Synthesis of star and H-shape polymersvia a combination of cobalt-mediated radical polymerization and nitrone-mediated radical coupling reactions†
Christophe
Detrembleur
*a,
Antoine
Debuigne
a,
Ozcan
Altintas
b,
Matthias
Conradi
c,
Edgar H. H.
Wong‡
b,
Christine
Jérôme
a,
Christopher
Barner-Kowollik
*b and
Tanja
Junkers
*c
aCenter for Education and Research on Macromolecules (CERM), University of Liege, Belgium. E-mail: Christophe.Detrembleur@ulg.ac.be; Fax: (+32) 4 3663497; Tel: (+32) 4 3663465
bPreparative Macromolecular Chemistry, Institut für Technische Chemie und Polymerchemie, Karlsruhe Institute of Technology (KIT), Engesserstr. 18, 76128, Karlsruhe, Germany. E-mail: christopher.barner-kowollik@kit.edu; Fax: (+49) 721 608-45740; Tel: (+49) 721 608-45642
cInstitute for Materials Research (IMO), Polymer Reaction Design Group, Universiteit Hasselt, Agoralaan, Gebouw D, BE-3590, Diepenbeek, Belgium. E-mail: tanja.junkers@uhasselt.be; Fax: (+32) 11 26-8399; Tel: (+32) 11 26-8318
First published on 12th September 2011
Abstract
Via consecutive cobalt-mediated radical polymerization (CMRP), nitrone-mediated radical coupling (NMRC) and copper catalyzed azide-alkynecycloaddition (CuAAC), polymers with mikto-arm star and H-shape architecture were synthesized. Poly(vinyl acetate)40-block-poly(acrylonitrile)78-Co(acac)2polymers were synthesized viaCMRC and subsequently coupled using an alkyne functional nitrone. The coupling efficiency of the NMRC process was assessed employing N-tert-butyl α-phenyl nitrone (PBN), which is structurally very similar to the later employed coupling agent. Generally, coupling efficiencies of close to 90% or higher were observed in all cases. Since the coupling reaction yields triblock copolymers bearing an alkoxyamine functionality (and thus also an alkynegroup) in the middle of the chain, well defined PEG conjugates could be obtained viaCuAAC. Miktoarm star polymers of the structure (PVAc-b-PAN)2-PEG were generated as well as H-shaped material of the structure (PVAc-b-PAN)2-PEG-(PVAc-b-PAN)2viaconjugation with bifunctional PEG. In all cases, very narrow molecular weight material was obtained. Molecular weight analysis of the intermediate and the final products reveals that the hydrodynamic volume of the miktoarm star and the H-shaped materials is not substantially increased during the final conjugation reaction despite the fact that the absolute molecular weight increases by more than a factor of two in the latter case. Success of the conjugation reactions was confirmed via composition analysis viaNMR.
Introduction
Modular design approaches have—in the last few years—revolutionized the design strategies applied in polymer synthesis.1 From the combination of controlled/living (radical) polymerization and so-called click-chemistry,2 a large number of variable polymer architectures and materials are obtained.3–8 In the realm of radical polymerization techniques,9 the most employed processes are the classical living/controlled polymerization procedures such as atom transfer radical polymerization (ATRP),10nitroxide-mediated polymerization (NMP)11 and reversible addition fragmentation chain transfer (RAFT) polymerization.12Polymerconjugations are mostly achieved via the copper catalyzed azide-alkynecycloaddition (CuAAC),13thiol-ene,14 and (hetero) Diels–Alder reaction15,16 to name the most prominent examples. The toolbox available to synthetic polymer chemists is, however, wider and offers a much larger number of useful reactions that can be utilized to form complex macromolecules. For example, radical coupling methods are a reliable alternative for the assembly of molecules and are able to adequately substitute click-type conjugation reactions. Examples for radical coupling techniques are the atom transfer radical coupling ATRC,17 atom transfer nitroxide radical coupling (ATNRC),18cobalt-mediated radical coupling (CMRC)19–21 and nitrone-mediated radical coupling (NMRC).22,23 All these methods have in common that macromolecular chains that are in a dormant form are activated and transformed into macroradicals which can subsequently undergo coupling reactions. In the simplest case—ATRC—these radicals recombine via conventional termination reactions. Yet, cross-coupling approaches with stable radicals (in ATNRC) are possible as well as the mediation of the coupling reaction using dedicated coupling agents such as dienes in CMRC or nitrones in NMRC. Although all radical in nature, high reaction efficiencies can be achieved with the various coupling methods and even the synthesis of dendrimers is achievable via such routes.In the NMRC reaction, nitrones mediate the radical coupling reaction. Such compounds are excellent radical spin traps and are often used in ESR studies to transform transient species into stable counterparts that can be analyzed over extended times. Since they add rapidly to transient, propagating chains in radical polymerizations, nitrones can be employed to mediate polymerizations, either via in situ formation of a nitroxide mediating agent in in situ NMP24–29 or via direct control of the average chain length in so-called enhanced spin capturing polymerization (ESCP).30,31
In NMRC, macroradicals react readily with the mediator to form a macronitroxide which subsequently quenches other macroradicals via a persistent radical effect instead of undergoing bimolecular termination, eventually forming a symmetric coupling adduct with an alkoxyamine functionality in the middle of the chain.22 As an activation step to generate the macroradicals, abstraction of the terminal brominegroup from an ATRP-made polymer by Cu0 species is a suitable reaction pathway. In principle, however, no prerequisite is given for the activation and other reactions may be compatible with NMRC. In CMRC19–21polymers are coupled in a very similar fashion. In this case, macromolecules prepared by organometallic-mediated radical polymerization (OMRP)32 based on cobalt complexes, namely the cobalt-mediated radical polymerization (CMRP),33 present a cobalt-carbon end-group that can be activated to form macroradicals. Similarly to NMRC, combination of chains is thereby promoted by the addition of isoprene (or another conjugated diene), which will be built in a mid-chain position.19CMRC (and therewith the underpinning polymerization process CMRP)33 has the advantage over other coupling techniques that it allows for activation of radical chains that are not accessible via the ATRP route. Monomers such as vinyl acetate (VAc)34 and N-vinyl pyrrolidone (NVP)35 are not readily controlled via the conventional polymerization techniques such as ATRP, but are readily polymerizable in a controlled fashion viacobalt-mediated polymerizations. Thus, PVAc and PNVP with defined molecular weight are accessible that are available for radical coupling reactions. So far, these polymers but also poly(acrylonitrile) (PAN)37 were only coupled via the CMRC route.19–21 While an insertion of distinct chemical functionality into the chains is possible in principle, a combination with the nitrone technique seems highly advantageous since with nitrones it was already shown that secondary functionalities can be introduced into the product at a defined mid-chain location. Polyacrylates and polystyrene were synthesized according to the nitrone-mediated radical coupling procedure while introducing alkynegroups.36 In addition—in an attempt to transform the coupling reaction into a synthesis tool for multi-sequential polymers in a step growth approach—alkynes were introduced in regular intervals on the polymer backbone via a nitrone serving as a functional carrier.37
In here, we report the combination of both CMRP and NMRC as a novel synthetic tool for providing unprecedented copolymers with precise architectures. PVAc and PAN, obtained from CMRP, are coupled by means of nitrone-mediation. Homopolymers are obtained as well as block copolymers starting from PVAc-block-PAN precursors. An alkyne moiety is introduced into the products via functional nitrones, which subsequently served as an additional anchor point for further conjugation reactions. Via the alkyne, azide-functional polyethylene glycol (PEG) chains are conjugated resulting in complex three-armed polymer structures as well as H-shaped polymers when bifunctional PEG was employed (see Scheme 1). The results will be presented and discussed in a bottom-up approach: First, we will describe the activation step of model oligomers end-capped with cobalt followed by the nitrone coupling reactions of the polymeric species. In the last step, the assembly of the building blocks viaCuAAc is discussed to demonstrate the efficiency of the underlying reactions.
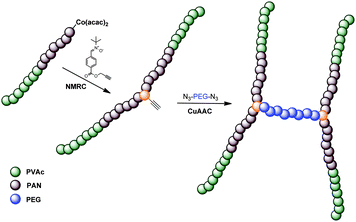 | ||
| Scheme 1 Schematic representation of the synthesis of H-shaped polymersvia combination of CMRP and NMRC followed by CuAACconjugation. | ||
Experimental section
Materials
Vinyl acetate (>99%, Aldrich) and acrylonitrile (>99%, Aldrich) were dried over calcium hydride, degassed by several freeze-thawing cycles before being distilled under reduced pressure and stored under argon. 2,2′–Azo-bis-(4-methoxy-2,4-dimethyl valeronitrile) (V-70) (Wako) and cobalt(II) acetylacetonate (Co(acac)2) (>98%, Merck), 2,2,6,6-tetramethylpiperidine-1-oxy (TEMPO) (98%, Aldrich) were used as received. The synthesis and the complete characterization of the low molecular weight alkyl-cobalt(III) adduct [Co(acac)2(–CH(OCOCH3)CH2)<4–R0] have been described elsewhere.9 Two stock solutions in dichloromethane of the alkyl cobalt(III) adduct initiator ([Co(acac)2(–CH(OCOCH3)CH2)<4–R0)]; R0 being the primary radical generated by V-70) were prepared and stored at −20 °C under argon.20 The cobalt content of these solutions was evaluated by Inductively Coupled Plasma Mass Spectrometry (ICP-MS) ([Co] = 0.0992 M and 0.148 M). N-tert-Butyl phenyl nitrone (PBN) as well as the alkyne-functional nitrones were synthesized as described previously.31,36 Polyethylene glycol materials, tosyl chloride and sodium azide were obtained from Aldrich and used as received. CuBr (99.9%, Acros) N,N,N′,N′′,N′′-pentamethyldiethyltriamine (PMDETA) (99.9%, Merck) and N,N-dimethylformamide extra dry (DMF) (99.8%, Acros) were also used as received.Size exclusion chromatography (CMRP and nitrone coupling)
Size exclusion chromatography (SEC) of poly(vinyl acetate) was carried out in tetrahydrofuran (THF) (flow rate: 1 mL min−1) at 40 °C with a Waters 600 liquid chromatograph equipped with a 410 refractive index detector and styragel HR columns (four columns HP PL gel 5μm 105Å, 104Å, 103Å, 102Å). The molar mass of PVAc determined by SEC with PS calibration was in good agreement with that determined by 1H NMR whenever the end group of the initiator (–OCH3 at δ = 3.13 ppm) could be observed and compared to the –C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) OCOCH3proton at δ = 4.8 ppm of the monomer unit, as reported elsewhere.20,21SEC of PVAc, PAN, PVAc-b-PAN and PVAc-b-PAN-b-PVAc was carried out in dimethylformamide (DMF) containing LiBr (0.025 mol L−1) at 55 °C (flow rate: 1 mL min−1), with a Waters 600 liquid chromatograph equipped with a 410 refractive index detector and styragel HR columns (HR1, 100–5000; HR3, 500–30000; HR4, 5000–500000; HR5, 2000–40000000). In all cases, polystyrene standards ranging from 580 to 225 × 104 g mol−1 were used for calibration. Absolute molecular weight parameters (Mn, Mp, Mw/Mn) of PAN based (co)polymers were determined by size exclusion chromatography (SEC) equipped with a Multi-Angle Laser Light Scattering (MALLS) detector, using (dn/dc) values determined for each sample by refractometry analysis.
OCOCH3proton at δ = 4.8 ppm of the monomer unit, as reported elsewhere.20,21SEC of PVAc, PAN, PVAc-b-PAN and PVAc-b-PAN-b-PVAc was carried out in dimethylformamide (DMF) containing LiBr (0.025 mol L−1) at 55 °C (flow rate: 1 mL min−1), with a Waters 600 liquid chromatograph equipped with a 410 refractive index detector and styragel HR columns (HR1, 100–5000; HR3, 500–30000; HR4, 5000–500000; HR5, 2000–40000000). In all cases, polystyrene standards ranging from 580 to 225 × 104 g mol−1 were used for calibration. Absolute molecular weight parameters (Mn, Mp, Mw/Mn) of PAN based (co)polymers were determined by size exclusion chromatography (SEC) equipped with a Multi-Angle Laser Light Scattering (MALLS) detector, using (dn/dc) values determined for each sample by refractometry analysis.
Size exclusion chromatography (CuAAC)
For the determination of molecular weight distributions (MWD) of the mikto-armed and H-shaped polymers, a Varian PL50 system, comprising an auto injector, a Polymer Laboratories 5.0 μm bead-size guard column, followed by three linear PL columns (PLgel 5 μm MIXED-C) and a differential refractive index detector using DMAc/0.03% LiBr as the eluent at 50 °C with a flow rate of 1 mL min−1 was used. The SEC system was calibrated using narrow polystyrene standards ranging from 160 to 6 × 106 g mol−1.Size exclusion chromatography coupled to electrospray ionization mass spectrometry detection (SEC-ESI-MS)
SEC /ESI-MS spectra were recorded on an LXQmass spectrometer (ThermoFisher Scientific, San Jose, CA, USA) equipped with an atmospheric pressure ionization source operating in the nebulizer assisted electrospray mode. The instrument was calibrated in the m/z range 195–1822 using a standard containing caffeine, Met-Arg-Phe-Ala acetate (MRFA) and a mixture of fluorinated phosphazenes (Ultramark 1621) (all from Aldrich). A constant spray voltage of 4.5 kV and a dimensionless sweep gas flow rate of 2 and a dimensionless sheath gas flow-rate of 12 were applied. The capillary voltage, the tube lens offset voltage and the capillary temperature were set to 60 V, 110 V and 275 °C respectively. The LXQ was coupled to a Series 1200 HPLC-system (Agilent, Santa Clara, CA, USA) consisting of a solvent degasser (G1322A), a binary pump (G1312A), a high-performance autosampler (G1367B), followed by a thermostatted column compartment (G1316A). Separation was performed on two mixed bed size exclusion chromatography columns (Polymer Laboratories, Mesopore 250 × 4.6mm, particle dia. 3μm) with pre-column (Mesopore 50 × 4.6mm) operating at 30 °C. THF at a flow rate of 0.30 mL min−1 was used as eluent. The mass spectrometer was coupled to the column in parallel to an RI-detector (G1362A with SS420x A/D) in a setup described previously.38 0.27 mL min−1 of the eluent were directed through the RI-detector and 30 μL min−1 infused into the electrospray source after post-column addition of a 100 μM solution of sodium iodide in methanol at 20 μL min−1 by a micro-flow HPLC syringe pump (Teledyne ISCO, Model 100DM). 20 μL of a polymer solution with a concentration of approximately 3 mg mL−1 were injected onto the HPLC system.NMR characterization
1H NMR spectra of the PVAc macroinitiators were recorded at 298 K with a Bruker Spectrometer (400 MHz) in CDCl3. (D1 = 2s, 16 scans, 5 wt % of polymer). 1H NMR spectra of PAN, PVAc-b-PAN and PVAc-b-PAN-b-PVAc were recorded with the same spectrometer at 298 K in deuterated dimethylsulfoxide (DMSO-d6). (D1 = 5 s, 32 scans, 5 wt % of polymer). 1H NMR spectra of the PEG polymers were recorded with two NMR spectrometers (300 and 400 MHz) from Oxford Instruments Ltd. using a Varian probe (9 mm-4-nucleus AutoSWPFG).Typical procedure for the synthesis of PVAc-Co(acac)2 and radical coupling with a nitrone
A solution of alkyl-cobalt(III) initiator ((Co(acac)2(–CH(OCOCH3)CH2)<4–R0) 0.0992M; 1.5 mL, 1.5 10−4 mol) in CH2Cl2 was introduced under argon in a Schlenk tube and the solvent (CH2Cl2) was evaporated to dryness under reduced pressure at room temperature (rt). The residue was then placed under argon at 0 °C and added with dry VAc (2 mL, 0.162 mol). The reaction medium was subsequently stirred for 4.5 h at 40 °C. An aliquot was withdrawn for the determination of the conversion by 1H NMR spectroscopy (30% conversion), and the molecular weight and polydispersity by SEC in THF (Mn = 3900 g mol−1; Mw/Mn = 1.04). The residual monomer was then evacuated under vacuum at rt.To the PVAc-Co(acac)2 a solution of N-tert-butyl phenyl nitrone (PBN, 0.053 g PBN in 1 mL of toluene; 3 × 10−4 mol) was added under argon. Once the polymer was dissolved, the reaction mixture was heated at 40 °C overnight. An aliquot was subsequently withdrawn to determine the molecular weight and polydispersity of the coupled polymer (Mn = 7200 g mol−1; Mw/Mn = 1.07; coupling efficiency = 92%). The final product was precipitated 2 times in heptane and dried at 40 °C under vacuum overnight.
Typical procedure for the synthesis of PAN-Co(acac)2 and radical coupling with a nitrone
A solution of alkyl-cobalt(III) initiator ((Co(acac)2(–CH(OCOCH3)CH2)<4–R0) 0.148M; 1.25 mL, 1.85 10−4 mol) in CH2Cl2 was introduced to a Schlenk tube under argon and the solvent (CH2Cl2) was evaporated to dryness under reduced pressure at ambient temperature (rt). The residue was then placed under argon at 0 °C and dry and degassed DMF (1 mL) and DMSO (1 mL) was added. Dry and degassed acrylonitrile (AN, 2ml, 0.162 mol) was subsequently added and the reaction medium was stirred for 18 h at 0 °C. An aliquot was withdrawn for the determination of the conversion by 1H NMR spectroscopy (55% conversion) and the molecular weight and polydispersity by SEC in DMF/LiBr using a PS calibration (Mn = 41400 g mol−1; Mw/Mn = 1.02). The residual monomer was finally removed under vacuum at rt. Absolute molecular weight (Mn,abs = 5300 g mol−1) and polydispersity (Mw/Mn = 1.02) of PAN were obtained by SEC equipped with a MALLSdetector, using a dn/dc value determined on a purified PAN sample prepared in an identical fashion (dn/dcPAN = 0.076) and deactivated by TEMPO to avoid any possible side reaction (such as coupling reaction) during sampling and exposure to air.To the PAN-Co(acac)2 solution, a solution of N-tert-butyl phenyl nitrone (PBN; 0.065 g PBN in 1 mL of DMF; 3.7 10−4 mol) was added under argon at 0 °C. The reaction mixture was stirred at 40 °C overnight. An aliquot was then withdrawn to determine the molecular weight and polydispersity of the coupled polymer by SECDMF/LiBr calibrated by PS standards (Mn = 66200 g mol−1; Mw/Mn = 1.03). The final product was deactivated by adding an excess TEMPO, precipitated 2 times in methanol and dried at 40 °C under vacuum overnight. Absolute molecular parameters were determined by SEC/MALLS (Mn,abs = 9700 g mol−1; Mw/Mn = 1.02; coupling efficiency = 92%).
Synthesis of PVAc-b-PAN-b-PVAc by radical coupling of PVAc-b-PAN-Co(acac)2 with nitrone bearing alkynegroup (nitrone 3)
A solution of alkyl-cobalt(III) initiator ((Co(acac)2(–CH(OCOCH3)CH2)<4–R0) 0.148M; 3 mL, 4.44 10−4 mol) in CH2Cl2 was introduced to a Schlenk tube under argon and the solvent (CH2Cl2) was evaporated under reduced pressure at room temperature (rt). The residue was subsequently placed under argon at 0 °C and dry VAc (4 mL, 0.324 mol) was added. The reaction medium was then stirred for 6 h at 40 °C. An aliquot was withdrawn for the determination of the conversion by 1H NMR spectroscopy (37% conversion) and the molecular weight and polydispersity by SEC in THF (Mn = 3400 g mol−1; Mw/Mn = 1.05) and in DMF/LiBr (Mn,DMF = 15900 g mol−1; Mw/Mn = 1.04). The residual monomer was subsequently removed under vacuum at rt.To the PVAc-Co(acac)2 dry and degassed DMF (4 mL) was then added under argon at 0 °C followed by AN (4 mL). The mixture was subsequently stirred at 0 °C until the conversion reached close to 50% (after about 24h). An aliquot was withdrawn for the determination of the molecular weight and polydispersity by SEC in DMF/LiBr (Mn = 50700 g mol−1; Mw/Mn = 1.03). The residual monomer was removed under vacuum at rt.
To the PVAc-b-PAN-Co(acac)2 solution a solution of the nitrone (0.23 g PBN in 2 mL of DMF; 8.88 10−4 mol) was added under argon. The reaction mixture was subsequently heated at 40 °C overnight. An aliquot was then withdrawn to determine the molecular weight and polydispersity of the coupled polymer by SECDMF/LiBr using PS calibration (Mn = 81400 g mol−1; Mw/Mn = 1.04). The final product was precipitated 3 times in a methanol/water (20/80) mixture heptane and dried at 40 °C under vacuum overnight.
Coupling reaction of Co(acac)2(–CH(OAc)CH2)∼4–R0 oligomers with nitrone1 (N-tert-butyl α-phenyl nitrone) for MS study
An alkyl-cobalt(III) compound, i.e. [Co(acac)2(–CH(OCOCH3)CH2)<4–R0], was prepared following a previously reported procedure. In order to guarantee the purity of the desired product, first and last millilitres of the alkyl cobalt(III) fraction were eliminated during the chromatographypurification. Subsequently a solution of [Co(acac)2(–CH(OCOCH3)CH2)<4–R0] in CH2Cl2 (1.0 mL of a 0.25 M stock solution, 0.25 mmol) was introduced to a Schlenk tube under argon and evaporated to dryness under reduced pressure at room temperature. The residue was subsequently combined with a PBN solution in degassed DMSO (1.0 mL of a 0.40 M stock solution, 0.40 mmol). After 72 h stirring at 40 °C, the crude mixture was analyzed by ESI-MS (see Fig. 3). Theoretical and measured mass-to-charge ratios of the PVAc coupling product are provided in the supporting information (Table S1, ESI†).Synthesis of tosylated poly(ethylene glycol) and poly(ethylene glycol)monomethyl ether
A typical procedure is described. To a stirred solution of poly(ethylene gylcol) monomethyl ether (5000 g mol−1, 5.00 g, 1.00 mmol) in anhydrous pyridine (20 mL) p-toluenesulfonyl chloride (3.82 g, 20.0 mmol) was added dropwise and the reaction mixture was then allowed to stir at room temperature for 24 h and subsequently added to cold distilled water (60 mL). The polymer was extracted with dichloromethane. The organic phase was dried over MgSO4 and concentrated to a volume of about 20 mL. Precipitation in cold diethyl ether afforded a white solid (4.54 g, 91%). 1H-NMR (CDCl3) (ppm): 7.79 (m, arom.), 4.13 (t, CH2CH2OTs, J = 4.8 Hz), 3.62 (s, O–(CH2)2–O), 3.36 (s, O–CH3), 2.43 (s, CH3-aryl).Synthesis of azide-functional poly(ethylene glycol) and azide-functional poly(ethylene glycol)monomethyl ether
A typical procedure is described. To a stirred solution of tosylated poly(ethylene gylcol) monomethylether (∼5000 g mol−1, 2.20 g, 0.5 mmol) in N,N-dimethyl formamid (25 mL) sodium azide (10 eq., 0.35 g, 5.0 mmol) was added. The mixture was stirred at room temperature for 3 days and then added to cold distilled water (60 mL). The polymer was extracted three times with DCM. The organic phase was dried over MgSO4 and concentrated to a volume of about 20 mL and subsequently precipitated in cold diethyl ether. A white solid was obtained (2.0 g, 92%). 1H-NMR (CDCl3) (ppm): 3.64 (s, O–(CH2)2–O), 3.60 (t, O–CH2–CH2–N3), 3.46 (t, O–CH2–CH2–N3), 3.36 (s, O–CH3).Typical procedure for the CuAAc reaction to obtain miktoarm star polymers
In a Schlenk tube were placed PVAc-b-PAN-alkyne-PAN-b-PVAc (100 mg, 0.0063 mmol, 1 equiv), PEG-N3 (40 mg, 0.0063 mmol, 1 equiv), PMDETA (6.7 μL, 0.032 mmol, 5 equiv), and DMF (5 mL). The tube was fitted with a rubber septum. The solution was further degassed by three freeze–thaw–pump cycles and left under argon. CuBr (4.6 mg, 0.032 mmol, 5 equiv) was added to the solution under argon. The tube was stirred at room temperature for 24 h. Subsequently the solution was passed through a neutral alumina column to remove the CuBr-PMDETA complex. The obtained crude product was concentrated, precipitated in methanol/water (1/4), filtrated and dried under vacuum at room temperatureTypical procedure for the CuAAc reaction to obtain H-shaped polymers
In a Schlenk tube were placed PVAc-b-PAN-alkyne-PAN-b-PVAc (100 mg, 0.0064 mmol, 1 equiv) N3-PEG-N3 (16.3 mg, 0.0032 mmol, 0.5 equiv), PMDETA (6.7 μL, 0.032 mmol, 5 equiv), and DMF (5 mL). The tube was fitted with a rubber septum. The solution was further degassed by three freeze–thaw–pump cycles and left under argon. CuBr (4.6 mg, 0.032 mmol, 5 equiv) was added to the solution under argon. The tube was stirred at room temperature for 24 h. Later the solution was passed through a neutral alumina column to remove CuBr-PMDETA. The obtained crude product was concentrated, precipitated in methanol/water (1/4), filtrated and dried under vacuum at room temperature overnight.Results and discussion
Radical coupling of poly(vinyl acetate) and poly(acrylonitrile) end-capped by Co(acac)2 with various nitrones (Scheme 1)
To assess the coupling efficiency of NMRC with Co-capped polymers, model oligomers were synthesized in order to obtain coupling products that can be readily analyzed viaSEC and NMR. A preformed alkyl-cobalt(III) adduct (R0–(CH2–CHOAc)<4–Co(acac)2; where R0 = primary radical from the decomposition of V-70) is used to initiate the polymerization of vinyl acetate at 40 °C by CMRP and to provide poly(vinyl acetate) chains end-capped by the cobalt complex (PVAc-Co(acac)2). For more details on the CMRP, the reader is referred to literature.33 After synthesis, the residual monomer is removed under vacuum and PVAc-Co(acac)2 is added to a solution of a nitrone (1, see Scheme 2). The coupling reaction occurs by generating PVAc macroradicals by thermal homolysis of the C–Co bond. The nitrone subsequently traps a PVAc macroradical, leading to the corresponding macronitroxide that further traps another macroradical by spin annihilation according to Scheme 3. Effectively, PVAc that is mid-chain functionalized by an alkoxyamine is formed (see Scheme 3).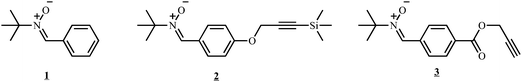 | ||
| Scheme 2 Nitrones employed for coupling polymers end-capped by Co(acac)2. | ||
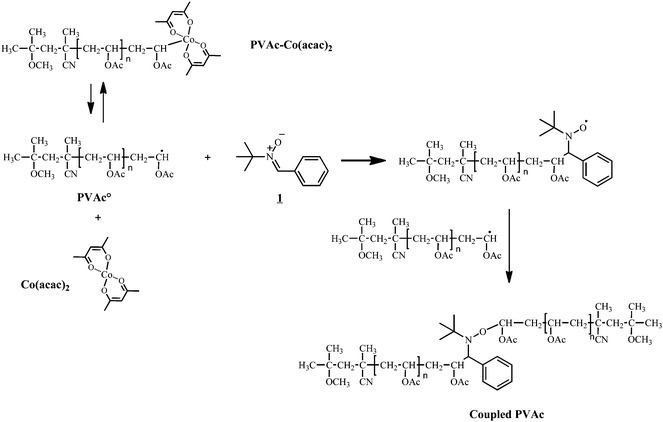 | ||
| Scheme 3 Radical coupling of PVAc-Co(acac)2 by nitrone1. | ||
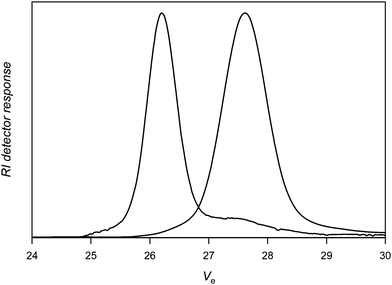
Using a two molar excess of nitrone1 (N-tert-butyl α-phenyl nitrone, PBN) compared to PVAc-Co(acac)2, Fig. 1 shows that the coupling reaction is effective indicated by the shift of the SEC chromatogram of the PVAc-Co(acac)2 precursor to higher molecular weights. The extent of coupling is calculated as xc = 2[1 − (Mn,0/Mn)] in which Mn,0 and Mn are the molar masses of the polymer precursor and the coupled product, respectively. The extent of coupling is high and reaches 92% (Table 1, entry 1), the small tailing at the low molar mass side of the SEC chromatogram accounts for some small amounts of unreacted PVAc. It should be noted that the nitrone coupling, at least for polyacrylates, was shown to proceed also under equimolar conditions.36 Nevertheless, higher amounts can be used to increase the efficiency of the coupling.
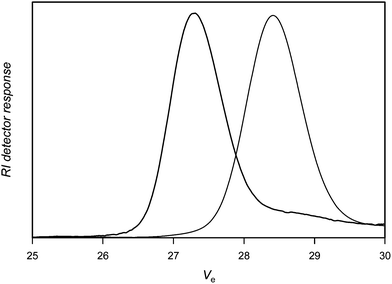 | ||
| Fig. 1 Size exclusion chromatograms of poly(vinyl acetate) before (right) and after (left) reaction with nitrone1. | ||
| Entry | PVAc-Co(acac)2 | Radical coupling | |||||
|---|---|---|---|---|---|---|---|
| M n a (g mol−1) | M w/Mna | [PVAc-Co(acac)2]/[nitrone] | Nitrone | M n b (g mol−1) | M w/Mnb | x c c | |
| a Number-average molecular weight and polydispersity of PVAc-Co(acac)2 determined by SEC analysis in THF calibrated by polystyrene standards. b Number-average molecular weight and polydispersity of coupled PVAc determined by SEC analysis in THF calibrated by polystyrene standards. c The extent of coupling calculated as xc = 2[1 − (Mn,0/Mn)] in which Mn,0 and Mn are the molar masses of the polymer precursor and the coupled product, respectively. d Solvent = toluene. e Solvent = DMSO. | |||||||
| 1d | 3900 | 1.04 | 1/2 | 1 | 7200 | 1.07 | 0.92 |
| 2e | 3450 | 1.04 | 1/2 | 1 | 6400 | 1.06 | 0.93 |
| 3d | 4700 | 1.05 | 1/4 | 1 | 8700 | 1.10 | 0.92 |
| 4d | 5500 | 1.07 | 1/2 | 2 | 9200 | 1.17 | 0.84 |
| 5d | 6300 | 1.06 | 1/2 | 3 | 11050 | 1.14 | 0.88 |
| 6d | 5650 | 1.06 | 1/4 | 3 | 10900 | 1.08 | 0.88 |
Two approaches are envisaged to improve the coupling efficiency. The first one consists of carrying out the coupling reaction in dimethylsulfoxide (DMSO) instead of in toluene. Indeed, it is known that DMSO activates the C–Co bond cleavage and should therefore accelerate the generation of PVAc macroradicals that might affect the coupling efficiency. Table 1 shows that identical radical coupling efficiencies are observed in both solvents (comparison of entries 1 and 2). The second strategy consists of increasing the amount of nitrone compared to PVAc-Co(acac)2 since the increase of nitrone concentration compared to the macroradical precursor has been shown to be beneficial to the coupling reaction. Higher nitrone concentrations increase the likelihood of macronitroxides formation and thus reduce the rate of random termination events. Since a persistent radical effect controls the formation of AA′ macroalkoxyamines, higher nitrone and thus slightly larger nitroxide concentrations may be used without problems in a relatively broad concentration window. However, the coupling efficiency remains identical irrespective of the PVAc-Co(acac)2/PBN molar ratio (comparison of entries 1 and 3, Table 1).
1H NMR analysis of PVAc (see Fig. 2) coupled by 1 (Table 1, entry 2) shows the typical peaks of PVAc at 4.8 ppm (CH2–C![[H with combining low line]](https://www.rsc.org/images/entities/i_char_0048_0332.gif) –OCOCH3), 1.9 ppm (CH2–CH–OCOC3) and at 1.75 ppm (C2–CH–OCOCH3) but also the methoxy group of the initiator at the α- and ω-positions at 3.15 ppm. Importantly, the presence of the alkoxyamine in the polymer chain is observed by the presence of the phenyl group originating from 1 at 7.1–7.55 ppm. The comparison of the integration of this peak corresponding to 5 protons with that of the methoxy groups of the initiator at both chain ends corresponding to 6 protons allows to confirm the presence of one phenyl group per polymer chain. The degree of polymerization can also be calculated by 1H-NMR by comparing the integral of CH2–C–OCOCH3 of the polymer backbone with the methoxy group of the initiator at both chain-ends at 3.15 ppm. This degree of polymerization (DPNMR = 70) is very close to that determined by SEC (DPSEC = 74).
–OCOCH3), 1.9 ppm (CH2–CH–OCOC3) and at 1.75 ppm (C2–CH–OCOCH3) but also the methoxy group of the initiator at the α- and ω-positions at 3.15 ppm. Importantly, the presence of the alkoxyamine in the polymer chain is observed by the presence of the phenyl group originating from 1 at 7.1–7.55 ppm. The comparison of the integration of this peak corresponding to 5 protons with that of the methoxy groups of the initiator at both chain ends corresponding to 6 protons allows to confirm the presence of one phenyl group per polymer chain. The degree of polymerization can also be calculated by 1H-NMR by comparing the integral of CH2–C–OCOCH3 of the polymer backbone with the methoxy group of the initiator at both chain-ends at 3.15 ppm. This degree of polymerization (DPNMR = 70) is very close to that determined by SEC (DPSEC = 74).
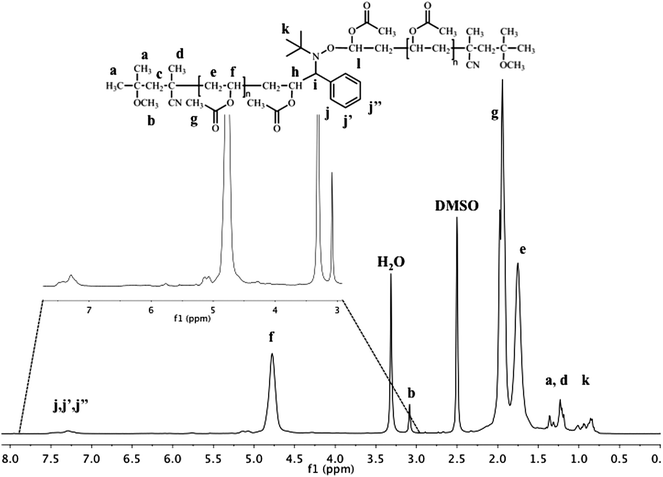 | ||
| Fig. 2 400MHz 1H-NMRspectrum of the coupling product from PVAc-Co(acac)2 mediated by PBN (Table 1, entry 2). | ||
To dispel any doubt about the insertion of nitrone1 during the coupling process, the alkyl-cobalt(III) adduct that was described above to start the VAcpolymerization and that is in fact just a very short PVAc-chain end-capped by Co(acac)2 itself, is coupled by nitrone1. Thereby a product is formed that is of sufficiently small molecular weight to be analyzed by soft ionization mass spectrometry such as electrospray ionization mass spectrometryESI-MS.
For the ease of the analysis, the ESI measurement was carried out on the crude mixture of the coupling reaction of very low molecular weight PVAc-[CoIII] precursors, i.e. [Co(acac)2(–CH(OCOCH3)CH2)<4–R]. In this case, 1.6 equivalents of PBN were used compared to the organometallic species. Fig. 3 shows a typical ESI spectrum acquired using DCM/methanol 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 as solvent and no specific cationization agent resulting in Na adducts. Highly uniform oligomers are detected with a repeat unit corresponding to a 86 Da interval and thus according to the mass of VAc. The measured m/z ratios demonstrate that the PVAc coupling product incorporates one PBN unit. For instance, the most intense signal (X6Na+) is observed at m/z 996 and can be attributed to sodium-cationized oligomers containing six VAc units, one PBN moiety and two V-70 initiating residues and thus the expected product. No signal corresponding to a PVAc chain without PBN unit (e.g.X6Na+, m/z 819, X7Na+, m/z 905) is detected, which confirms that the PVAc precursors do not undergo a direct coupling by combination. Also, uncoupled PVAc is not detected in significant amounts.
:1 as solvent and no specific cationization agent resulting in Na adducts. Highly uniform oligomers are detected with a repeat unit corresponding to a 86 Da interval and thus according to the mass of VAc. The measured m/z ratios demonstrate that the PVAc coupling product incorporates one PBN unit. For instance, the most intense signal (X6Na+) is observed at m/z 996 and can be attributed to sodium-cationized oligomers containing six VAc units, one PBN moiety and two V-70 initiating residues and thus the expected product. No signal corresponding to a PVAc chain without PBN unit (e.g.X6Na+, m/z 819, X7Na+, m/z 905) is detected, which confirms that the PVAc precursors do not undergo a direct coupling by combination. Also, uncoupled PVAc is not detected in significant amounts.
![ESI-MS
spectrum of a PVAc sample prepared by the coupling reaction of [Co(acac)2(–CH(OCOCH3)CH2)<4–R0]) with PBN.](/image/article/2012/PY/c1py00297j/c1py00297j-f3.gif) | ||
| Fig. 3 ESI-MS spectrum of a PVAc sample prepared by the coupling reaction of [Co(acac)2(–CH(OCOCH3)CH2)<4–R0]) with PBN. | ||
In order to introduce an alkynegroup at the middle of the PVAc chain, nitrone2 functionalized by a protected alkynegroup was then tested for the radical coupling. A protected alkyne was first used to avoid any suspected side reactions between alkyne and radicals. The radical coupling occurs in similar conditions established for 1, i.e. in toluene using a two molar excess of nitrone2 compare to PVAc-Co(acac)2 at 40 °C. The coupling efficiency tends to decrease when using the functional nitronevs. the unsubstituted one since xc is limited to 0.84 (entry 4, Table 1). Steric hindrance might account for this loss of coupling efficiency when nitrone1 is replaced by the more bulky nitrone2. Such a hindrance may result in lower addition rates of macroradicals to the nitrone as well as reduced combination rates of the macronitroxides towards other radicals. With polyacrylates, no reduction in efficiency was observed when exchanging 1 for 2,23,36 however PVAc macroradicals have a different reactivity and lead to more profound changes in the addition rate due to steric hindrance.
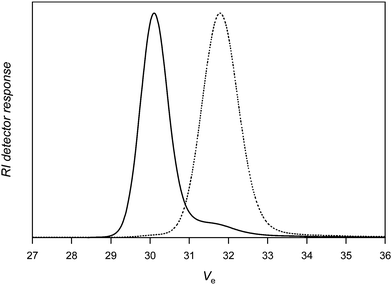
For the sake of comparison, a nitrone with an alkynegroup that is not protected (3) was subsequently tested under identical conditions. The coupling efficiency remains high (around 90%; Table 1, entry 5), suggesting that side reactions between the PVAc macroradical and alkynegroup are limited. Purification of the polymer by dialysis against methanol (SpectraPore 3000 dialysismembrane) allows removing excess nitrone and most of the uncoupled PVAc (see Fig. 4). A clear shift of the distribution towards double molecular weight is observed, whereby small amounts of material are apparently not reacted in the coupling reaction as is seen from the tailing towards higher elution volumes of the product distribution. Doubling the amount of nitrone in the reaction medium does not increase nor decrease further the extent of coupling and also similar distributions are obtained (Table 1, entry 6).
 | ||
| Fig. 4 Size exclusion chromatography of poly(vinyl acetate) before (right) and after (left) reaction with nitrone 3 followed by dialysis against methanol (Table 1, entry 5). | ||
1H NMR analysis evidences the presence of the alkoxyamine in the middle of the polymer chain by the signals typical of protons of the phenyl group at 7.5–8.0 ppm (see Figure S1 in the supporting information section). Comparison of the integral of this signal corresponding to 4 protons to that of methoxy groups of chain-ends corresponding to 6 protons at 3.15 ppm confirms the presence of one phenyl group per polymer chain. The methylene group (O–C2–C≡CH) of the alkoxyamine is also observed at 4.94 ppm and its integral perfectly fits to the expected structure.
In the next step, the radical coupling of poly(acrylonitrile) end-capped by Co(acac)2 (PAN-Co(acac)2) was explored employing nitrone1 and using a PAN-Co(acac)2 to nitrone molar ratio = 1:2. In contrast to the previous experiments, the reaction is carried out in a DMF/DMSO (2/1) mixture, as PAN is insoluble in toluene. At 40 °C, the coupling is efficient as shown by the SEC chromatogram of PAN-Co(acac)2 that is almost completely shifted towards the higher molecular weight side (see Fig. 5). Only a small amount of unreacted PAN is present at the low molar mass side of the SEC chromatogram. Analysis of the PAN precursor and the coupled product by SEC equipped with a multi-angle laser light scattering (MALLS) detector allows determining the absolute molar masses of the polymers and thus to precisely calculate the extent of coupling to xc = 0.91. As observed for PVAc, the doubling of the PBN amount does not affect the coupling efficiency (Table 2, entry 2). 1H NMR spectroscopy shows the typical signals of the methyne and methyleneprotons of the PAN main chain at 3.14 ppm and 2.04 ppm, respectively. Typical peaks of the phenyl group of nitrone1 inserted at the middle of the polymer chain are observed at 7.3–7.65 ppm, clearly confirming the incorporation of PBN in the chain (see Fig. S2, ESI†). It should be noted that based on the results obtained on PVAc, very similar coupling efficiencies are also expected for PAN employing the nitrones 1–3, given that the type of activation as well as the general structure of the nitrones is very similar.
 | ||
| Fig. 5 Size exclusion chromatography of poly(acrylonitrile) before (dotted line) and after (solid line) reaction with nitrone1. | ||
| Entry | PAN-Co(acac)2 | Radical coupling | ||||
|---|---|---|---|---|---|---|
| M n a (g mol−1) | M w/Mna | [PAN-Co(acac)2]/[nitrone] | M n b (g mol−1) | M w/Mnb | x c c | |
| a Absolute number-average molecular weight and polydispersity of PAN-Co(acac)2 determined by SEC analysis in DMF/LiBr using a MALLSdetector with the specific refractive index increment (dn/dc) of PAN = 0.076 ml g−1. b Absolute number-average molecular weight and polydispersity of coupled PAN determined by SEC analysis in DMF/LiBr using a MALLSdetector with the specific refractive index increment (dn/dc) of PAN = 0.076 ml g−1. c The extent of coupling calculated as xc = 2[1 − (Mn,0/Mn)] in which Mn,0 and Mn are the molar masses of the polymer precursor and the coupled product, respectively. | ||||||
| 1 | 5300 | 1.02 | 1/2 | 9700 | 1.02 | 0.91 |
| 2 | 5400 | 1.02 | 1/4 | 9800 | 1.02 | 0.91 |
Preparation of mid-chain functionalized poly(vinyl acetate)-b-poly(acrylonitrile)-b-poly(vinyl acetate), building blocks for mikto-arm and H-shape block copolymers
After the coupling of both PVAc and PAN macroradicals was proven successful, more complex structures could be targeted. The coupling of polymer chains end-capped by Co(acac)2 with the nitrone bearing alkynegroup (nitrone3) is very useful for designing new symmetrical and mid-chain functionalized ABA triblock copolymers, which resemble interesting building blocks for mikto-arm block copolymers. Indeed, alkynegroups in mid-chain position can be used to graft single molecules or polymers to the chains through the highly efficient and selective copper catalyzed azide alkyne cycloaddition (CuAAC) reaction, the most prominent representative of the so-called click reactions.39As an illustration, when nitrone3 bearing the unprotected alkynegroup is added to a PVAc40-b-PAN78-Co(acac)2diblock copolymer prepared by CMRP, the symmetrical PVAc40-b-PAN156-b-PVAc40triblock copolymer mid-chain functionalized by the alkynegroup is formed (see Scheme 4).
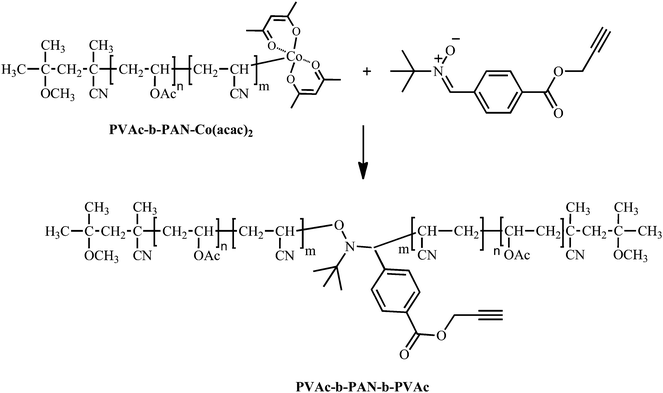
Again, the high efficiency of the coupling reaction is also here demonstrated by the clear shift of the SEC chromatogram of the diblock copolymer towards the higher molecular weight side with almost no residual diblock copolymer being left over. Moreover, a very low polydispersity (Mw/Mn = 1.04) is maintained after this reaction. Fig. 6 depicts the successive growth of the polymer chains. On the right, the PVAc-Co(acac)2polymer is shown with the dashed line representing the distribution of the chain extended PVAc-PANdiblock copolymer. Finally, the ABA triblock copolymer that is directly obtained from the nitrone coupling reaction is shown at the lowest retention time.
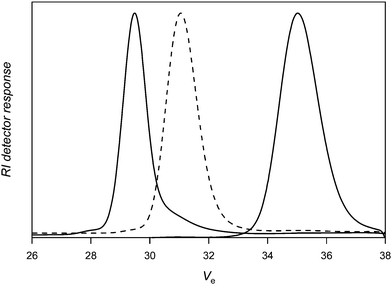 | ||
| Fig. 6 Size exclusion chromatography of PVAc-Co(acac)2 (right; Mn,SEC DMF = 15900 g mol−1; Mw/Mn = 1.04), PVAc-b-PAN-Co(acac)2 (middle; Mn,SEC DMF = 50700 g mol−1; Mw/Mn = 1.03) and PVAc-b-PAN-b-PVAc (left; Mn,SEC DMF = 81400 g mol−1; Mw/Mn = 1.04) formed by coupling PVAc-b-PAN-Co(acac)2 with nitrone3. | ||
1H NMR analysis of the coupled copolymer evidences the presence of signals that are typical of PVAc and PAN but also the presence of protons of the phenyl group of the alkoxyamine at 7.6–7.9 ppm (see Fig. S3, ESI†), attesting the successful incorporation of the functional nitrone in the copolymer. Comparison of the integrals of C–O(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)–CH3 of PVAc at 4.78 ppm to that of the phenylprotons at 7.6–7.8 ppm allows to determine the experimental VAc unit/alkoxyamine ratio in the copolymer. This VAc/alkoxyamine molar ratio is 90 and thus close to the theoretical value ([VAc]/[alkoxyamine]th = 80) determined from the absolute molecular weight of PVAc used for the synthesis of the copolymer (Mn,abs = 3400 g mol−1) and taking into account that one alkoxyamine is formed per chain during coupling as depicted in Scheme 4. This corresponds to a coupling efficiency of approximately 90% (assuming a perfect block extension).
O)–CH3 of PVAc at 4.78 ppm to that of the phenylprotons at 7.6–7.8 ppm allows to determine the experimental VAc unit/alkoxyamine ratio in the copolymer. This VAc/alkoxyamine molar ratio is 90 and thus close to the theoretical value ([VAc]/[alkoxyamine]th = 80) determined from the absolute molecular weight of PVAc used for the synthesis of the copolymer (Mn,abs = 3400 g mol−1) and taking into account that one alkoxyamine is formed per chain during coupling as depicted in Scheme 4. This corresponds to a coupling efficiency of approximately 90% (assuming a perfect block extension).
Preparation of azide-functional polyethylene glycol chains and formation of mikto-arm and H-shape block copolymers
To create mikto-armed star polymers and H-shape materials, azide functional counterparts for the above described polymer building blocks were synthesized. Therefore, three different commercially available hydroxy-functional polyethylene glycol compounds were modified to convert the hydroxyl into azide functionalities. First, the PEG were reacted with an excess of tosyl chloride to create a suitable leaving group which was subsequently replaced by N3 following literature procedures.40–42 Mono- and bifunctional PEGs were obtained as listed in Table 3. Overall, good functional fidelities of the polymers were obtained, with more than 90% of all chains carrying the desired azidegroups (or two azides, respectively). The identity and purity of PEG 1 and PEG 2 was additionally checked viaESI-MSspectroscopy giving consistent results with the NMR characterization. It should be noted that the obtained functional fidelities are well in line with literature reports, especially regarding that PEG 2 carries two azide endgroups. Molecular weights of the PEGs were checked viaSEC and NMR. Since the employed SEC setup was directly calibrated with polystyrene standards, Mn based on the NMR evaluation is more reliable and polymer concentrations in solution were based on this value.| Sample | Endgroups | M n, SEC THF a | M n/Mw | M n, NMR b | Functionalityc |
|---|---|---|---|---|---|
| a THF GPC directly calibrated with polystyrene standards. b Calculated based on integration of –CH2–N3NMR peak compared to backbone –CH2–. c Calculated based on residual tosylate observed in spectrum. | |||||
| PEG 1 | CH3O, N3 | 7040 | 1.03 | 6400 | 93% |
| PEG 2 | 2x N3 | 5860 | 1.03 | 5120 | 92% |
To assess the coupling efficiency of the CuAAC reaction, first conjugations were carried out on the simplest available system, i.e. the midchain functional PVAc as also shown in Fig. 4. Fig. 7 depicts the molecular weight distributions before and after polymerconjugation. The dotted line represents the PVAcpolymer with the embedded alkyne functionality ion midchain location. The dashed line shows PEG 1. As already seen in the elugram discussed above in the context of PVAcNMRC, a small amount of uncoupled starting material is still visible in the distribution, which consequently also does not react in the CuAAC reaction. The main distribution, however, shifted and a number average molecular weight of 10800 g mol−1 is obtained by SEC in dimethylacetamide solution (for a list of all molecular weights involved in the CuAAC reactions, refer to Table 4). It should be noted that no match of the measured molecular weight with the addition of the individual building blocks must be expected since block copolymers and star-shaped materials in particular do not allow for accurate molecular weight distributions when the universal calibration principle is applied. Nevertheless, the clear shift of the distribution is indicative of a successful click reaction. Due to the overlapping low-molecular weight shoulder of the PVAc material, it is unclear if some of traces of PEG 1 are still present in the polymer after reaction. Inspection of the product distribution, however, suggests that an almost quantitative conjugation has occurred.
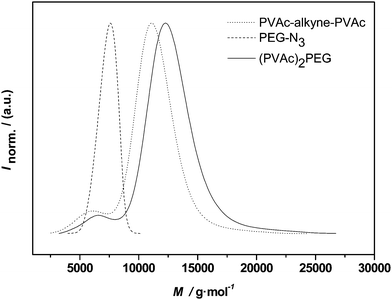 | ||
| Fig. 7 Distributions of alkyne-midchain functional PVAc, PEG-1 and the conjugation product (PVAc)2-PEG. | ||
| Polymer | M n,GPC/g mol−1a | M w/Mn |
|---|---|---|
| a Measured on a SEC with DMAc as the eluent, calibrated directly with polystyrene standards. | ||
| PVAc-alkyne-PVAc | 9400 | 1.10 |
| PVAc-b-PAN-alkyne-PAN-b-PVAc | 38700 | 1.05 |
| (PVAc)2PEG | 10800 | 1.09 |
| (PVAc-b-PAN)2PEG | 45200 | 1.05 |
| (PVAc)2PEG(PVAc)2 | 12600 | 1.09 |
| (PVAc-b-PAN)2PEG(PVAc-b-PAN)2 | 43200 | 1.05 |
With the success of the above reaction largely confirmed, the mid-chain functionalized ABA triblock copolymer was subjected to the CuAACconjugation. The outcome of such reaction is depicted in Fig. 8. Again, a clear shift of the whole distribution is observed with the apparent Mn changing from 38700 to 45200 g mol−1, indicating an efficient polymerconjugation reaction. Since the PEG 1 is significantly smaller in size than the PVAc-b-PAN-b-PVAc triblock, no overlap of the product distribution is observed and complete disappearance of the PEG can be confirmed. Thus, it can be concluded that the reaction—as expected—proceeds well in equimolar conditions.
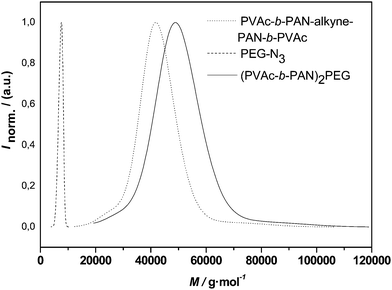 | ||
| Fig. 8 Distributions of alkyne-midchain functional pVAc-b-PAN-b-PVAc, PEG-1 and the conjugation product (PVAc-b-PAN)2-PEG. | ||
In the present case relatively small PEG chains were attached to the PVAc-PAN triblocks. Even though not tested, there is, however, only little doubt that also larger chains could be coupled since CuAAC usually allows for conjugations also between larger macromolecules. Instead of conjugation of higher molecular weight compounds, we choose a different route and aimed at conjugation of the same PVAc and PVAc-PAN blocks with bifunctional PEG 2, thus creating H-shaped polymers. Analogue reactions to PEG 1 were carried out and the resulting molecular weight distributions are given in Fig. 9 and Fig. 10.
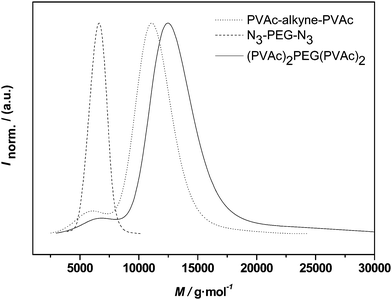 | ||
| Fig. 9 Distributions of alkyne-midchain functional PVAc, PEG 2 and the conjugation product (PVAc)2-PEG-(PVAc)2. | ||
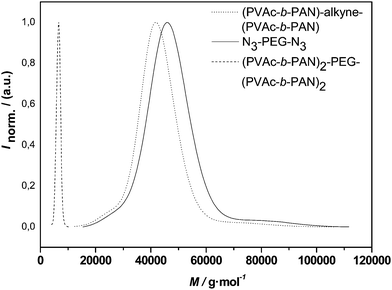 | ||
| Fig. 10 Distributions of alkyne-midchain functional PVAc-b-PAN-b-PVAc, PEG 2 and the conjugation product (PVAc-b-PAN)2-PEG-(PVAc-b-PAN)2. | ||
Overall, a very similar outcome to the conjugations with PEG 1 is observed for the PVAchomopolymer as well as the triblock copolymer. Uniform molecular weight distributions with low polydispersities are obtained. While the molecular weight distributions indicate a success of the reaction based alone on occurrence of a symmetrical shift of the distributions, closer inspection of the apparent molecular weights raises questions. As detailed in Table 4, only a slightly higher molecular weight is observed for the PVAc H-shaped polymer when compared to the three-arm star. With the coupling of the triblocks, even a slightly smaller apparent Mn is observed for the H-shaped vs. the miktostar product. Thus, at a first glance, failure of the reaction might be expected. Closer analysis indicates, however, that hydrodynamic volume effects are most likely responsible for the lack of larger shifts in the apparent molecular weight. By comparison of the midchain functionalized PVAc with the corresponding 3-arm star, one can see that the addition of PEG has only a small effect on the apparent average molecular weight, an effect that was observed before, in some cases even causing an apparent reduction of molecular weight after conjugation.43 Thus, when discussing the H-shaped material, one may assume in a good approximation that the polymer might be regarded as a four-arm star with respect to the hydrodynamic volume of the molecule. A reduction in hydrodynamic volume of chains in solution at constant molecular weight is well known to occur when going from linear polymers to n-armed star polymers. Radke et al. found that a correction factor of 1.4 or higher applies (depending on the type of polymer) when a 4-arm star (homo)polymer is analyzed using universal calibration with linear standards.44 Taking such correction into account, almost a doubling in molecular weight is observed when going from the alkyne-functional starting material to the H-shaped product. Another indication for the success of the reaction is the practically unchanged polydispersity of the product distribution. If only partial conjugation would have occurred (resulting in a mixture of unreacted precursor, three-arm and H-shape material), then a significant broadening of the distribution would need to be observed. The lack of such thus is highly indicative for an efficient conjugation.
With the larger PVAc-b-PAN-b-PVAcconjugation, similar considerations apply, even though it needs to be noted that a significantly larger correction factor would need to be applied (∼1.8) in order to explain the comparatively low apparent molecular weight of the final product. While such a high value might in principle be possible (the PVAc-PAN block composition adds to the complexity of the hydrodynamic volume effect), it cannot be proven by the present data. Thus, in order to confirm the target structure, the monomer composition of the product was analyzed viaNMR. Comparison of characteristic peaks in the 1H-NMRspectrum of the miktoarm star polymer reveals a composition (in monomer units) of PVAc:PAN:PEG of 1:1.86:2.6 (by integrating the peaks around 4.8 ppm (VAc), 3.5 (PEG) and 3.2 (AN), see supporting information section for details). Based on the number of VAc monomer units present in the polymer (see NMR analysis above), a molecular weight of 6550 g mol−1 is calculated for the PEG arm, which is in almost perfect agreement with the analysis of the starting materials (6400 g mol−1, see Table 3). For the H-shape polymer, a composition of 1:0.87:2.6 is obtained, confirming the constant composition of VAc:AN and concomitantly the expected reduction of the number of PEG units in the product. Calculation of the molecular weight of the PEG block yields a molecular weight of 6100 g mol−1 and thus a result that is only slightly larger than the molecular weight of the bifunctional starting material (5120 g mol−1, see Table 3). Considering the increasing inaccuracy of the calculation method and the partial overlap of solvent peaks, also in this case a good agreement between both values is obtained. It should be noted, that while both the miktoarm and the H-shaped polymer show a constant VAc:AN content, the number of AN units is slightly overestimated. Closer inspection of this peak reveals, however, the presence of small peaks overlapping with the characteristic peak region, which might stem from the cycloaddition product. This overestimation is however inconsequential for the relative determination of the PEG content. In summary, for both polymers—despite the only small change in apparent molecular weight derived by conventional SEC analysis—successful formation of the target structures can hence be largely confirmed.
Conclusions
Mikto-arm star polymers and H-shaped polymers bearing PVAc or PVAc-b-PAN arms as well as a PEG arm or bridge were successfully synthesized by a cascade of living/controlled cobalt-mediated radical polymerization, nitrone-mediated radical coupling and copper catalyzed azide alkyne cycloaddition reactions. Initially, PVAc and PVAc-b-PANpolymers endcapped with Co(acac)2 were obtained from CMRP. After isolation of the polymers, macroradicals were freed from the cobalt complex and trapped by functional nitrones in the coupling reaction. In the final step, the so-obtained midchain functional building blocks were conjugated with mono- or bi-functional PEG chains to arrive at the synthesis target. Each reaction step was carefully monitored by means of size exclusion chromatography, NMR and soft-ionization mass spectrometry to assess the success of the individual reactions. For the final products, materials with very narrow molecular weight distributions were obtained, thus exemplifying that CMRP and NMRC are highly complementary and allow for generation of precisely defined mid-chain functional materials.Acknowledgements
C.B.-K. acknowledges financial support from the Karlsruhe Institute of Technology (KIT) in the context of the Excellence Initiative for leading German universities, the German Research Council (DFG) and the Ministry for Science and Arts of the state of Baden-Württemberg. O. A. acknowledges the Islamic Development Bank (IDB) for a Ph.D. scholarship. T. J. is grateful for financial support from the Fonds Wetenschappelijk Onderzoek (FWO) via the project G.0491.11N. M. C. acknowledges additional support from the Universiteit Hasselt. The authors from Liège are grateful to the “Belgian Science Policy” for financial support in the frame of the Interuniversity Attraction Pole Program (PAI VI/27) and to the National Funds for Scientific Research (F.R.S.-FNRS). They also thank Grégory Cartigny for his skilful assistance.References
- C. Barner-Kowollik and A. J. Inglis, Macromol. Chem. Phys., 2009, 201, 987–992 CrossRef.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- C. R. Becer, R. Hoogenboom and U. S. Schubert, Angew. Chem., Int. Ed., 2009, 48, 4900–4908 CrossRef CAS.
- R. K. Iha, K. L. Wooley, A. M. Nystrom, D. J. Burke, M. J. Kade and C. J. Hawker, Chem. Rev., 2009, 109, 5620–5686 CrossRef CAS.
- W. H. Binder and R. Sachsenhover, Macromol. Rapid Commun., 2008, 29, 952–98 CrossRef CAS.
- C. J. Hawker and K. L. Wooley, Science, 2005, 309, 1200–1205 CrossRef CAS.
- B. S. Sumerlin and A. P. Vogt, Macromolecules, 2010, 43, 1–13 CrossRef CAS.
- O. Altintas, A. P. Vogt, C. Barner-Kowollik and U. Tunca, Poly. Chem., 2011 10.1039/C1PY00249J.
- W. A. Braunecker and K. Matyjaszewski, Prog. Polym. Sci., 2007, 32, 93–146 CrossRef CAS.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661–3688 CrossRef CAS.
- G. Moad and C. Barner-Kowollik, in Handbook of RAFT Polymerization, ed. C. Barner-Kowollik, Wiley-VCH, Weinheim, 2008 Search PubMed.
- R. A. Evans, Aust. J. Chem., 2006, 60, 384–395 CrossRef.
- A. B. Lowe, Polym. Chem., 2010, 1, 17–36 RSC.
- (a) S. Sinnwell, A. J. Inglis, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, Chem. Commun., 2008, 2052–2054 RSC; (b) A. J. Inglis, S. Sinnwell, M. H. Stenzel and C. Barner-Kowollik, Angewandte Chemie Int. Ed., 2009, 48, 2411–2414 CAS.
- A. Dag, H. Durmaz, U. Tunca and G. Hizal, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 178–187 CrossRef CAS.
- T. Sarbu, K.-Y. Lim, J. Ell, D. J. Siegwart, J. Spanswick and K. Matyjaszewski, Macromolecules, 2004, 37, 3120–3127 CrossRef CAS.
- J. Kulis, C. A. Bell, A. S. Micallef, Z. Jia and M. J. Monteiro, Macromolecules, 2009, 42, 8218–8227 CrossRef CAS.
- A. Debuigne, C. Jerome and C. Detrembleur, Angew. Chem., Int. Ed., 2009, 48, 1422–1424 CrossRef CAS.
- A. Debuigne, R. Poli, J. De Winter, P. Laurent, P. Gerbaux, P. Dubois, J.-P. Wathelet, C. Jerome and C. Detrembleur, Chem.–Eur. J., 2010, 16, 1799–1811 CrossRef CAS.
- A. Debuigne, R. Poli, J. De Winter, P. Laurent, P. Gerbaux, J.-P. Wathelet, C. Jerome and C. Detrembleur, Macromolecules, 2010, 43, 2801–2813 CrossRef CAS.
- E. H. H. Wong, C. Boyer, M. H. Stenzel, C. Barner-Kowollik and T. Junkers, Chem. Commun., 2010, 46, 1959–1961 RSC.
- E. H. H. Wong, O. Altintas, M. H. Stenzel, C. Barner-Kowollik and T. Junkers, Chem. Commun., 2011, 47, 5491–5493 RSC.
- C. Detrembleur, V. Sciannamea, C. Koulic, M. Claes, M. Hoebeke and R. Jérôme, Macromolecules, 2002, 35, 7214–7223 CrossRef CAS.
- V. Sciannamea, A. Guerrero-Sanchez, U. S. Schubert, J.-M. Catala, R. Jerome and C. Detrembleur, Polymer, 2005, 46, 9632–9641 CrossRef CAS.
- V. Sciannamea, M. Bernard, J.-M. Catala, R. Jerome and C. Detrembleur, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7273–7279 CrossRef.
- V. Sciannamea, J.-M. Catala, R. Jerome and C. Detrembleur, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 1219–1235 CrossRef CAS.
- V. Sciannamea, J.-M. Catala, R. Jerome, C. Jerome and C. Detrembleur, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 1085–1097 CrossRef CAS.
- V. Sciannamea, R. Jerome and C. Detrembleur, Chem. Rev., 2008, 108, 1104–1126 CrossRef CAS.
- E. H. H. Wong, T. Junkers and C. Barner-Kowollik, Polym. Chem., 2011, 2, 1008–1017 RSC.
- (a) E. H. H. Wong, T. Junkers and C. Barner-Kowollik, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7273–7279 CrossRef CAS; (b) E. H. H. Wong, M. H. Stenzel, T. Junkers and C. Barner-Kowollik, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 1098–1107 CrossRef CAS; (c) T. Junkers, E. H. H. Wong, M. H. Stenzel and C. Barner-Kowollik, Macromolecules, 2009, 42, 5027–5035 CrossRef CAS; (d) L. Zang, E. H. H. Wong, C. Barner-Kowollik and T. Junkers, Polymer, 2010, 51, 3821–3825 CrossRef CAS; (e) T. Junkers, L. Zang, E. H. H. Wong, N. Dingenouts and C. Barner-Kowollik, J. Polym. Sci. – Polym. Chem., 2011 Search PubMed , in press.
- M. Hurtgen, C. Detrembleur, C. Jerome and A. Debuigne, Polym. Rev., 2011, 51, 188–213 CrossRef CAS.
- A. Debuigne, R. Poli, C. Jerome, R. Jerome and C. Detrembleur, Prog. Polym. Sci., 2009, 34, 211–239 CrossRef CAS.
- (a) A. Debuigne, J.-R. Caille and R. Jerome, Angew. Chem., Int. Ed., 2005, 44, 1101–1104 CrossRef CAS; (b) S. Maria, H. Kaneyoshi, K. Matyjaszewski and R. Poli, Chem.–Eur. J., 2007, 13, 2480–2492 CrossRef CAS; (c) A. Debuigne, Y. Champouret, R. Jerome, R. Poli and C. Detrembleur, Chem.–Eur. J., 2008, 14, 4046–4059 CrossRef CAS.
- (a) A. Debuigne, N. Willet, R. Jerome and C. Detrembleur, Macromolecules, 2007, 40, 7111–7118 CrossRef CAS; (b) H. Kaneyoshi and K. Matyjaszewski, Macromolecules, 2006, 39, 2757–2763 CrossRef CAS.
- E. H. H. Wong, M. H. Stenzel, T. Junkers and C. Barner-Kowollik, Macromolecules, 2010, 43, 3785–3793 CrossRef CAS.
- E. H. H. Wong, M. H. Stenzel, T. Junkers and C. Barner-Kowollik, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 2118–2126 CrossRef CAS.
- T. Gruendling, M. Guilhaus and C. Barner-Kowollik, Anal. Chem., 2008, 80, 6915–6927 CrossRef CAS.
- C. Barner-Kowollik, F. E. Du Prez, P. Espeel, C. J. Hawker, T. Junkers, H. Schlaad and W. Van Camp, Angew. Chem., Int. Ed., 2011, 50, 60–62 CrossRef CAS.
- A. J. Inglis, S. Sinnwell, M. H. Stenzel and C. Barner-Kowollik, Angew. Chem. Int. Ed., 2009, 48, 2411–2414 CAS.
- S. Hiki and K. Kataoka, Bioconjugate Chem., 2007, 18, 2191–2196 CrossRef CAS.
- G. Floudas, P. Papadopoulos, H.-A. Klok, G. W. M. Vandermeulen and J. Rodriguez-Hernandez, Macromolecules, 2003, 36, 3673–3683 CrossRef CAS.
- O. Altintas, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 5699–5707 CrossRef CAS.
- W. Radke, J. Gerber and G. Wittmann, Polymer, 2003, 44, 519–525 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: NMR spectra of the building blocks and the final products. See DOI: 10.1039/c1py00297j |
| ‡ Current address: Department of Chemical and Biomolecular Engineering, The University of Melbourne, Parkville, Victoria 3010, Australia. |
| This journal is © The Royal Society of Chemistry 2012 |