degree of C60 functionality. The “click” chemistry has indeed provided a powerful means to assure a high degree of functionalization and least influence on the molecular weight and polydispersity of the parent polymer after functionalization. Several PEO-b-PS-C60polymers have been synthesized and their molecular characterizations are summarized in Table 1.
| Label | M n all /kg mol−1 | M n PEO /kg mol−1 | M n PS /kg mol−1 | M n PVBC /kg mol−1 | PDIb | f C60 | C60 d (wt%) | |
|---|---|---|---|---|---|---|---|---|
| 1H NMR | UV-Vis | |||||||
| a Molecular weight was calculated by 1H NMR. b Polydispersity of the precursors, measured by SEC. c Degree of C60 functionality as determined by 1H NMR and UV-Visspectrometry. d C60 weight content in the polymer, calculated according to eqn (5). | ||||||||
| PEO-b-PS-C60-1 | 14.7 | 8.4 | 5.5 | — | 1.07 | — | 1 | 5.7 |
| PEO-b-PS-C60-2 | 18.2 | 8.4 | 9.0 | — | 1.06 | — | 1 | 4.3 |
| PEO-b-PS-C60-3 | 25.4 | 11.0 | 13.6 | — | 1.06 | — | 1 | 3.3 |
| PEO-(C60)-PS-1 | 19.5 | 8.4 | 10.3 | — | 1.09 | — | 1 | 4.3 |
| PEO-(C60)-PS-2 | 19.0 | 9.0 | 9.2 | — | 1.08 | — | 1 | 4.4 |
| PEO-(C60)-PS-3 | 31.8 | 11.0 | 20.0 | — | 1.10 | — | 1 | 2.6 |
| PEO-b-PS/C60-1 | 19.8 | 9.0 | 6.3 | 0.7 | 1.17 | 4.5 | 5.8 | 19.0 |
| PEO-b-PS/C60-2 | 24.8 | 9.0 | 7.2 | 1.3 | 1.15 | 8.7 | 8.2 | 29.7 |
Giant lipids with asymmetric composition [PEO-(C60)-PS]: achieving quantitative in-chain functionality
Compared to small-molecule surfactants, lipids in nature are much more versatile in their structures and self-assemblies. Typical lipids include fatty acids, waxes, sterols, phospholipids, and cholesterol, composed of a polar head with various numbers of tails of different composition.50 In analogy to such lipids, the “giant lipids” refer to nanoparticles tethered with two or more polymer tails of different composition. A block copolymer with a nanoparticle at the junction point, such as PEO-(C60)-PS, can thus be viewed as a prototype giant lipid. The synthetic scheme is shown in Scheme 3. To tether an azidegroup at the junction point of two blocks, hetero-functional groups need to be attached to the PEO chain-end to allow orthogonal chain growth and “click” functionalization. The epoxy group was thus introduced by treating PEO-OH with epichlorohydrin under basic conditions in 90% yield followed by a quantitative ring opening reaction to give an azide and a hydroxylgroup [PEO-(N3)-OH].51 The synthesis of the macroinitiator [PEO-(N3)-Br] is similar to that of PEO-Br by esterification. ATRP was performed at a much lower temperature of 45 °C over an extended time. This is critical to obtain a high functionality of azide which is known to decompose to reactive intermediates such as nitrene at elevated temperatures, leading to the partial loss of the azide functionality.33,52 To avoid thermal decomposition, a low-temperature polymerization was executed at 45 °C. A control experiment was performed to confirm the integrity of the azidegroup by using 1H NMR spectrometry to monitor the change of 4-methoxybenzyl azide under identical reaction conditions, i.e. in the presence of styrene, CuBr, and PMDETA at 45 °C. The azide functionality was followed based on the integration ratio of protons on the ethylene group adjacent to azide (4.20 ppm), and those on the methoxy group (3.75 ppm). The normalized integration area does not decrease over time even after 36 hours, indicating that no side reaction takes place between the azide and vinyl group under these conditions (see the description in the ESI and Fig. S11†). Although it requires 2–3 days for polymerization of styrene to a reasonable molecular weight, it affords a quantitative azide functionality which is important in obtaining a well-defined “clicked” product with high degree of C60 functionality. The “click” reaction was then performed in a similar manner as in the case of PEO-b-PS-C60, again exemplifying the modular feature of this approach.![Synthesis of PEO-b-PS with C60 tethered at the junction point [PEO-(C60)-PS]: (i) epichlorohydrin, NaOH, rt, 90%; (ii) NaN3, NH4Cl, DMF, 50 °C, 76%; (iii) 2-bromoisobutyryl bromide, TEA, CH2Cl2, rt, 87%; (iv) styrene, CuBr, PMDETA, toluene, 45 °C, 35%; and (v) Fulleryne01, CuBr, PMDETA, toluene, rt, 71%.](/image/article/2012/PY/c1py00435b/c1py00435b-s3.gif) | ||
| Scheme 3 Synthesis of PEO-b-PS with C60 tethered at the junction point [PEO-(C60)-PS]: (i) epichlorohydrin, NaOH, rt, 90%; (ii) NaN3, NH4Cl, DMF, 50 °C, 76%; (iii) 2-bromoisobutyryl bromide, TEA, CH2Cl2, rt, 87%; (iv) styrene, CuBr, PMDETA, toluene, 45 °C, 35%; and (v) Fulleryne01, CuBr, PMDETA, toluene, rt, 71%. | ||
In the 1H NMR spectra, the successful installment of epoxy group is evidenced by the signals at δ 3.16, 2.79, and 2.61 ppm (Fig. 4a). The degree of chain-end functionality is determined by the integration ratio between the peaks at δ 3.16, 2.79, 2.61, and 1.20 ppm, which is 0.99:0.91:0.96:9.04, very close to 1:1:1:9 and consistent with a quasi-quantitative conversion. After ring opening with NaN3, the signals at δ 3.17, 2.79, and 2.61 ppm in the 1H NMR spectra completely disappeared (Fig. 4b) and a strong band near 2100 cm−1 characteristic of the azide appeared in the FT-IR spectrum (Fig. S7†). Since the new signals from methylene near the azide and hydroxylgroup have very similar shifts to the methyleneprotons on the PEO backbone, no new peaks can be observed (Fig. 4b).51 The PEO-(N3)-OH was also used to synthesize PEO-C60 as discussed earlier and described in the ESI†. After esterification, the protons (d and c) of PEO-(N3)-Br can be seen at δ 1.92 and 5.15 ppm, respectively (Fig. 4c). The SEC overlay of these polymers shows that their elution volumes and polydispersities are essentially unchanged, since the small chain-end functional groups exert little influence on the overall hydrodynamic radius of the polymers. With the macro-initiatorPEO-(N3)-Br, ATRP was performed (Scheme 3) and the conversion was kept less than 40% by controlling the polymerization time so as to control the molecular weight of the PS block while maintaining a low polydispersity. The polymerization was carried out at a low temperature of 45 °C to avoid thermal degradation of the azide functionality.33,52 After the polymerization, the FT-IR spectrum of PEO-(N3)-PS shows an absorption band at 2100 cm−1, characteristic of the azide functionality (Fig. S12†). Therefore, the azide functionalities should be intact during the polymerization, though the degree of azide functionality is still difficult to determine due to the low signal intensity of chain-end groups in the 1H NMR spectrum (Fig. 4d). Nevertheless, the UV-Visspectrometry provides an effective way to measure the C60 functionality of PEO-(C60)-PS after C60 was attached, which is indirect evidence of the degree of azide functionality on the precursor polymer.
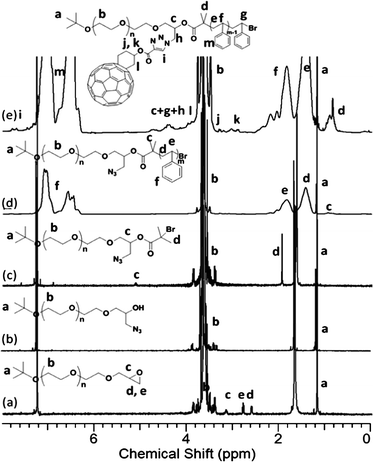 | ||
| Fig. 4 1H NMR spectra of (a) PEO-Epoxy; (b) PEO-(N3)-OH; (c) PEO-(N3)-Br; (d) PEO-(N3)-PS; and (e) PEO-(C60)-PS. The results were based on the samples with MnPEO = 9.0 kg mol−1 and MnPS = 9.2 kg mol−1. | ||
The success of this “click” reaction is supported by NMR, FT-IR spectroscopy, SEC, and quantitative UV analysis. The signals from the Fulleryne01 component at δ 2.9 ppm (protonk), 3.2 ppm (protonj), 4.0 ppm (protonl), the hydrogen atom at the junction carbon at δ 4.35 ppm (protonc, and h), and the triazole ring at δ 7.8 ppm (protoni) were observed as indicated in Fig. 4e. Due to the high molecular weight of the diblock copolymer and the low intensity of the characteristic peaks, it is difficult to measure their integration ratio quantitatively and accurately. In the 13C NMR spectra (Fig. S13†), the sp2 carbons of the C60 moiety (135–155 ppm overlapping with aromatic carbons) can be clearly observed. The FT-IR spectrum of PEO-(C60)-PS exhibits the complete disappearance of azide vibration at 2100 cm−1, suggesting a complete transformation (Fig. S12†). Similar to that observed in PEO-C60 and PEO-b-PS-C60, the retention volume essentially remains the same after C60 functionalization while a shoulder appears at the high molecular weight side (Fig. 5), which can also be attributed to the aggregation of C60 in THF. Again, a quantitative functionality of 100.3% was determined for PEO-(C60)-PS (Fig. S14†), confirming that only mono-addition takes place under the reaction conditions.
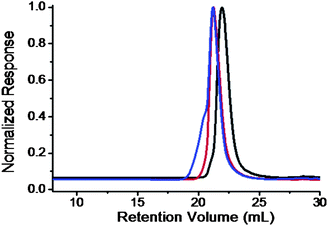 | ||
| Fig. 5 SEC overlay of PEO-(N3)-Br (black); PEO-(N3)-PS (red); and PEO-(C60)-PS (blue) chromatograms. The results were based on the samples with MnPEO = 9.0 kg mol−1 and MnPS = 9.2 kg mol−1. | ||
Crosslinking-free attachment of multiple C60s onto PS block (PEO-b-PS/C60)
One drawback in functionalizing polymers with fullerene is the limited content of C60 in the final materials, compromising the electronic properties. This can be partially compensated by attaching multiple C60s onto a polymer. The so-called “double-cable” polymer is a typical example.41 Traditionally, it is composed of a conjugated polymer backbone as the donor and C60s tethered as acceptors.53,54 Such polymers were expected to show a great promise in improving organic solar cell's performance by forming two separate channels to transport holes and electrons, respectively.41 However, most of the “double cable” polymers developed so far failed the expectation, showing low efficiencies in the organic photovoltaic devices. Attaching many fullerenes to one block will surely affect the self-assembled phase behaviors, since not only does it add significant C60–C60 interactions into the system, but also it increases the volume fraction of C60 significantly. To gain insight into such systems, PEO-b-PS/C60 was chosen as the model system to study the synthesis and self-assembled phase behaviors, in the hope of elucidating the basic underlying physics using well-defined samples of high purity prepared by the “click” chemistry. Majority of the works reported so far is based on the reaction between pristine C60 and functional polymers.35,37–40 The biggest challenge in attaching multiple C60s onto polymers lies in the efficiency and the potential crosslinking due to multi-addition, which makes it difficult to achieve high fullerene loading. Esterification has been used to attach carboxylic acid functionalized C60s onto polymer, yet the efficiency is relatively low.55 Here, we demonstrate a crosslinking-free attachment of C60s onto the PS blocks of PEO-b-PS utilizing “click” chemistry, which provides a viable solution through precise functionalization.To prepare PEO-b-PS with multiple reactive azide functionalities on the PS block, vinyl benzyl chloride (VBC) was chosen as a comonomer with styrene. Hence, it is no longer suitable to use ATRP because benzyl chloride is also a good initiator for ATRP. Instead, RAFT-mediated polymerization is compatible with the monomer and was chosen as the method to prepare PEO-b-PS/Cl. The synthetic scheme is shown in Scheme 4. The diblock copolymer, PEO-b-PS/Cl, was first polymerized by RAFT with PEO-TC as the macro-chain transfer agent. The azidegroup was readily introduced by reacting PEO-b-PS/Cl with NaN3 in DMF. “Click” reaction was then applied to link fullerene moieties onto the PS blocks with a controllable loading in the absence of crosslinking. By continuously increasing the C60 loading on the PS block, the PS coils may become more and more stretched due to the increasing bulky volume of C60.56
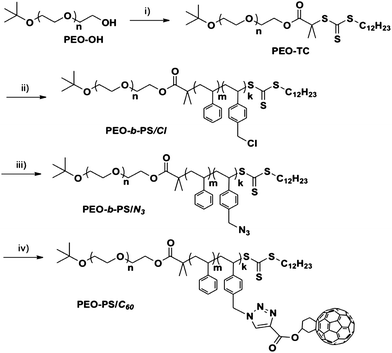 | ||
| Scheme 4 Synthesis of multiple-C60s-functionalized PEO-b-PS (PEO-b-PS/C60): (i) S-1-dodecyl-S′-(r,r′-dimethyl-r′′-carbonyl chloride)trithio carbonate, TEA, CH2Cl2, rt, 85%; (ii) styrene, vinyl benzyl chloride, AIBN, toluene, 110 °C, 38%; (iii) NaN3, DMF, rt, 88%; and (iv) Fulleryne01, CuBr, PMDETA, toluene, rt, 73%. | ||
PEO-OH was converted to a macro-chain transfer agent, PEO-TC, a light yellow powder in 85% yield. Fig. 6a shows the 1H NMR spectrum of this product. The characteristic signal of PEO at 3.64 ppm (protonb) and the signals from TC at 1.69 (d), 1.65 (f), 1.26 (h), and 0.90 (i) ppm are apparent. A chemical shift at 4.24 ppm can be assigned to the protons on the methylene adjacent to the as-formed estergroup (protonc), further confirming the structure of the product. A SEC trace of PEO-TC is identical to its precursor, indicating the integrity of the backbone (data not shown). Copolymerization was carried out with PEO-TC (Mw = 9.0 kg mol−1) as the macro-chain transfer agent, and AIBN as the initiator. The conversion was controlled below 40% so as to keep a narrow polydispersity. The overall molecular weight was controlled by polymerization time while the ratio between styrene segments and VBC segments on the polymer chain was controlled by their feed ratio. This molar fraction of VBC incorporation can be determined from 1H NMR spectra (Fig. 6b) by the integration ratio between the broad chemical shifts between 7.40 and 6.30 ppm (phenyl rings on the PS blocks) and the chemical shift at 4.51 ppm (hydrogen on the ethylene group adjacent to the chloride, protong) (eqn (2)). The degree of polymerization and number average molecular weight of the PVBC segments and the PS segments can also be calculated accordingly. The SEC diagrams (Fig. S15†) show narrow polydispersity with different molecular weights and segment ratios.
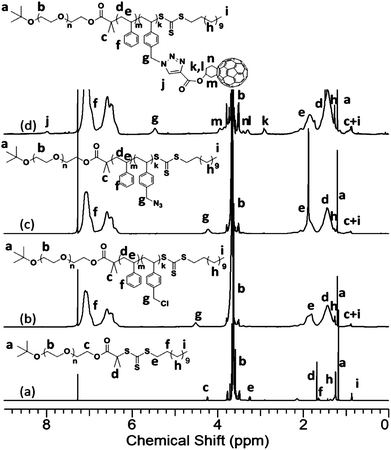 | ||
| Fig. 6 1H NMR spectra of (a) PEO-TC; (b) PEO-b-PS/Cl; (c) PEO-b-PS/N3; and (d) PEO-b-PS/C60. The samples were based on the samples with MnPEO = 9.0 kg mol−1 and MnPS = 7.0 kg mol−1. | ||
PEO-b-PS/N3 can then be readily obtained by reacting with NaN3 in DMF.35,38 The azidegroup was characterized by FT-IR and 1H NMR. In the IR spectrum, a typical azide absorption band at 2100 cm−1 is observed with relatively strong intensity (Fig. S16†). The signal of the azidegroup in a high molecular weight polymer is usually weak (Fig. S2 and S12†). A relatively strong absorption indicates the existence of multiple azidegroups on the PS block. A complete shift from 4.51 ppm to 4.25 ppm, corresponding to the protons on the methyleneproton close to the chloridegroup (protong), is observed, which indicates full conversion with no chloride residual.
“Click” chemistry was utilized here to connect Fulleryne01 onto the PS block to avoid multi-addition which may result in crosslinking. The product was collected in 73% yield as a dark brown powder. The loss is mainly due to residual adsorption onto column during purification to remove the coppercatalyst and excess Fulleryne01. After “click” reaction, the absorption band of the azidegroup around 2100 cm−1 on the FT-IR spectrum disappears completely, indicating the success of the reaction (Fig. S16†). A new strong band appears at 1728 cm−1 due to the existence of a carbonyl group on Fulleryne01. The characteristic absorption of fullerene is also found at 525 cm−1. The 1H NMR spectrum (Fig. 6d) exhibited broad chemical shifts at δ 2.90 ppm (k), 3.35 ppm (l), 3.40 ppm (n), and 3.97 ppm (m) assignable to the protons on the Fulleryne01 moiety. The signal from protons near the triazole (protong) and the one on the triazole (protonj) appears at δ 5.45 ppm and 7.98 ppm, respectively. It also clearly shows the sp3 carbons (60.8, 63.6 ppm) and sp2 carbons (135–155 ppm) of the C60 unit in the 13C NMR spectrum (Fig. S17†). Fig. 7 is a SEC trace overlay of the final product and its precursors. After polymerization, a symmetric peak with lower retention volume is observed. After the “click” reaction, large shoulder peaks appear on the high molecular weight side, which is much more significant than in previous cases. This may in part be due to the increase of overall molecular volume as a result of multiple-C60 attachment and/or the aggregation of C60s, since the molecules possess much higher C60 loading. Since it was carried out under mild conditions where the thermal [3 + 2] cycloaddition between the azide and pristine C60 core should not take place, the reaction only occurred between the azide and the alkyne under the coppercatalyst, which rules out the possibility of multi-addition and crosslinking. Therefore, the product can now be reversibly dissolved in most common organic solvents such as toluene, THF, CHCl3, CH2Cl2, and others, even after long-term storage. The degree of C60 functionality was also measured by UV-Visspectrometry. Solutions in toluene with a series of concentrations were prepared and their absorbance was measured at 330 nm to get a standard curve which is fitted to eqn (1). The linear fitting curves for PEO-b-PS/C60-1 (MnPEO = 9.0 kg mol−1, MnPS = 6.3 kg mo−1) and PEO-b-PS/C60-2 (MnPEO = 9.0 kg/mol, MnPS = 7.2 kg mol−1) yield the average number of C60 per chain (NC60) to be 5.8 and 8.2, respectively (Fig. S18 and S19†). This is similar to the C60 loading per chain (4.5 and 8.7, respectively) obtained from the 1H NMR spectra using eqn (2). The C60 content in the polymer, C60 wt%, can then be estimated using eqn (5). The C60 wt% in this case can be as high as ∼30%. The characterizations are summarized in Table 1.
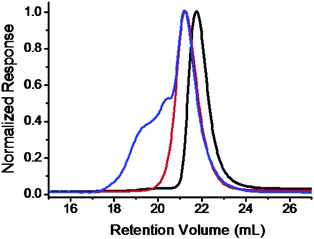 | ||
| Fig. 7 SEC overlay of PEO-OH (black), PEO-b-PS/Cl (red), and PEO-b-PS/C60 (blue) chromatograms. The samples were based on the PEO molecular weight of 9.0 kg mol−1 and the PS/PVBC molecular weight of 7.0 kg mol−1. | ||
Conclusions
In summary, “click” chemistry provides an efficient and modular approach to constructfullerene-containing polymers and highlights a quantitative functionality, mild reaction condition, and crosslinking-free process. This is demonstrated by the synthesis of C60-containing diblock copolymers with different compositions and architectures, such as chain-end-functionalized PEO-b-PS (PEO-b-PS-C60), junction-point-functionalized PEO-b-PS [PEO-(C60)-PS], and multiple-C60s-functionalized PEO-b-PS (PEO-b-PS/C60). These fullerenepolymers resemble the structure of giant surfactants and giant lipids and may serve as model compounds for shape amphiphiles. The method can also be applied to the synthesis of “double-cable” polymers to improve the control of their primary chemical structure and avoid crosslinking to facilitate processing. The work currently ongoing in our group involves using the self-assembly of these polymers, such as single crystal growth, micellization, and block copolymer phase separation in the bulk solution and thin film, as templates to manipulate the ordered structure formation of C60 in multi-dimensions across different length scales.Acknowledgements
This work was supported by NSF (DMR-0906898). W.-B. Z. acknowledges the Lubrizol Corporation for a fellowship. We thank Dr Xiaopeng Li and Prof. Chrys Wesdemiotis for their help with the MALDI-TOF mass characterizations.Notes and references
- R. W. Date and D. W. Bruce, J. Am. Chem. Soc., 2003, 125, 9012–9013 CrossRef CAS.
- S. C. Glotzer, M. A. Horsch, C. R. Iacovella, Z. L. Zhang, E. R. Chan and X. Zhang, Curr. Opin. Colloid Interface Sci., 2005, 10, 287–295 CrossRef CAS.
- S. C. Glotzer and M. J. Solomon, Nat. Mater., 2007, 6, 557–562 CrossRef.
- W.-B. Zhang, Y. Li, X. Li, X. Dong, X. Yu, C.-L. Wang, C. Wesdemiotis, R. P. Quirk and S. Z. D. Cheng, Macromolecules, 2011, 44, 2589–2596 CrossRef CAS.
- Z. Zhang, M. A. Horsch, M. H. Lamm and S. C. Glotzer, Nano Lett., 2003, 3, 1341–1346 CrossRef CAS.
- W.-B. Zhang, J. He, X. Dong, C.-L. Wang, H. Li, F. Teng, X. Li, C. Wesdemiotis, R. P. Quirk and S. Z. D. Cheng, Polymer, 2011, 52, 4221–4226 CrossRef CAS.
- W.-B. Zhang, Y. Tu, R. Ranjan, R. M. V. Horn, S. Leng, J. Wang, M. J. Polce, C. Wesdemiotis, R. P. Quirk, G. R. Newkome and S. Z. D. Cheng, Macromolecules, 2008, 41, 515–517 CrossRef CAS.
- X. Yu, S. Zhong, X. Li, Y. Tu, S. Yang, R. M. V. Horn, C. Ni, D. J. Pochan, R. P. Quirk, C. Wesdemiotis, W.-B. Zhang and S. Z. D. Cheng, J. Am. Chem. Soc., 2010, 132, 16741–16744 CrossRef CAS.
- H.-J. Sun, Y. Tu, C.-L. Wang, R. M. Van Horn, C.-C. Tsai, M. J. Graham, B. Sun, B. Lotz, W.-B. Zhang and S. Z. D. Cheng, J. Mater. Chem., 2011, 21, 14240–14247 RSC.
- K. M. Kadish and R. S. Ruoff, Fullerenes: Chemistry, Physics, and Technology, Wiley-Interscience, New York, 2000 Search PubMed.
- N. Martín, F. Giacalone and M. Prato, Fullerene Polymers: Synthesis, Properties and Applications, Wiley-VCH, Weinheim, 2009 Search PubMed.
- A. Hirsch and M. Brettreich, Fullerenes: Chemistry and Reactions, Wiley-VCH, Weinheim, Great Britain, 2005 Search PubMed.
- N. Martin, Chem. Commun., 2006, 2093–2104 RSC.
- M. Horsch, Z. Zhang and S. Glotzer, Phys. Rev. Lett., 2005, 95, 056105-1 CrossRef.
- S. C. Glotzer, M. A. Horsch, C. R. Iacovella, Z. Zhang, E. R. Chan and X. Zhang, Curr. Opin. Colloid Interface Sci., 2005, 10, 287–295 CrossRef CAS.
- H.-J. Sun, Y. Tu, C.-L. Wang, R. M. Van Horn, C.-C. Tsai, M. J. Graham, B. Sun, B. Lotz, W.-B. Zhang and S. Z. D. Cheng, J. Mater. Chem., 2011, 21, 14240–14247 RSC.
- O. Stoilova, C. Jerome, C. Detrembleur, A. Mouithys-Mickalad, N. Manolova, I. Rashkov and R. Jerome, Chem. Mater., 2006, 18, 4917–4923 CrossRef CAS.
- C. J. Hawker, Macromolecules, 1994, 27, 4836–4837 CrossRef CAS.
- C. Weis, C. Friedrich, R. Mmaupt and H. Frey, Macromolecules, 1995, 28, 403–405 CrossRef CAS.
- P. Zhou, G.-Q. Chen, H. Hong, F.-S. Du, Z.-C. Li and F.-M. Li, Macromolecules, 2000, 33, 1948–1954 CrossRef CAS.
- T. Kawauchi, J. Kumaki and E. Yashima, J. Am. Chem. Soc., 2005, 127, 9950–9951 CrossRef CAS.
- I. W. Hamley, The Physics of Block Copolymers, Oxford University Press, Oxford, 1998 Search PubMed.
- N. Hadjichristidis, S. Pispas and G. Floudas, Block Copolymers: Synthetic Strategies, Physical Properties, and Applications, Wiley-Interscience, New York, 2002 Search PubMed.
- B. Lotz and A. J. Kovacs, Kolloid. Z. Z. Polym., 1966, 209, 97–114 CAS.
- R. M. Van Horn, J. X. Zheng, H.-J. Sun, M.-S. Hsiao, W.-B. Zhang, X.-H. Dong, J. Xu, E. L. Thomas, B. Lotz and S. Z. D. Cheng, Macromolecules, 2010, 43, 6113–6119 CrossRef CAS.
- M.-S. Hsiao, W. Y. Chen, J. X. Zheng, R. M. Van Horn, R. P. Quirk, D. A. Ivanov, E. L. Thomas, B. Lotz and S. Z. D. Cheng, Macromolecules, 2008, 41, 4794–4801 CrossRef CAS.
- L. Zhang and A. Eisenberg, Polym. Adv. Technol., 1998, 9, 677–699 CrossRef CAS.
- P. Bhargava, J. X. Zheng, P. Li, R. P. Quirk, F. W. Harris and S. Z. D. Cheng, Macromolecules, 2006, 39, 4880–4888 CrossRef CAS.
- F. S. Bates, Science, 1991, 251, 898–904 CAS.
- F. S. Bates and G. H. Fredrickson, Annu. Rev. Phys. Chem., 1990, 41, 525–557 CrossRef CAS.
- Y.-L. Liu, Y.-H. Chang and W.-H. Chen, Macromolecules, 2008, 41, 7857–7862 CrossRef CAS.
- X. Wang, Y. Zhang, Z. Zhu and S. Liu, Macromol. Rapid Commun., 2008, 29, 340–346 CrossRef CAS.
- C. Li, J. Hu, J. Yin and S. Liu, Macromolecules, 2009, 42, 5007–5016 CrossRef CAS.
- K. Tajima, S. Miyanishi, Y. Zhang and K. Hashimoto, Chem. Commun., 2010, 46, 6723–6725 RSC.
- S. Barrau, T. Heiser, F. Richard, C. Brochon, C. Ngov, K. v. d. Wetering, G. Hadziioannou, D. V. Anokhin and D. A. Ivanov, Macromolecules, 2008, 41, 2701–2710 CrossRef CAS.
- X. Chen, B. Gholamkhass, X. Han, G. Vamvounis and S. Holdcroft, Macromol. Rapid Commun., 2007, 28, 1792–1797 CrossRef CAS.
- M. Dante, C. Yang, B. Walker, F. Wudl and T.-Q. Nguyen, Adv. Mater., 2010, 22, 1835–1839 CrossRef CAS.
- F. Richard, C. Brochon, N. Leclerc, D. Eckhardt, T. Heiser and G. Hadziioannou, Macromol. Rapid Commun., 2008, 29, 885–891 CrossRef CAS.
- U. Stalmach, B.d. Boer, C. Videlot, P. F.v. Hutten and G. Hadziioannou, J. Am. Chem. Soc., 2000, 122, 5464–5472 CrossRef CAS.
- M. H.v. d. Veen, B.d. Boer, U. Stalmach, K. I.v. d. Wetering and G. Hadziioannou, Macromolecules, 2004, 37, 3673–3684 CrossRef.
- H. Spanggaard and F. C. Krebs, Sol. Energy Mater. Sol. Cells, 2004, 83, 125–146 CrossRef CAS.
- M. A. Fazio, O. P. Lee and D. I. Schuster, Org. Lett., 2008, 10, 4979–4982 CrossRef CAS.
- J.-F. Nierengarten, J. Iehl, V. Oerthel, M. Holler, B. M. Illescas, A. Muñoz, N. Martín, J. Rojo, M. Sánchez-Navarro, S. Cecioni, S. Vidal, K. Buffet, M. Durka and S. P. Vincent, Chem. Commun., 2010, 46, 3860 RSC.
- N. F. Steinmetz, V. Hong, E. D. Spoerke, P. Lu, K. Breitenkamp, M. G. Finn and M. Mancheste, J. Am. Chem. Soc., 2009, 131, 17093–17095 CrossRef CAS.
- J. Iehl and J.-F. Nierengarten, Chem.–Eur. J., 2009, 15, 7306–7309 CrossRef CAS.
- J. Iehl, I. Osinska, R. Louis, M. Holler and J.-F. Nierengarten, Tetrahedron Lett., 2009, 50, 2245–2248 CrossRef CAS.
- A. J. Inglis, P. Pierrat, T. Muller, S. Bräse and C. Barner-Kowollik, Soft Matter, 2010, 6, 82 RSC.
- C.-Y. Hong and C.-Y. Pan, Macromolecules, 2006, 39, 3517–3524 CrossRef CAS.
- J. D. Ingle and S. R. Crouch, Spectrochemical Analysis, Prentice Hall, Englewood Cliffs, NJ, 1988 Search PubMed.
- O. G. Mouritsen, Life—as a Matter of Fat: the Emerging Science of Lipidomics, Springer, Berlin, 2005 Search PubMed.
- J. Li, W.-D. He, N. He, S.-C. Han, X.-L. Sun, L.-Y. Li and B.-Y. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 1450–1462 CrossRef CAS.
- V. Ladmiral, T. M. Legge, Y. Zhao and S. Perrier, Macromolecules, 2008, 41, 6728–6732 CrossRef CAS.
- F. Giacalone and N. Martin, Chem. Rev., 2006, 106, 5136–5190 CrossRef CAS.
- A. Cravino, Polym. Int., 2007, 56, 943–956 CrossRef CAS.
- J. U. Lee, A. Cirpan, T. Emrick, T. P. Russell and W. H. Jo, J. Mater. Chem., 2009, 19, 1483 RSC.
- N. Fujita, T. Yamashita, M. Asai and S. Shinkai, Angew. Chem., Int. Ed., 2005, 44, 1257–1261 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of PEO-C60; additional results and discussions; and detailed characterization data of new compounds. See DOI: 10.1039/c1py00435b |
| This journal is © The Royal Society of Chemistry 2012 |