Aqueous RAFT/MADIXpolymerisation of N-vinyl pyrrolidone at ambient temperature†
Aymeric
Guinaudeau
a,
Stéphane
Mazières
a,
D. James
Wilson
b and
Mathias
Destarac
*a
aUniversicPaul Sabatier, Laboratoire Hétérochimie Fondamentale et Appliquée, UMR-CNRS 5069, Bât 2R1, 118 route de Narbonne, 31062, Toulouse Cedex 9, France. E-mail: destarac@chimie.ups-tlse.fr; Fax: +33 (0)5 6155 8204; Tel: +33 (0)5 6155 6354
bRhodia Opérations, Centre de Recherches et Technologies d'Aubervilliers, 52 rue de la Haie Coq, 93308, Aubervilliers Cedex, France
First published on 14th October 2011
Abstract
The RAFT/MADIXpolymerisation of N-vinyl pyrrolidone was successfully carried out in homogeneous water medium at 25 °C using the tert-butyl hydroperoxide/ascorbic acidredoxinitiator. The controlled character of the polymerisation was confirmed by MALDI-TOF mass spectrometry, SEC and NMR analyses. This finding paves the way for the preparation of novel all-hydrophilic complex copolymer architectures based on poly(N-vinyl pyrrolidone).
The seventieth birthday of poly(N-vinyl pyrrolidone) (PVP) was celebrated two years ago.1,2 Over the years, PVP has attracted the constant attention of researchers from both academic and industrial worlds. Its exceptional film-forming and adhesive characteristics, pH-stability, strong resistance to thermal decomposition in water, non-toxicity and biocompatibility undoubtedly positions PVP as one of the most interesting water-soluble polymers.3
Polymerisation of N-vinyl pyrrolidone (NVP) has only been reported via a free-radical process, mostly in a diazo-,4 photo-5 and redox-initiated6 fashion. Following the advent of radical polymerisation by reversible deactivation (RPRD) techniques,7 it became possible to synthesise PVPs with predetermined molecular weights (Mn) and narrow dispersities (Đ = Mw/Mn).8Nitroxide-mediated polymerisation (NMP),9,10 organocobalt-mediated radical polymerisation (OCRP),11 atom transfer radical polymerisation (ATRP)12 and iodine transfer polymerisation (ITP)13 have so far met with very limited or no success in the control of NVPpolymerisation. On the other hand, organoheteroatom-mediated polymerisation (OHRP)14–16 and reversible addition–fragmentation/macromolecular design by interchange of xanthates (RAFT/MADIX)18–25polymerisation were found to be suitable approaches to deactivate PVP chain growth in a reversible manner, thereby giving access to PVP-based controlled architectures. In the latter approach, O-ethyl xanthates26–30 were by far the most employed RAFT/MADIX agents due to their high efficiency in controlling both molecular weights and yielding narrowly dispersed PVPs up to high conversions with a limited effect on the overall rate of polymerisation. However, Pound et al.22,23 thoroughly investigated diazo-initiated RAFT/MADIXpolymerisation of NVP with O-ethyl xanthates and found that several unexpected by-products were formed during polymerisation. For instance, NVP monomer undergoes side reactions in the presence of acid-functional xanthates and impurities. In the same study, it was also demonstrated that the O-ethyl xanthategroup at the end of PVP chains is thermally unstable and can be eliminated during polymerisation to give a sulfur-free, unsaturated chain end. In another contribution, the same authors31 heated a xanthate-terminated PVP in water at 40 °C for 16 h and obtained a hydroxyl terminal group after hydrolysis. Water is also well-known for reacting with NVP generating several by-products—most of the side reactions being acid-catalysed—if the former is used as a polymerisation medium.22,32–35
Therefore, it was suggested to work at moderated temperatures (<60 °C) and to avoid water as a polymerisation medium.22
Consequently, all the successful RDRP processes for NVPpolymerisation were thus far carried out either in bulk or in organic solvents such as methanol, 1,4-dioxane, fluorinated alcohols and toluene.14–25 From a practical standpoint, working in the absence of solvent implies dealing with an excessively high viscosity during the reaction, especially at high conversions. Moreover, for the numerous applications of PVP in aqueous formulations, the use of a polymer synthesised in an organic medium requires either an additional step of precipitation/redissolution in water or a solvent exchange. These process steps are costly from both energetic and environmental points of view. For all the aforementioned reasons, it appears that the development of a RAFT/MADIX process for polymerising NVP in water at low temperature represents a major scientific and industrial challenge.
In this communication, we report our results on the redox-initiated RAFT/MADIXpolymerisation of NVP at room temperature in water (Scheme 1).
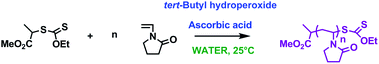 | ||
| Scheme 1 Synthetic scheme for redox-initiated RAFT/MADIXpolymerisation of N-vinyl pyrrolidone in water at ambient temperature. | ||
Historically, the first free-radical polymerisation of NVP was reported by Fikentscher and Herrle36 employing an aqueous redox initiating system comprising hydrogen peroxide, ammonia and metal ions. Later, only a few other redox couples such as K2H2P2O8/Ag+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 6 and H2O2/ascorbic acid37 (AscAc) were reported to initiate NVPhomopolymerisation in pure water. This limited development can be ascribed to the presence of side reactions between NVP and water (vide supra).22,32–35 We carried out a screening study of many different redox couples likely to initiate aqueous NVPpolymerisation at 25 °C. The following prerequisites should be fulfilled before considering a redox couple for initiating aqueous NVPpolymerisation. Firstly, the difference between the oxidizing and reducing potentials of respectively oxidant and reducing agents must be great enough to generate radicals at a sufficient rate. Then, the oxidizing agent potential should be low enough in order to prevent the direct oxidation of NVP. Importantly, as side reactions between NVP and water are catalyzed in acidic media, it is crucial to maintain a sufficiently high pH and to avoid the presence of strong acids in the redox couple. We decided to rule out metallic reducing agents (e.g.Ag+) to avoid any regulatory issues in some of the envisioned fields of application. The previously reported H2O2/AscAc pair37 failed at initiating aqueous RAFT/MADIXpolymerisation of NVP in an efficient manner, yielding less than 10% polymer. Furthermore, a large amount of NVP by-products were formed when the concentration of AscAc was increased.
6 and H2O2/ascorbic acid37 (AscAc) were reported to initiate NVPhomopolymerisation in pure water. This limited development can be ascribed to the presence of side reactions between NVP and water (vide supra).22,32–35 We carried out a screening study of many different redox couples likely to initiate aqueous NVPpolymerisation at 25 °C. The following prerequisites should be fulfilled before considering a redox couple for initiating aqueous NVPpolymerisation. Firstly, the difference between the oxidizing and reducing potentials of respectively oxidant and reducing agents must be great enough to generate radicals at a sufficient rate. Then, the oxidizing agent potential should be low enough in order to prevent the direct oxidation of NVP. Importantly, as side reactions between NVP and water are catalyzed in acidic media, it is crucial to maintain a sufficiently high pH and to avoid the presence of strong acids in the redox couple. We decided to rule out metallic reducing agents (e.g.Ag+) to avoid any regulatory issues in some of the envisioned fields of application. The previously reported H2O2/AscAc pair37 failed at initiating aqueous RAFT/MADIXpolymerisation of NVP in an efficient manner, yielding less than 10% polymer. Furthermore, a large amount of NVP by-products were formed when the concentration of AscAc was increased.
Among all the oxidant/reducing agent pairs which were tested, the tert-butyl hydroperoxide/ascorbic acid (t-BuOOH/AscAc) system met these criteria and allowed the formation of PVP with high conversion after 24 h without side reactions (vide infra). A 1:1 molar ratio between t-BuOOH and AscAc was applied. The polymerisation was conducted at an initial NVP concentration of 6.2 mol L−1 (66 wt%). Under these conditions at pH = 6.5, the RAFT/MADIX agent used in this study, namely O-ethyl-S-(1-methoxycarbonyl)ethyl dithiocarbonate (Rhodixan A1, XA1) was soluble in the aqueous NVP solution. NVP was first polymerised in the absence of XA1. After 24 h, a PVP with Mn equal to 40500 g mol−1 and Đ of 1.72 was obtained with 75% NVP conversion. RAFT/MADIXpolymerisations were conducted at different concentrations in XA1 in order to demonstrate the influence of the xanthate transfer agent. The obtained results are reported in Table 1 and Fig. 1 and 2. As can be observed in Table 1, high NVP conversions were achieved after 24 h of reaction in all cases (89% and above). Mn is inversely proportional to the initial XA1 concentration at the end of the polymerisation (Table 1 and Fig. 1), with values determined by 1H NMR which are in excellent agreement with those expected for an efficient RAFT/MADIXpolymerisation. In addition, the presence of the O-ethyl dithiocarbonate fragment of XA1 in the polymer chains was confirmed by SEC using a UV-Visdetector at λ = 290 nm. Indeed, the observed strong UV response in SEC chromatograms of PVP is due to the well-known, excellent chromophore properties of the xanthate at this wavelength.
| [XA1]/mol L−1 | x (%) | M n,th /g mol−1 | M n,NMR /g mol−1 | M n,SEC /g mol−1 | Đ |
|---|---|---|---|---|---|
| a Determined by 1H NMR. b M n,th = ([NVP]0/[XA1]0) × (conv.) × MNVP + MXA1. c Determined by 1H NMR. d Determined by SEC in DMF/LiCl with PMMA standards. | |||||
| — | 75 | — | — | 40500 |
1.72 |
| 1.89 × 10−1 | 93 | 3400 | 3400 | 5500 | 1.15 |
| 7.16 × 10−2 | 95 | 6800 | 5400 | 9400 | 1.15 |
| 6.96 × 10−2 | 97 | 9600 | 9600 | 14600 |
1.30 |
| 3.51 × 10−2 | 89 | 13000 |
12000 |
16900 |
1.25 |
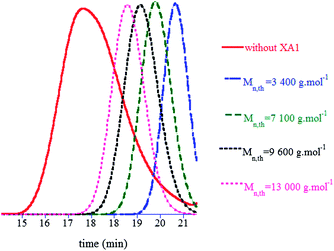 | ||
| Fig. 1 Overlay of SEC chromatograms of PVPs synthesised at 25 °C in water using different initial XA1 concentrations (entries 1–5 of Table 1). | ||
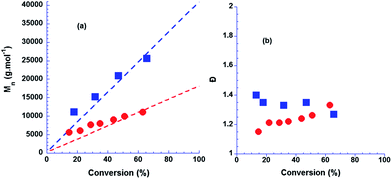 | ||
| Fig. 2 Dependence of (a) Mn and (b) Đ = Mw/Mn on monomer conversion in the aqueous RAFT/MADIXpolymerisation of NVP at 25 °C at different initial XA1 concentrations. (●) 3.80 × 10−2 mol L−1 and (■) 1.68 × 10−2 mol L−1 (■). Dashed lines represent the theoretical evolution of Mn for a controlled polymerisation. Mn was determined by SEC in DMF/LiCl with PMMA standards. | ||
M n,NMR was determined after PVP precipitation in diethyl ether by comparing the signal integration of the methineprotons of the main chain (3.4–4.0 ppm) to that of the methine of the terminal NVP unit adjacent to the dithiocarbonategroup (5.6–5.8 ppm, see Fig. S1 in the ESI†). Mn values measured by SEC in DMF/LiCl using PMMA standards followed the same trend although they tended to be slightly higher than expected, which may be ascribed to slight differences in hydrodynamic volumes between PVP and PMMA in the eluent phase. Dispersity values are very low (1.15 < Đ < 1.30) and in the same range as those for RAFT/MADIXpolymerisation of NVP in bulk or organic media.
Fig. 2 shows the evolution of Mn,SEC and Đ with conversion for two different initial XA1 concentrations. Mn values increase linearly with conversion and are in good agreement with the theoretical values even at low conversions. Đ is less than 1.4 early in the reaction and remains below this value during polymerisation. These results suggest a high transfer constant to XA1 and a high exchange constant Cex (=kex/kp) during polymerisation.38
The microstructure of the synthesised PVPs was then analysed by 1H NMR spectroscopy (see Fig. S1 in the ESI†). We observed that the known by-products22 due to hydrolysis or coupling of NVP were absent. The methineprotons of the NVP unit next to the thiocarbonylthio group (proton i in Fig. S1 of ESI†) and the methylprotons in the O-ethyl terminal group are clearly present in the purified polymer, which confirms that the xanthate moiety is attached to the polymer backbone. As mentioned earlier, the good agreement between the theoretical Mn and those calculated by 1H NMR confirms the excellent chain-end fidelity of the xanthategroup and the living character of the polymerisation.
To confirm these results, a low molar mass PVP was synthesised (Mn,th = 1500 g mol−1, Mn,NMR = 1600 g mol−1 with 97% conversion after 24 h). A MALDI-TOF mass spectrometry analysis was performed on this PVP sample taken after 24 h of reaction (Fig. 3). The reaction mixture was kept at 25 °C for an additional five days and a sample was analyzed by MALDI-TOF MS after this time (Fig. S2 in the ESI†). Both spectra are identical, which means that there is no evolution of the PVP sample after five days in water at ambient temperature. The mass spectrum is symmetrical and shows one major and two minor populations. For each population distribution, the peaks are separated by 111.1 g mol−1, the molar mass of NVP. The structures corresponding to the major population and one of the minor populations are CH3OCOCH3CH–(NVP)n−1–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH(C4H6NO), A being cationised with sodium and B with potassium. This observation brings additional evidence that the R substituent of the xanthate agent fragments and efficiently reinitiates the polymerisation. However, the xanthate moiety of the polymer has disappeared in favor of a double bond. The loss of the xanthate moiety is due to its fragmentation by the MALDI-TOF laser, as previously reported by Destarac et al.26 The C–S bond is often too fragile to be observed even at low MALDI-TOF laser intensity depending on the monomer/RAFT agent couple. 1H NMR analysis and the strong absorption of the PVP–XA1 samples in the UV range at 290 nm, which is characteristic of the presence of the –S(CS)– group in the polymer, support this assumption. The expected population CH3OCOCH3CH–(NVP)n–S(CS)OCH2CH3 cationised with potassium was nevertheless observed and corresponds to the second minor population C. It is noteworthy that no tert-butylhydroperoxy-initiated chain populations are visible in the spectrum, which means that nearly all the chains were initiated by the methylpropionyl radical coming from XA1. This suggests that the fraction of initiating radicals was low for the chosen polymerisation conditions considering the high [initiator]0/[XA1]0 ratios that were used. In addition, no evidence of termination and hydrogen transfer reactions was found. Therefore, MALDI-TOF MS further demonstrated that the combination of t-BuOOH and AscAc could efficiently initiate a well-controlled RAFT/MADIXpolymerisation of NVP in water. Furthermore, the PVP–XA1 aqueous solutions remain stable over time and could therefore, if needed, be easily stored at ambient temperature for several days.
CH(C4H6NO), A being cationised with sodium and B with potassium. This observation brings additional evidence that the R substituent of the xanthate agent fragments and efficiently reinitiates the polymerisation. However, the xanthate moiety of the polymer has disappeared in favor of a double bond. The loss of the xanthate moiety is due to its fragmentation by the MALDI-TOF laser, as previously reported by Destarac et al.26 The C–S bond is often too fragile to be observed even at low MALDI-TOF laser intensity depending on the monomer/RAFT agent couple. 1H NMR analysis and the strong absorption of the PVP–XA1 samples in the UV range at 290 nm, which is characteristic of the presence of the –S(CS)– group in the polymer, support this assumption. The expected population CH3OCOCH3CH–(NVP)n–S(CS)OCH2CH3 cationised with potassium was nevertheless observed and corresponds to the second minor population C. It is noteworthy that no tert-butylhydroperoxy-initiated chain populations are visible in the spectrum, which means that nearly all the chains were initiated by the methylpropionyl radical coming from XA1. This suggests that the fraction of initiating radicals was low for the chosen polymerisation conditions considering the high [initiator]0/[XA1]0 ratios that were used. In addition, no evidence of termination and hydrogen transfer reactions was found. Therefore, MALDI-TOF MS further demonstrated that the combination of t-BuOOH and AscAc could efficiently initiate a well-controlled RAFT/MADIXpolymerisation of NVP in water. Furthermore, the PVP–XA1 aqueous solutions remain stable over time and could therefore, if needed, be easily stored at ambient temperature for several days.
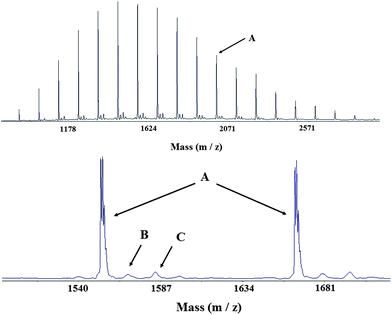 | ||
| Fig. 3
MALDI-TOF mass spectrum of a PVP synthesised by XA1-mediated RAFT/MADIXpolymerisation in water at 25 °C. Reaction time = 24 h. Mn,NMR = 1600 g mol−1. Matrix: 4-(4-nitrophenylazo)resorcinol. No cationisation agent. A = CH3OCOCH3CH–(NVP)n−1–CHCH(C4H6NO) (Na+), B = CH3OCOCH3CH–(NVP)n−1–CHCH(C4H6NO) (K+), C = CH3OCOCH3CH–(NVP)n–S(CS)OCH2CH3 (K+). See Fig. S2 in the ESI† for the detailed experimental procedure. | ||
RAFT/MADIX technology is particularly suited for macromolecular engineering based on NVP. The high reactivity of the PVP radical compared to styrenics, acrylic and acrylamido radicals imposes that the PVP block be synthesised as the second block in order to ensure efficient reinitiation. Until now, only organosoluble PVP diblock copolymers could be afforded through an all organic solvent synthesis approach. The case where a double hydrophilic NVPblock copolymer is desired in which the first block is exclusively water soluble imposes that the second NVP block be synthesised in water.
The access to redox initiated RAFT/MADIXpolymerisation of NVP with O-ethyl xanthates in water therefore paves the way for the synthesis of a wide variety of PVP-based, all-hydrophilic controlled macromolecular architectures.
As an example, a first low molar mass poly(acrylamide) block controlled with XA1 in a water/ethanol mixture39,40 was synthesised at 25 °C with the t-BuOOH/AscAcinitiator. A Mn,th equal to 1000 g mol−1 was targeted in order to be able to solubilise and analyse the resulting PAm by SEC in DMF. After 24 h, conversion reached 99% with a molecular weight determined by 1H NMR of Mn,NMR = 1200 g mol−1. Ethanol was removed under vacuum and two PAm–PVP block copolymers were synthesised in pure water with targeted PVP blocks of 2000 and 9000 g mol−1. NVP was fully converted to PVP after 24 h. PAm–PVP diblock copolymers with Mn values of 8600 and 12200 g mol−1 were respectively obtained by SEC in DMF/LiCl with a PMMA calibration, with dispersities equal to 1.09 and 1.25 (Fig. 4). It is interesting to note that PAm1K–PVP9Kblock copolymer, which contains ∼90 wt% of PVP, exhibits a Mn value which is very close to theory, in contrast to the PAm1K–PVP2K which is much richer in PAm. This can be attributed to a larger hydrodynamic volume of PAm compared to that of PVP and PMMA in DMF/LiCl solution. Indeed, the molecular weight of the PAm1K–XA1 sample determined with PMMA standards equalled Mn,SEC = 3600 g mol−1 (Đ = 1.07). The representation of SEC chromatograms of the PAm–XA1 precursor and final copolymer confirms that block copolymerisation occurred efficiently, with a characteristic shift of the monomodal polymer distribution to high molar mass region after VPpolymerisation and no visible trace of unreacted PAm precursor.
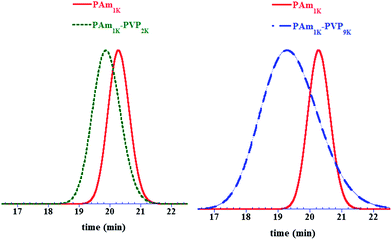 | ||
| Fig. 4
SEC chromatograms of PAm–PVP diblock copolymers synthesised by RAFT/MADIXpolymerisation in water at 25 °C. Left: PAm–XA1 (Mn,NMR = 1200 g mol−1, Mn,SEC = 3600 g mol−1, Đ = 1.07) and PAm1K–PVP3Kdiblock copolymer (Mn,th = 2900 g mol−1, Mn,SEC = 8600 g mol−1, Đ = 1.09). Right: PAm–XA1 (Mn,NMR = 1200 g mol−1, Mn,SEC = 3600 g mol−1, Đ = 1.07) and PAm1K–PVP9Kdiblock copolymer (Mn,th = 10900 g mol−1, Mn,SEC = 12200 g mol−1, Đ = 1.25). | ||
Conclusions
The control of RAFT/MADIXpolymerisation of NVP was achieved in pure water at ambient temperature by means of a redox initiation with the tert-butyl hydroperoxide/ascorbic acid couple and the Rhodixan A1 xanthate transfer agent. SEC, 1H NMR and MALDI-TOF MS analysis supported the control of macromolecular characteristics of the obtained PVPs up to high conversions. No by-products of NVP and water were detected, the xanthate end-group remained stable during polymerisation, and for at least a further five days in solution at ambient temperature. PAm–PVP diblock copolymers were successfully synthesised for the first time using the same reaction conditions. These first results undoubtedly open many interesting perspectives for the synthesis and application of novel all-hydrophilic block copolymers and more complex architectures that include PVP.Acknowledgements
The authors would like to thank Rhodia Operations and CNRS for the permission to publish this work.Notes and references
- Polyvinylpyrrolidone (PVP)-A “multi-talent” turns seventy, http://www.btc-europe.com/en/aktuell-pvp/.
- W. Reppe and S. Curt, US Pat., 2265450, I.G. Farbenindustrie Aktiengesellschaft, 1941.
- Y. E. Kirsh, Water Soluble Poly-N-vinylamides: Synthesis and Physicochemical Properties, Wiley, Chichester, UK, 1998 Search PubMed.
- J. C. Cizravi, T. Y. Tay and E. C. Pon, J. Appl. Polym. Sci., 2000, 75, 239–246 CrossRef CAS.
- M. A. Tasdelen, B. Karagoz, N. Bicak and Y. Yagci, Polym. Bull., 2008, 59, 759–766 CrossRef CAS.
- K. C. Gupta, J. Appl. Polym. Sci., 1994, 53, 71–78 CrossRef CAS.
- Terminology recently proposed by IUPAC for “Controlled radical polymerisation”: D. A. Jenkins, R. G. Jones and G. Moad, Pure Appl. Chem., 2010, 82, 483–491 CrossRef.
- R. G. Gilbert, M. Hess, A. D. Jenkins, R. G. Jones, P. Kratochvil and R. F. T. Stepto, Pure Appl. Chem., 2009, 81, 351 CrossRef CAS.
- P. Bilalis, M. Pitsikalis and N. Hadjichristidis, J. Polym. Sci., Part A: Polym. Chem., 2005, 44, 659–665 CrossRef.
- P. Bilalis, G. Zorba, M. Pitsikalis and N. Hadjichristidis, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 5719–5728 CrossRef CAS.
- A. Debuigne, N. Willet, R. Jérôme and C. Detrembleur, Macromolecules, 2007, 40, 7111–7118 CrossRef CAS.
- X. Lu, S. Gong, L. Meng, C. Li, S. Yang and L. Zhang, Polymer, 2007, 48, 2835–2842 CrossRef CAS.
- G. David, C. Boyer, J. Tonnar, B. Ameduri, P. Lacroix-Desmazes and B. Boutevin, Chem. Rev., 2006, 106, 3936–3962 CrossRef CAS.
- S. Yamago, B. Ray, K. Iida, J-i. Yoshida, T. Tada, K. Yoshizawa, Y. Kwak, A. Goto and T. Fukuda, J. Am. Chem. Soc., 2004, 126, 13908–13909 CrossRef CAS.
- B. Ray, M. Kotani and S. Yamago, Macromolecules, 2006, 39, 5259–5265 CrossRef CAS.
- S. Yamago, E. Kayahara, M. Kotani, B. Ray, Y. Kwak, A. Goto and T. Fukuda, Angew. Chem., Int. Ed., 2007, 46, 1304–1306 CrossRef CAS.
- S. Deroo, M. Destarac and M. Morvan, Fr Pat., 2838653, Rhodia Chimie, 2003.
- D. Wan, K. Satoh, M. Kamigaito and Y. Okamoto, Macromolecules, 2005, 38, 10397–10405 CrossRef CAS.
- G. Pound, J. B. McLeary, J. M. McKenzie, R. F. M. Lange and B. Klumperman, Macromolecules, 2006, 39, 7796–7797 CrossRef CAS.
- T. L. U. Nguyen, K. Eagles, T. P. Davis, C. Barner-Kowollik and M. H. Stenzel, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 4372–4383 CrossRef CAS.
- A. Postma, T. P. Davis, G. Li, G. Moad and M. S. O'Shea, Macromolecules, 2006, 39, 5307–5318 CrossRef CAS.
- G. Pound, Z. Eksteen, R. Pfukwa, J. M. McKenzie, R. F. M. Lange and B. Klumperman, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6575–6593 CrossRef CAS.
- G. Pound, PhD thesis, Stellenbosch University, 2008.
- M. Benaglia, J. Chiefari, Y. K. Chong, G. Moad, E. Rizzardo and S. H. Thang, J. Am. Chem. Soc., 2009, 131, 6914–6915 CrossRef CAS.
- C.-F. Huang, R. Nicolay, Y. Kwak, F.-C. Chang and K. Matyjaszewski, Macromolecules, 2009, 42, 8198–8210 CrossRef CAS.
- M. Destarac, C. Brochon, J.-M. Catala, A. Wilczewska and S. Z. Zard, Macromol. Chem. Phys., 2002, 203, 2281–2289 CrossRef CAS.
- M. Destarac, W. Bzducha, D. Taton, I. Gauthier-Guillaizeau and S. Z. Zard, Macromol. Rapid Commun., 2002, 23, 1049–1054 CrossRef CAS.
- M. Adamy, A. M. Van Herk, M. Destarac and M. J. Monteiro, Macromolecules, 2003, 36, 2293–2301 CrossRef CAS.
- M. Destarac, D. Taton, S. Z. Zard, T. Saleh and Y. Six, Advances in Controlled/Living Radical Polymerization, in ACS Symposium Series, ed. K. Matyjaszewski, American Chemical Society, 2003, vol. 854, pp. 536–550 Search PubMed.
- D. Taton, M. Destarac and S. Z. Zard, Handbook of RAFT Polymerization, ed. C. Barner-Kowollik, Wiley-VCH, Weinheim, 2008, pp. 373–421 Search PubMed.
- G. Pound, J. M. McKenzie, R. F. M. Lange and B. Klumperman, Chem. Commun., 2008, 3193–3195 RSC.
- J. W. Breitenbach, J. Polym. Sci., 1957, 23, 949–953 CrossRef CAS.
- G. Kaupp and D. Matthies, Chem. Ber., 1987, 120, 1897–1903 CrossRef CAS.
- J.-C. Zhuo, Molecules, 1999, 4, M117 CAS.
- A. Madl and S. Spange, Macromolecules, 2000, 33, 5325–5335 CrossRef CAS.
- H. Fikentscher and K. Herrle, Mod. Plast., 1945, 23, 157–161 Search PubMed.
- M. T. Taghizadeh and M. T. Salehi, Iran. J. Polym. Sci. Technol., 1995, 4, 262–267 CAS.
- M. Destarac, Polym. Rev., 2011, 51, 163–187 CrossRef CAS.
- D. Taton, A. Wilczewska and M. Destarac, Macromol. Rapid Commun., 2001, 22, 1497–1503 CrossRef CAS.
- M. Destarac, A. Guinaudeau, R. Geagea, S. Mazières, E. Van Gramberen, C. Boutin, S. Chadel and J. Wilson, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5163–5171 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR Spectra of reaction mixture and PVP purified, MALDI-TOF MSspectrum of reaction mixture after 5 days. See DOI: 10.1039/c1py00373a |
| This journal is © The Royal Society of Chemistry 2012 |