Preparation and applications of novel fluoroalkyl end-capped oligomeric nanocomposites
Hideo
Sawada
*
Department of Frontier Materials Chemistry, Graduate School of Science and Technology, Hirosaki University, Bunkyo-cho, Hirosaki 036-8561, Japan. E-mail: hideosaw@cc.hirosaki-u.ac.jp; Fax: +81-172-39-3578; Tel: +81-172-39-3578
First published on 15th September 2011
Abstract
Fluoroalkyl end-capped oligomers [RF–(M)n–RF] were prepared by reaction of fluoroalkanoyl peroxide [RF–C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)OO(OC)–RF] with traditional radical polymerizable monomers (M) under very mild conditions. These fluoroalkyl end-capped oligomers can form nanometre size controlled self-assembled molecular aggregates with the aggregation of terminal fluoroalkyl segments in aqueous and organic media. Due to the poor radical polymerizable characteristic of fluoroalkanoyl peroxide toward traditional radical polymerizable monomers, fluoroalkanoyl peroxide was applied to the preparation of cross-linked fluoroalkyl end-capped oligomeric nanoparticles. Furthermore, fluoroalkyl end-capped oligomeric aggregates can interact with a variety of guest molecules such as silica gel, magnetite, gold, silver, copper, zinc oxide, titanium oxide, calcium carbonate, nanodiamond, and hydroxyapatite to afford the corresponding fluorinated oligomer/guest molecule nanocomposites. The preparation and applications of these cross-linked fluorinated oligomeric nanoparticles and fluorinated oligomer/guest molecule nanocomposites are reviewed in this article.
O)OO(OC)–RF] with traditional radical polymerizable monomers (M) under very mild conditions. These fluoroalkyl end-capped oligomers can form nanometre size controlled self-assembled molecular aggregates with the aggregation of terminal fluoroalkyl segments in aqueous and organic media. Due to the poor radical polymerizable characteristic of fluoroalkanoyl peroxide toward traditional radical polymerizable monomers, fluoroalkanoyl peroxide was applied to the preparation of cross-linked fluoroalkyl end-capped oligomeric nanoparticles. Furthermore, fluoroalkyl end-capped oligomeric aggregates can interact with a variety of guest molecules such as silica gel, magnetite, gold, silver, copper, zinc oxide, titanium oxide, calcium carbonate, nanodiamond, and hydroxyapatite to afford the corresponding fluorinated oligomer/guest molecule nanocomposites. The preparation and applications of these cross-linked fluorinated oligomeric nanoparticles and fluorinated oligomer/guest molecule nanocomposites are reviewed in this article.
 Hideo Sawada | H. Sawada received his MS degree in 1980 from Tokyo Metropolitan University. He was associated with NOF Corporation from 1980 and received his PhD degree in 1986 from Tokyo Metropolitan University. In 1993, he accepted a position as an Associate Professor at Nara National College of Technology, and he became a Professor of Chemistry at the Nara National College of Technology in 2000. In 2002, he moved to Hirosaki University as a Professor of Polymer Chemistry. His current research interest concerns the synthesis and applications of fluorinated oligomeric nanocomposites by the use of fluoroalkanoyl peroxides. He received the Progress Award of the Japan Oil Chemists' Society in 1992, the Award of the Japan Society of Colour Materials in 1994, the Award of the Japan Research Institute of Material Technology in 1996 and 2010, and the SPSJ Mitsubishi Chemical Award in 2006. He is a member of the Editorial Board of the Journal of Fluorine Chemistry. |
1. Introduction
Hitherto, considerable effort has been devoted to the design and controlled fabrication of nanostructured materials with a wide variety of unique properties, which result from a function of nanoscale materials' size, composition, and structure order.1–3 Of these, organic polymeric nanoparticles have a higher potential for practical applications to a wide variety of fields, such as nanocoatings, nanostructure-supported catalysts, and biomedical and pharmaceutical materials.4–10Polymer-coated particles offer interesting prospects of broad applications in numerous fields.11–18 In these polymer-coated particles, surface modification of silicananoparticles by chemically bound polymers is of great interest due to their potential applications in a variety of fields such as coatings, electronics, catalysts, optics, and diagnosis.19–26 In general, preparations of polymer grafted silicananoparticles can be classified according to their preparative methods into the following:(I) Free radical polymerization.27–29
(II) Cationic and anionic polymerization.30,31
(III) Miniemulsion polymerization.32
(IV) Living radical polymerization:
(a) RAFT (reversible addition–fragmentation chain transfer) polymerization with click reaction.33–36
(b) Nitroxide-mediated polymerization.37,38
(c) Atom-transfer radical polymerization.39,40
Inorganic nanoparticles are usually utilized as the cores of grafted polymers to combine the superior properties of the organic and inorganic materials. For example, silicananoparticles containing methacryoyloxypropylgroups can copolymerize with styrene as comonomer catalyzed by potassium persulfate to afford polystyrene grafted silicananoparticles (Scheme 1).41 Similarly, a diblock copolymer brush consisting of poly(methyl methacrylate)-block-poly(pentafluoropropyl acrylate) was prepared on a porous silica substrate.42–46 Ober et al. reported the preparation of planar silicon oxide surface-grafted styrene-based diblock copolymer brushes bearing semifluorinated alkyl side groups by nitroxide-mediated controlled radical polymerization.47
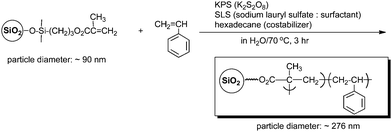 | ||
| Scheme 1 Preparation of polystyrene grafted silicananoparticles. | ||
Hitherto, there has been increasing interest in material sciences toward the development of partially fluoroalkylated polymers, especially fluoroalkyl end-capped oligomers [RF–(M)n–RF; RF = fluoroalkyl groups; M = radical polymerizable monomers] are attractive functional materials, because they exhibit various unique properties such as high solubility, surface active properties, biological activities and nanometre size-controlled molecular aggregates which cannot be achieved by the corresponding non-fluorinated, randomly or block-type fluoroalkylated polymers, and low-molecular weight fluorinated surfactants.48–51 Thus, from the developmental viewpoint of new fluorinated polymeric materials, it is of particular interest to develop fluoroalkyl end-capped oligomeric nanoparticles and fluoroalkyl end-capped oligomer-coated nanoparticles possessing a variety of unique characteristics imparted by fluorine.
Fluoroalkyl end-capped oligomers can form nanometre size-controlled self-assembled oligomeric aggregates through the aggregations of end-capped fluoroalkyl groups (see Fig. 1).52–54 Fluoroalkyl end-capped oligomeric aggregates can also interact with guest molecules to afford fluorinated aggregate/guest molecule nanocomposites (see Fig. 2); although the corresponding non-fluorinated oligomers cannot form such molecular aggregates to interact with guest molecules.55
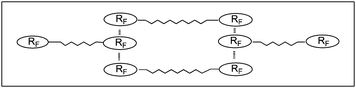 | ||
| Fig. 1 Schematic model of micelles formed with fluoroalkyl end-capped acrylic acid–trimethylvinylsilane cooligomers. | ||
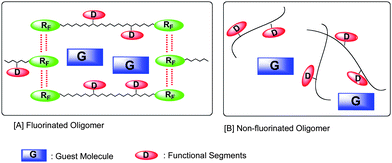 | ||
| Fig. 2 Schematic illustration for the interaction of self-assembled fluoroalkyl end-capped oligomers with guest molecules. | ||
Especially, it is suggested that silicananoparticles should act as guest molecules in fluorinated oligomeric aggregate cores to give new fluorinated oligomer-coated silicananoparticles (see Fig. 3).56
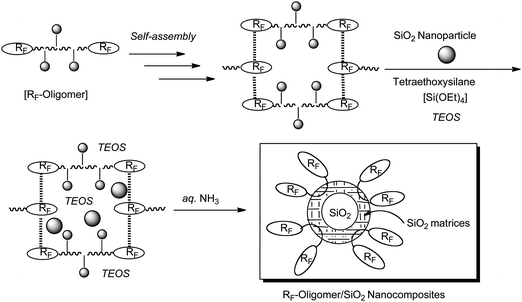 | ||
| Fig. 3 Preparation of fluoroalkyl end-capped oligomer/SiO2 nanocomposites. | ||
In this way, fluoroalkyl end-capped oligomers have high potential for the preparation of novel fluorinated oligomeric nanoparticles and fluorinated oligomer/guest molecule nanocomposites. In this paper, we review the recent development of preparation and applications of fluorinated polymeric nanoparticles and fluorinated polymeric nanocomposites by the use of fluoroalkyl end-capped oligomers as key intermediates.
2. Synthesis of fluoroalkyl end-capped oligomers
In general, it is well known that randomly fluoroalkylated macromolecules such as (meth)acrylate and acrylamidepolymers containing longer perfluoroalkyl groups exhibit interesting characteristics imparted by fluorine that set them apart from the usual alkylated macromolecules.57 A considerable interest has also been devoted to block copolymers containing longer perfluoroalkyl groups owing to exhibiting the lower surface energy and self-assembled polymeric aggregates resembling micelle in aqueous and organic media, which cannot be achieved by the randomly perfluoroalkylated polymers.58–60 These fluorinated block copolymers have recently been prepared through RAFT (reversible-addition–fragmentation-transfer) and ATRP (atom transfer radical polymerization) techniques.61–67 However, these longer perfluoroalkyl groups are in general introduced into the polymeric main chain through ester or amidegroups owing to the synthetic difficulty of direct perfluoroalkylation with carbon–carbon bond formation into polymeric molecules.50,51 These perfluoroalkylated polymers have some difficulties in exhibiting surface active characteristics imparted by perfluoroalkyl groups on the surface for a long time, because these polymers are likely to suffer the hydrolysis of the ester or amidegroups due to the strong electron negativity of fluorine.50,51From this point of view, it is very important to develop new fluorinated block copolymers with an excellent surfactant property imparted by longer fluoroalkyl groups for a long time, the same as for low-molecular weight fluorinated surfactants, in which fluoroalkyl groups should be directly introduced into polymeric molecules through carbon–carbon bond formation.50,51
It is well known that fluoroalkanoyl peroxides [RF–C(O)–O–O–(O)C–RF; RF = fluoroalkyl groups] decompose homolytically through the three bond radical fissions to afford RF˙ radicals, although the corresponding alkanoylperoxides [R–C(O)–O–O–(O)C–R; R = alkyl groups] decompose homolytically through the stepwise radical fissions to afford R–C(O)–O˙ radicals.48 This suggests that fluoroalkanoyl peroxide is a convenient tool for the direct introduction of the corresponding fluoroalkyl groups into organic molecules via a radical process. In fact, as shown in Scheme 2, fluoroalkanoyl peroxides reacted with acrylic acid monomer to give two fluoroalkyl end-capped acrylic acid oligomers in good isolated yields.68Acrylic acid oligomers with two fluoroalkyl end groups were obtained via primary radical termination or radical chain transfer to the peroxides.68 In this oligomerization process, primary radical termination of electron poor RF˙ radicals toward poly(acrylic acid) propagation radical [∼CH2–CH(COOH)˙] should predominately occur to produce RF end-capped oligomers due to the effective interaction between the HOMO of the propagation radical and the SOMO of the RF˙ radical, differing from the production of usual macromolecules by the use of a nonfluorinated R–(CO)–O˙ radical or R˙ radical.69 In contrast, it is well known that fluoroalkanoyl peroxide is an excellent radical initiator for tetrafluoroethylene (CF2CF2), which is an electron poor monomer, to produce extremely higher molecular weight poly(tetrafluoroethylene).70,71 In this case, since the poly(tetrafluoroethylene) propagation radical [∼CF2–CF2˙ radical] is electron poor, we have some difficulties in observing primary radical termination of the electron poor RF˙ radical toward the propagation radicals. Thus, higher molecular weight fluorinated polymers are produced under such conditions.
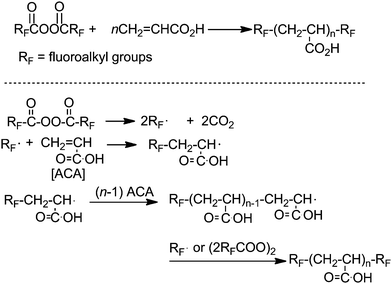 | ||
| Scheme 2 Preparation of fluoroalkyl end-capped acrylic acid oligomers. | ||
Two fluoroalkyl end-capped acrylic acid oligomers, which were prepared by using fluoroalkanoyl peroxide as a key intermediate, were found to be attractive functional materials due to their numerous unique properties and the formation of self-assembled molecular aggregates, not possible by the corresponding randomly fluoroalkylated polymers and fluoroalkylated block polymers, in which fluoroalkyl groups are introduced through the ester or amidegroups.72
3. Formation of self-assembled fluorinated oligomeric aggregates—application to the cross-linked fluoroalkyl end-capped cooligomeric nanoparticles
Amphiphilic fluoroalkyl end-capped trimethylvinylsilane–acrylic acid cooligomers [RF–(CH2CHSiMe3)x–(CH2CHCOOH)y–RF; RF = fluoroalkyl groups] can be prepared by the cooligomerization of fluoroalkanoyl peroxides with trimethylvinylsilane and acrylic acid.52 These obtained fluorinated cooligomers can form molecular assemblies possessing an ellipsoidal shape with the aggregations of terminal fluoroalkyl segments, as shown in Fig. 1.53,54These fluorinated molecular aggregates could interact with positively charged HIV-1 as a guest molecule to exhibit a potent and selective anti-HIV-1 activity by an electrostatic interaction (Fig. 4).73 However, the shape of these fluorinated oligomeric aggregates is in general easily changeable under a variety of conditions, and the encapsulated guest molecules should have an easy releasing characteristic from the aggregate cores to the outside region under these conditions (see Fig. 2).55 Therefore, it is of particular interest to develop new fluoroalkyl end-capped cooligomeric nanoparticles, in which their structures are fixed from the developmental viewpoint of new fluorinated functional materials.
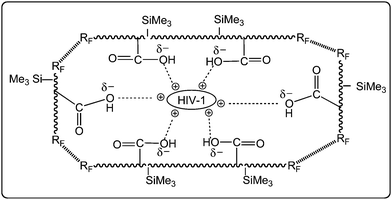 | ||
| Fig. 4 Schematic illustration for the interaction of HIV-1 and the aggregates formed by RF–(CH2–CHCO2H)x–(CH2–CHSiMe3)y–RF. | ||
As mentioned before, fluoroalkanoyl peroxides are useful for the preparation of fluoroalkyl end-capped oligomers, whose molecular weights are controlled to the few thousand level.68,69 Cross-linked fluoroalkyl end-capped cooligomeric nanoparticles can be prepared by using this lower polymerizable characteristic of fluoroalkanoyl peroxide toward a variety of radical polymerizable monomers. Fluoroalkanoyl peroxides reacted with the dimethylacrylate monomer containing poly(oxyethylene) units and acrylic acid to afford cross-linked fluoroalkyl end-capped cooligomeric nanoparticles without the formation of gels (see Scheme 3).74 These cross-linked fluorinated cooligomeric particles were nanometre size-controlled (42–363 nm) fine particles, and exhibited a good dispersibility and stability in a variety of common organic solvents including water. We can use a variety of fluoroalkanoyl peroxides possessing shorter (n = 0; m = 2, 3) or longer (n = 1) perfluorooxaalkyl groups [RF = CF(CF3){OCF2CF(CF3)}nO(CF2)mCF3; n = 0, 1; m = 2, 3] for the preparation of cross-linked fluorinated cooligomeric nanoparticles, as shown in Scheme 3. The cross-linked fluorinated nanoparticles possessing shorter perfluorooxafluoroalkyl groups thus obtained were found to exhibit a similar dispersibility and stability to that of longer ones toward numerous solvents, indicating that these perfluorooxaalkyl groups possess a flexible characteristic due to their ether linkages, quite different from usual longer stiff perfluoroalkyl groups [RF = (CF2)nCF3; n = 5–7].
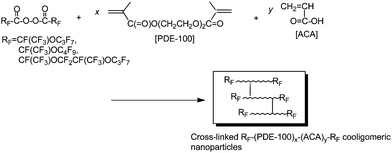 | ||
| Scheme 3 Preparation of cross-linked fluoroalkyl end-capped cooligomeric nanoparticles. | ||
Fluoroalkyl end-capped cooligomers that contain both oxime-blocked isocyanto and hydroxyadamantyl segments were prepared by the cooligomerization of fluoroalkanoyl peroxide with the corresponding monomers (see Scheme 3).75 These fluorinated cooligomers afforded new cross-linked fluoroalkyl end-capped cooligomeric nanoparticles that contain adamantane segments by deprotecting reactions of oxime-blocked isocyanato segments in cooligomers (see Scheme 4).75 These fluorinated nanoparticles exhibited a good dispersibility in methanol and fluorinated aliphatic solvents, and their size is around the 10 nm level. These cross-linked fluorinated cooligomeric nanoparticles were applied to the preparation of colloidal stable cross-linked fluorinated cooligomeric magnetic nanoparticles (Scheme 5).76
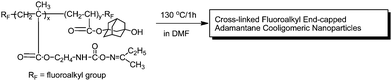 | ||
| Scheme 4 Preparation of cross-linked fluoroalkyl end-capped cooligomeric nanoparticles containing adamantane segments by deprotecting reactions of oxime-blocked isocyanato segments in cooligomers. | ||
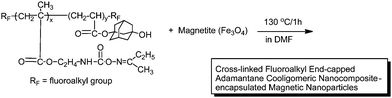 | ||
| Scheme 5 Preparation of colloidal stable cross-linked fluorinated cooligomeric magnetic nanoparticles. | ||
Cross-linked fluoroalkyl end-capped cooligomeric nanoparticle-encapsulated fullerene was prepared by the deprotecting reactions of fluoroalkyl end-capped isocyanatoethyl methacrylate 2-butanone oxime adduct–1-hydroxy-5-adamantylacrylate cooligomers in the presence of fullerene (Scheme 6).77 The size of these obtained fluorinated composites was nanometre-size controlled (28–82 nm), and exhibited a good dispersibility in a variety of solvents such as methanol, ethanol, isopropyl alcohol, tetrahydrofuran, N,N-dimethylformamide, dimethyl sulfoxide and 1,2-dichloroethane. Transmission electron microscopy (TEM) images showed that these particles are very fine nanoparticles with a mean diameter of 45 nm and fullerenes are tightly encapsulated into fluorinated nanoparticle cores.77 These fluorinated nanocomposite-encapsulated fullerenes were applied to the surface modification of poly(methyl methacrylate) to exhibit a good oleophobicity imparted by fluorine on the modified film surfaces.77 Fluorescence emission related to the encapsulated fullerene in nanoparticles was visibly observed on the only modified PMMA film surface by irradiating light (excitation: λmax 330–385 nm) on it.77 This finding also suggests that encapsulated fullerene was arranged regularly on the polymer surface due to the excellent surface-active characteristics imparted by end-capped fluoroalkyl segments in nanoparticles (see Fig. 5).77
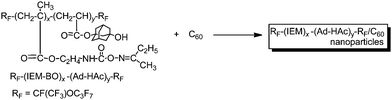 | ||
| Scheme 6 Preparation of cross-linked fluoroalkyl end-capped cooligomeric nanoparticle-encapsulated fullerene. | ||
 | ||
| Fig. 5 Fluorescent microscopy images of the cross-section of PMMA films treated with RF–(IEM)x–(Ad-HAc)y–RF/C60nanoparticles. | ||
Vinylsilanes are well known to possess a poor radical polymerizable characteristic, due to the small Q value of 0.08 (ref. 78) for vinyltrimethoxysilane. Thus, we cannot detect the radical polymerizable products of 1,3-divinyltetramethyldisiloxane by the use of usual azo-initiators such as 2,2′-azobis(2,4-dimethylvaleronitrile) under mild radical polymerization conditions below 100 °C.79 On the other hand, fluoroalkanoyl peroxide [RFC(O)OO(O)CRF] has been already applied to a radical initiator for the preparation of fluoroalkyl end-capped vinyltrimethoxysilane oligomer [RF–(CH2CHSi(OMe)3)n–RF; n = 2, 3] under very mild conditions (Scheme 7).80
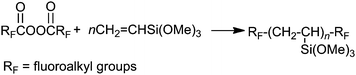 | ||
| Scheme 7 Preparation of fluoroalkyl end-capped vinyltrimethoxysilane oligomers. | ||
Fluoroalkanoyl peroxide was applied to the preparation of fluoroalkyl end-capped oligomers containing vinylsiloxane segments by using this radical polymerizable characteristic of vinylsilanes toward fluoroalkanoyl peroxides.79 These fluorinated vinylsilane oligomers containing vinylsiloxane segments reacted with a variety of radical polymerizable monomers in the presence of a traditional radical azo initiator to afford new fluorinated copolymeric nanoparticles (size of particles: around 200 nm) (Scheme 8).79
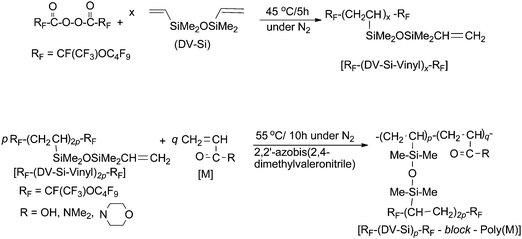 | ||
| Scheme 8 Preparation of fluorinated vinylsilane oligomers containing vinylsiloxane segments and fluorinated copolymeric nanoparticles by using RF–(DV–Si–Vinyl)x–RF. | ||
Fluoroalkanoyl peroxide reacted with 1,3,5,7-tetravinyl-1,3,5,7-tetramethylcyclotetrasiloxane (TRIV-Si) to afford fluoroalkyl end-capped oligomers containing some unreacted vinyl segments.81 Fluoroalkyl end-capped cyclosiloxane oligomers containing some vinyl segments thus obtained reacted with N,N-dimethylacrylamide and fluoroalkanoyl peroxide to afford new fluorinated dendrimer-type block copolymeric nanoparticles in good yields (Scheme 9). These fluorinated copolymeric nanoparticles had an excellent solubility not only in water but also in traditional organic solvents, including aliphatic fluorinated solvents, and the size of these particles was nanometre size-controlled (∼120 nm).81
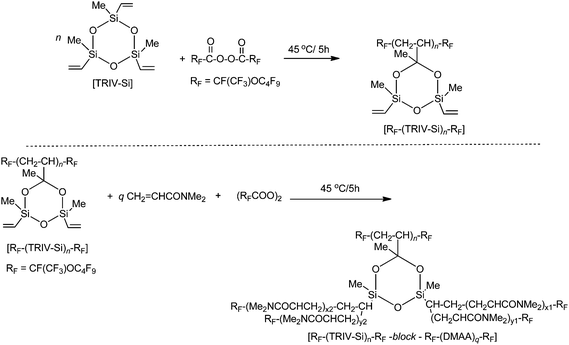 | ||
| Scheme 9 Preparation of fluoroalkyl end-capped oligomers containing some unreacted vinyl segments and fluorinated dendrimer-type block copolymeric nanoparticles by using RF–(TRIV–Si)n–RF. | ||
Interestingly, these fluorinated block copolymeric nanoparticles had an extremely higher dispersion ability toward not only single-walled carbon nanotubes and fullerenes but also magnetic nanoparticles in water, compared to that of the corresponding fluoroalkyl end-capped vinylsilanes-block-poly(N,N-dimethylacrylamide) copolymeric nanoparticles (see Scheme 8)79 or two fluoroalkyl end-capped oligomeric aggregates.82
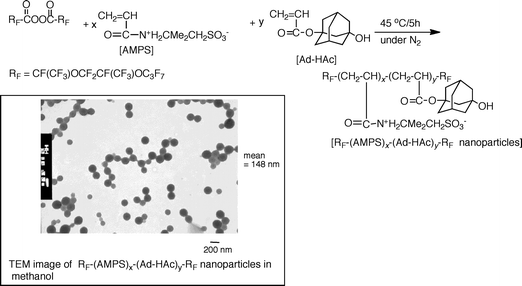
Fluoroalkyl end-capped 2-acrylamido-2-methylpropanesulfonic acid cooligomers containing adamantyl segments were prepared by reaction of fluoroalkanoyl peroxide with 2-acrylamido-2-methylpropanesulfonic acid (AMPS) and 1-hydroxy-5-adamantyl acrylate (Ad-HAc).83 These obtained fluorinated AMPS–Ad-HAc cooligomers can form nanometre size-controlled fine particles not only in water but also in a large variety of traditionally organic solvents.83
Fluorinated AMPS–Ad-HAc cooligomeric nanoparticles exhibited the lower critical solution temperature (LCST) around 50 °C in organic media (tert-butyl alcohol) (Fig. 6).83 The size of these fluorinated cooligomeric nanoparticles was extremely sensitive to the solvent dielectric constant and temperature changes.83 Hitherto, it is well-known that block copolymers such as poly(N-isopropylacrylamide)-block-poly(ethylene oxide) exhibit thermosensitive micellization because of their hydrophobic character of the poly(N-isopropylacrylamide) block above its LCST combined with the hydrophilic property of the poly(ethylene oxide) block in aqueous systems.84,85 However, this LCST behavior would be mainly related to the oleophilic–oleophobic balance in these cooligomeric nanoparticles corresponding to the oleophilic character from adamantyl segments and the oleophobic character from fluoroalkyl groups.83 Therefore, this LCST observation in organic media is strongly due to the oleophobic characteristic imparted by fluoroalkyl alkyl groups.
 | ||
| Fig. 6 Photographs of tert-butyl alcohol solutions of RF–(AMPS)x–(Ad-HAc)y–RF (6 g dm−3) possessing a LCST characteristic. | ||
As mentioned in Scheme 5, cross-linked fluoroalkyl end-capped cooligomeric nanocomposite-encapsulated magnetic nanoparticles can be prepared by the deprotecting reactions of the corresponding fluorinated cooligomers containing oxime-blocked isocyanato segments in the presence of magnetic nanoparticles.76 These fluorinated nanocomposite-encapsulated magnetic nanoparticles were applied to the surface modification of poly(methyl methacrylate) to exhibit not only a good oleophobicity imparted by fluorine but also a magnetic property related to the encapsulated magnetic nanoparticles (Fig. 7).76
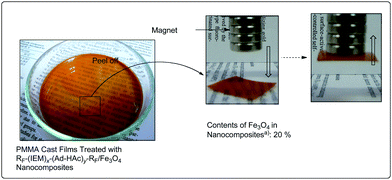 | ||
| Fig. 7 Photographs of the PMMA film treated with RF–(IEM)x–(Ad-HAc)y–RF/Fe3O4 nanocomposites: the concentration of RF–(IEM)x–(Ad-HAc)y–RF/Fe3O4nanoparticles on PMMA is 3% (m/m). | ||
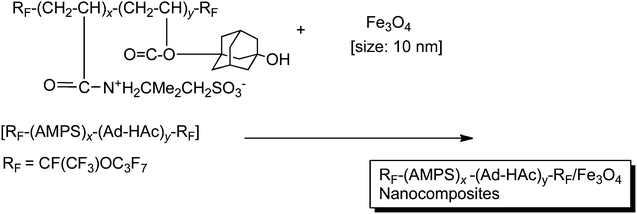
Fluoroalkyl end-capped 2-acrylamido-2-methylpropanesulfonic acid cooligomeric nanoparticles containing adamantyl segments were able to interact with magnetic nanoparticles to afford the corresponding fluorinated betaine-type cooligomeric nanocomposite-encapsulated magnetic nanoparticles (average particle size: 25–183 nm). These fluorinated betaine-type cooligomeric nanocomposite-encapsulated magnetic nanoparticles can exhibit the LCST characteristic in organic media such as tert-butyl alcohol and were found to decrease the LCST effectively through the encapsulation of magnetic nanoparticles into fluorinated cooligomeric nanoparticle cores (Fig. 8).76
![Photographs of tert-butyl alcohol solutions of RF–(AMPS)x–(Ad-HAc)y–RF/Fe3O4 [x : y = 44 : 56] (4 g dm−3) possessing the lower critical solution temperature (LCST).](/image/article/2012/PY/c1py00325a/c1py00325a-f8.gif) | ||
Fig. 8 Photographs of tert-butyl alcohol solutions of RF–(AMPS)x–(Ad-HAc)y–RF/Fe3O4 [x![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :y = 44:56] (4 g dm−3) possessing the lower critical solution temperature (LCST). :y = 44:56] (4 g dm−3) possessing the lower critical solution temperature (LCST). | ||
TEM images showed that magnetic nanoparticles in Scheme 10 can be encapsulated inside the cross-linked fluorinated cooligomeric nanocomposite cores; in contrast, magnetic nanoparticles in Scheme 11 can be adsorbed outside the fluorinated betaine-type cooligomeric nanocomposite cores (Fig. 9).76
 | ||
| Scheme 10 Preparation of fluoroalkyl end-capped 2-acrylamido-2-methylpropanesulfonic acid cooligomers containing adamantyl segments and their TEM image. | ||
 | ||
| Scheme 11 Preparation of fluorinated betaine-type cooligomeric nanocomposite-encapsulated magnetic nanoparticles. | ||
 | ||
| Fig. 9 Selective preparation of novel fluoroalkyl end-capped cooligomeric nanocomposites. | ||
4. Preparation and applications of fluoroalkyl end-capped oligomer-coated silica nanocomposites
Fluoroalkyl end-capped vinylsilane oligomer-coated silicananoparticles can be prepared by the sol–gel reaction of the corresponding fluorinated vinyltrimethoxysilane oligomer under alkaline conditions (Scheme 12).86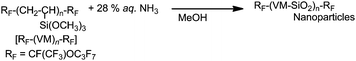 | ||
| Scheme 12 Preparation of fluoroalkyl end-capped vinylsilane oligomer-coated silicananoparticles. | ||
The modified glass surface treated with this fluorinated vinylsilane oligomeric nanoparticle exhibited a completely superhydrophobic characteristic (a water contact angle of 180°) with a non-wetting property against water droplets (Fig. 10).86
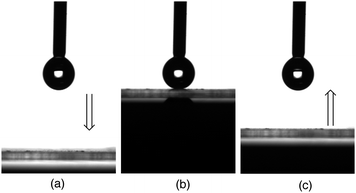 | ||
| Fig. 10 CCD (Charge Coupled Device) camera images of the water droplets: (a) water droplet which adhered in the needle tip (process before adhesion of the water droplet on the modified glass surface); (b) water droplet on the modified glass surface; and (c) the pull-up process of the needle from the modified glass surface. | ||
The topographic images of this modified glass surface by AFM showed that fluoroalkyl groups in nanoparticles are located at a rough surface to indicate the completely superhydrophobic characteristic by RF–(VM–SiO2)n–RF oligomeric nanoparticles.86
Fluoroalkyl end-capped oligomeric aggregates could interact with silicananoparticles as guest molecules in their aggregate cores. Thus, the expected fluoroalkyl end-capped oligomer/silica nanocomposites can be easily obtained by the sol–gel reactions of these fluorinated oligomeric aggregate-encapsulated silicananoparticles with tetraethoxysilane (TEOS) under alkaline conditions as shown in Fig. 3.56
Similarly, we prepared fluoroalkyl end-capped acrylic acid oligomer [RF–(ACA)n–RF]/silica nanocomposites containing vinyl groups by the sol–gel reactions of the corresponding fluorinated oligomer with tetraethoxysilane and silicananoparticles in the presence of trimethoxyvinylsilane under alkaline conditions, respectively.87 These fluorinated nanocomposites were applied to the surface initiated radical copolymerization, and polystyrene grafted RF–(ACA)n–RF/SiO2 nanocomposites were prepared by the surface copolymerization of RF–(ACA)n–RF/silica nanocomposites containing vinyl groups with styrene (see Scheme 13).87 These polystyrene grafted fluorinated silica nanocomposites were effective for the dispersion of fullerene and single-walled carbon nanotubes into water.87
![Preparation of fluoroalkyl end-caped acrylic acid oligomer [RF–(ACA)n–RF]/silica nanocomposites containing vinyl groups and polystyrene or poly(N,N-dimethylacrylamide) grafted RF–(ACA)n–RF/SiO2 nanocomposites.](/image/article/2012/PY/c1py00325a/c1py00325a-s13.gif) | ||
| Scheme 13 Preparation of fluoroalkyl end-caped acrylic acid oligomer [RF–(ACA)n–RF]/silica nanocomposites containing vinyl groups and polystyrene or poly(N,N-dimethylacrylamide) grafted RF–(ACA)n–RF/SiO2 nanocomposites. | ||
We previously reported that RF–(VM–Si)x–(ACA)y–RF cooligomers can form nanometre size-controlled self-assembled molecular aggregates, and these fluorinated molecular aggregates could interact with human immunodeficiency virus type 1 (HIV-1) as a guest molecule to exhibit a potent and selective anti-HIV-1 activity in vitro (see Fig. 4).73 Thus, it is of particular interest to prepare new fluoroalkyl end-capped oligomer-coated silicananoparticles possessing an anti-HIV-1 activity. In fact, fluoroalkyl end-capped trimethylvinylsilane–acrylic acid cooligomer [RF–(VM–Si)x–(ACA)y–RF] reacted with tetraethoxysilane and silicananoparticles under alkaline conditions to afford the corresponding fluorinated cooligomer/silicananoparticles.88 Fluoroalkyl end-capped N,N-dimethylacrylamide–2-methacryloyloxyethanesulfonic acid cooligomer/silica nanocomposites [RF–(DMAA)x–(MES)y–RF/SiO2] were also prepared under similar conditions (Scheme 14).88
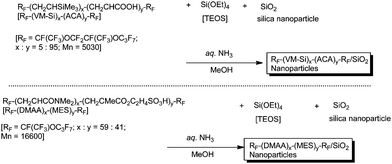 | ||
| Scheme 14 Preparation of fluoroalkyl end-capped cooligomer-coated silicananoparticles possessing an anti-HIV-1 activity. | ||
RF–(VM–Si)x–(ACA)y–RF/SiO2 nanocomposites were found to have a potent and selective anti-HIV-1 activity.88 In contrast, RF–(DMAA)x–(MES)y–RF/SiO2 nanocomposites were able to exhibit a potent and selective anti-SIVmac (simian immunodeficiency virus) activity.88 Therefore, these fluorinated cooligomeric silica nanocomposites are expected to be widely applicable not only for antiviral polymeric drugs but also in the fields of materials science.
5. Fluoroalkyl end-capped oligomers possessing nonflammable and flammable characteristics in silica gel matrices after calcination at 800 °C
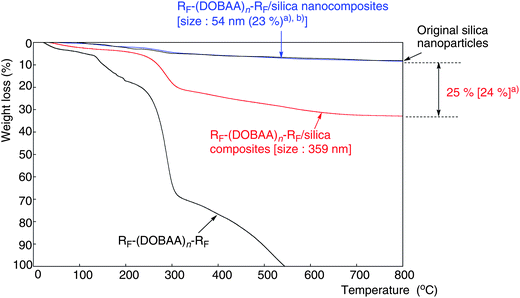
Traditional fluoropolymers such as poly(tetrafluoroethylene) (PTFE) possess an excellent thermal resistance.89,90 Hybridization of PTFE with silica gels is of particular interest from the developmental viewpoint of novel fluorinated functional polymeric materials. Chen et al. have already prepared PTFE/SiO2 hybrids; however, the parent PTFE in the silica gel hybrids decomposes completely around 700 °C.91 On the other hand, the molecular weights of fluoroalkyl end-capped oligomers are in general within 10000, and their thermal stability is extremely poor compared to that of usual perfluorinated polymers or perfluoroalkylated polymers.68,69 In fluoroalkyl end-capped N-(1,1-dimethyl-3-oxobutylacrylamide) oligomers [RF–(DOBAA)n–RF]/silica nanocomposites [the contents of oligomers: 18–72%], which were prepared under alkaline conditions, unexpectedly, weight loss behavior corresponding to the content of each fluorinated oligomer in the silica nanocomposites was not observed at all even after calcination at 800 °C (Scheme 15).92,93 However, RF–(DOBAA)n–RF/silica nanocomposites, in which the oligomer contents are above 75%, exhibited a clear weight loss after calcination at 800 °C (see Fig. 11).92,93
![Preparation of fluoroalkyl end-capped N-(1,1-dimethyl-3-oxobutylacrylamide) oligomer [RF–(DOBAA)n–RF]/silica nanocomposites.](/image/article/2012/PY/c1py00325a/c1py00325a-s15.gif) | ||
| Scheme 15 Preparation of fluoroalkyl end-capped N-(1,1-dimethyl-3-oxobutylacrylamide) oligomer [RF–(DOBAA)n–RF]/silica nanocomposites. | ||
![Thermogravimetric analyses of RF–(DOBAA)n–RF/silica nanocomposites [oligomer contents in composites: 18, 19, 33, 42, 72, 75, 94, 96%], RF–(DOBAA)n–RF oligomers and original SiO2nanoparticles.](/image/article/2012/PY/c1py00325a/c1py00325a-f11.gif) | ||
| Fig. 11 Thermogravimetric analyses of RF–(DOBAA)n–RF/silica nanocomposites [oligomer contents in composites: 18, 19, 33, 42, 72, 75, 94, 96%], RF–(DOBAA)n–RF oligomers and original SiO2nanoparticles. | ||
RF–(DOBAA)n–RF/silica nanocomposites prepared under acidic conditions showed a clear weight loss at 800 °C, a value quite similar to that of the elementary analyses of fluorine in the nanocomposites (see Fig. 12).92,93
 | ||
| Fig. 12 Thermogravimetric analyses of RF–(DOBAA)n–RF/silica composites, which were prepared under acidic conditions and the parent RF–(DOBAA)n–RF oligomer. (a) The oligomer content in composites determined by elementary analyses of fluorine and (b) nanocomposites were prepared under alkaline conditions. | ||
19F MAS NMR spectra of RF–(DOBAA)n–RF/silica nanocomposites before calcination showed CF3 and CF2 peaks related to the presence of the RF–(DOBAA)n–RF oligomer in the composites (Fig. 13).94 A relatively sharp peak at around −154.8 ppm was observed in the RF–(DOBAA)n–RF/silica nanocomposites prepared under alkaline conditions, which exhibited nonflammability; although such peak was not observed at all in the RF–(DOBAA)n–RF/silica nanocomposites possessing flammability. This peak should be ascribed to (NH4)2SiF6.94
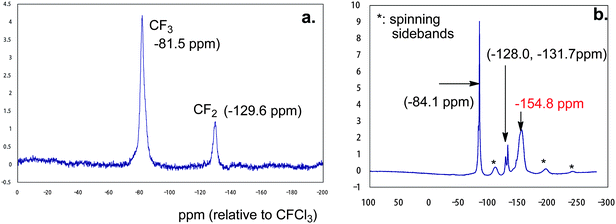 | ||
| Fig. 13 19F MAS NMR spectra of RF–(DOBAA)n–RF/silica gel composites possessing weight loss characteristic (a) and possessing no weight loss characteristic (b) before calcination at 800 °C. | ||
The room temperature FT-IR spectra of RF–(DOBAA)n–RF/silica gel nanocomposites possessing nonflammability showed the complete disappearance of the carbonyl bands (1709, 1655 and 1543 cm−1) related to the RF–(DOBAA)n–RF oligomer, even before calcination (Fig. 14).94 However, the treatment of this RF–(DOBAA)n–RF/silica gel nanocomposite with aqueous HF showed a clear band at around 700 cm−1 in addition to those of the parent RF–(DOBAA)n–RF oligomer, although we cannot detect such bands at all in the corresponding flammable RF–(DOBAA)n–RF oligomer.94 The presence of a clear band at around 700 cm−1 would be due to the presence of ammonium hexafluorosilicate in the composite. The RF–(DOBAA)n–RF/silica gel nanocomposite possessing a nonflammable characteristic after calcination at 800 °C had been treated with aqueous HF. The FT-IR spectra of this treated product show the formation of ammonium hexafluorosilicate.94 These findings suggest that the nanocomposite reaction of the RF–(DOBAA)n–RF oligomer with silicananoparticles should proceed smoothly under alkaline conditions to afford not only the expected RF–(DOBAA)n–RF/silica nanocomposite but also ammonium hexafluorosilicate as shown in the plausible reaction mechanism (see Scheme 16).94
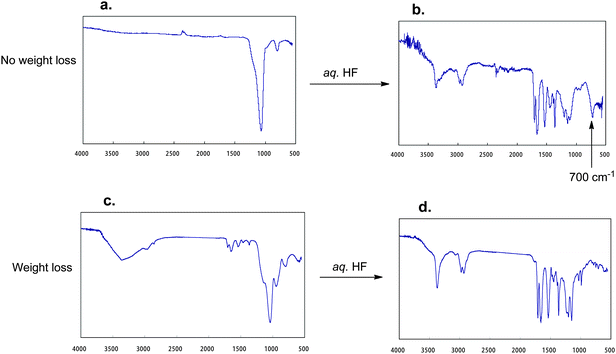 | ||
| Fig. 14 FT-IR spectra of the RF–(DOBAA)n–RF/silica gel nanocomposite possessing no weight loss characteristic (a), weight loss characteristic (c), and FT-IR spectra of (b) and (d), which were treated with aqueous HF with (a) and (c), respectively. | ||
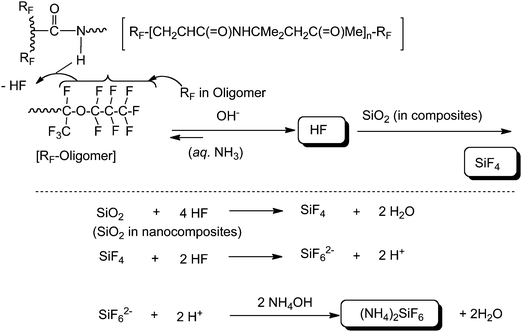 | ||
| Scheme 16 Plausible reaction mechanism for the formation of ammonium hexafluorosilicate. | ||
The formation of ammonium hexafluorosilicate during the composite reactions can afford a nonflammable characteristic toward RF–(DOBAA)n–RF oligomer. That is, the RF–(DOBAA)n–RF oligomer in the nanocomposite should be encapsulated quite effectively into the nanometre size-controlled silica gel matrices through the molecular-level synergistic combination, which is due to not only the strong interaction between fluorine in the oligomer and silicon in the silica gel nanocomposite but also the effective interaction between ammonium hexafluorosilicate and the RF–(DOBBA)n–RF oligomer in silica gel matrices, to afford a nonflammable characteristic for the oligomer.94 Such effective interactions between ammonium hexafluorosilicate and RF–(DOBAA)n–RF oligomer in silica gel matrices should enable the RF–(DOBAA)n–RF/silica gel nanocomposite to afford the complete disappearance of the amido band in the RF–(DOBAA)n–RF oligomer in FT-IR spectra measurements before and after calcination at 800 °C.941H MAS NMR spectra of the RF–(DOBAA)n–RF/silica nanocomposite [content of oligomer in the composite: 23% (determined by elementary analyses of fluorine)] possessing no weight loss characteristic even after calcination at 800 °C showed similar relatively sharp peaks around 50 to −20 ppm to those before calcination (see Fig. 15).95 These peak areas are almost the same before calcination at 800 °C.95
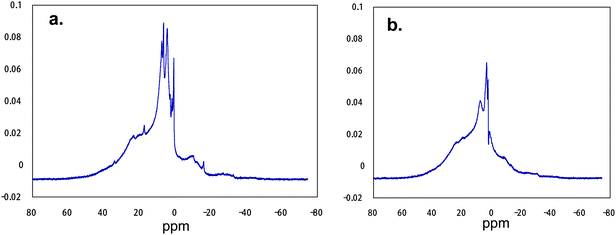 | ||
| Fig. 15 1H MAS NMR spectra of RF–(DOBAA)n–RF/SiO2 nanocomposites before (a) and after (b) calcination at 800 °C. | ||
On the other hand, fluoroalkyl end-capped acrylic acid oligomer/silica nanocomposites [RF–(CH2CHCOOH)n–RF/SiO2] have been shown to exhibit a clear weight loss at 800 °C corresponding to the content of the oligomer.951H MAS NMR spectra of the RF–(ACA)n–RF/silica nanocomposite possessing a clear weight loss at 800 °C, in which the content of the oligomer is 22% (determined by elementary analyses of fluorine), showed an extreme decrease of the peak areas for this nanocomposite by 90% after calcination at 800 °C (see Fig. 16).95
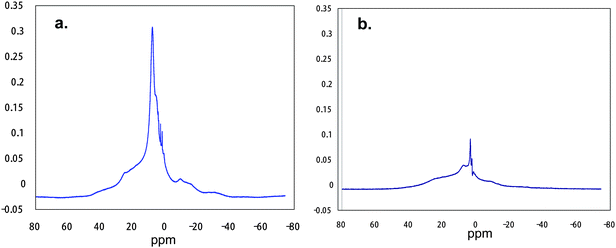 | ||
| Fig. 16 1H MAS NMR spectra of RF–(ACA)n–RF/silica gel nanocomposites before (a) and after (b) calcination at 800 °C. | ||
Fluoroalkyl end-capped oligomers possessing sulfo groups and fluoroalkyl end-capped oligomers containing carboxy groups possessing electron-withdrawing CF3 units as neighboring groups [RF–(CH2–CMeCOOH)x–(CH2–CCF3COOH)y–RF] can exhibit no weight loss behavior by the nanocomposite reactions.94 Similarly, poly(VDF-ter-TFMA-ter-HFP) terpolymers containing carboxy groups possessing electron-withdrawing CF3 units as neighboring groups in the silica nanocomposites were found to exhibit no weight loss behavior even after calcination at 800 °C (see Scheme 17).96
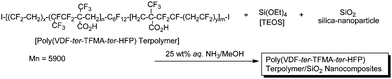 | ||
| Scheme 17 Preparation of poly(VDF-ter-TFMA-ter-HFP) terpolymer/SiO2 nanocomposites. | ||
In this way, fluoroalkyl end-capped oligomers containing amidoprotons or more acidic protons such as sulfo groups can exhibit a nonflammability, even after calcination at 800 °C, through the formation of ammonium hexafluorosilicate during nanocomposite reactions. In contrast, the lack of formation of ammonium hexafluorosilicate during the usual composite reactions affords a flammable behavior for the RF–(DOBAA)n–RF oligomer or RF–(ACA)n–RF oligomer in silica nanocomposites.95
Fluoroalkyl end-capped acrylic acid oligomer [RF–(ACA)n–RF] reacted with tetraethoxysilane (TEOS) and silicananoparticles in the presence of low molecular weight aromatic compounds [Ar–H] such as cetylpyridinium chloride (CPC) and bisphenol AF under alkaline conditions to afford RF–(ACA)n–RF/SiO2 nanocomposite-encapsulated Ar–H in excellent to moderate isolated yields (Scheme 18).97
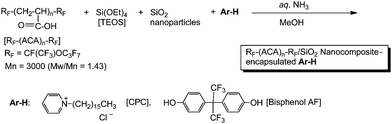 | ||
| Scheme 18 Preparation of RF–(ACA)n–RF/SiO2 nanocomposite-encapsulated Ar–H. | ||
These fluorinated silica nanocomposite-encapsulated Ar–H can exhibit no weight loss behavior corresponding to the contents of Ar–H after calcination at 800 °C, although the fluoroalkyl end-capped acrylic acid oligomer in the nanocomposites decomposed completely under similar conditions.97
Not only fluoroalkyl end-capped oligomers but also low molecular weight fluorinated surfactants such as perfluoro-1,3-propanedisulfonic acid are effective for the preparation of the corresponding fluorinated silica nanocomposites.98Perfluoro-1,3-propanedisulfonic acid/silica [PFPS/SiO2] nanocomposites were prepared by the sol–gel reactions of the corresponding disulfonic acid [PFPS] with tetraethoxysilane and silicananoparticles under alkaline conditions (Scheme 19).98 These fluorinated nanocomposites can exhibit no weight loss behavior corresponding to the contents of PFPS in the composites even after calcination at 800 °C, although the parent PFPS can decompose completely around 270 °C.98
![Preparation of perfluoro-1,3-propanedisulfonic acid/silica [PFPS/SiO2] nanocomposites.](/image/article/2012/PY/c1py00325a/c1py00325a-s19.gif) | ||
| Scheme 19 Preparation of perfluoro-1,3-propanedisulfonic acid/silica [PFPS/SiO2] nanocomposites. | ||
We also succeeded in the encapsulation of a variety of low molecular weight aromatic compounds such as bisphenol-A, bisphenol-AF, bisphenol-F, 4,4′-biphenol and 1,1′-bi-2-naphthol into PFPS/SiO2 nanocomposite cores (Scheme 20).98
 | ||
| Scheme 20 Encapsulation of low molecular weight aromatic compounds into PFPS/SiO2 nanocomposite cores. | ||
These encapsulated low molecular weight aromatic compounds were able to exhibit nonflammable characteristics at 800 °C in the fluorinated nanocomposite cores.98
Novel cross-linked fluoroalkyl end-capped trimethoxyvinylsilane oligomeric nanoparticle [RF–(VM–SiO2)n–RF]-encapsulated 1,1′-bi(2-naphthol) (BINOL) were prepared by the sol–gel reaction of the fluoroalkyl end-capped trimethoxyvinylsilane oligomer in the presence of BINOL under alkaline conditions (Scheme 21).99 RF–(VM–SiO2)n–RF/BINOL nanocomposites thus obtained were found to afford a clear weight loss at 800 °C, which corresponds to the content of the fluorinated oligomer in the composite.99 However, unexpectedly, RF–(VM–SiO2)n–RF/BINOL nanocomposites were found to exhibit no weight loss corresponding to the presence of BINOL in the composite at 800 °C under atmospheric conditions.99
![Preparation of fluoroalkyl end-capped trimethoxyvinylsilane oligomeric nanoparticle [RF–(VM–SiO2)n–RF]-encapsulated 1,1′-bi(2-naphthol) (BINOL).](/image/article/2012/PY/c1py00325a/c1py00325a-s21.gif) | ||
| Scheme 21 Preparation of fluoroalkyl end-capped trimethoxyvinylsilane oligomeric nanoparticle [RF–(VM–SiO2)n–RF]-encapsulated 1,1′-bi(2-naphthol) (BINOL). | ||
6. Fluoroalkyl end-capped oligomer/metal nanocomposites
Preparation of novel fluorinated polymer/metal nanoparticles possessing surface active properties imparted by fluorine is of particular interest from the developmental viewpoint of new tailored organic polymer/metal nanocomposites.100–105 Nanometre size-controlled gold particles were prepared in 1,2-dichloroethane under mild conditions by reducing the corresponding metal precursor in the presence of self-assembled molecular aggregates formed by fluoroalkyl end-capped N-(1,1-dimethyl-3-oxobutyl)acrylamide oligomers. In contrast, the corresponding non-fluorinated oligomer was not able to afford the gold nanoparticles, effectively, because this oligomer is not likely to form such molecular aggregates (see Fig. 17).106![UV-vis spectra of colloidal gold prepared by the use of fluoroalkyl end-capped oligomers (2.1 g dm−3) in CH2ClCH2Cl: (a) RF–(DOBAA)n–RF [RF = CF(CF3)OC3F7; Mn = 3710], (b) RF–(DOBAA)n–RF [RF = CF(CF3)OCF2CF(CF3)OC3F7; Mn = 6770], and (c) –(DOBAA)n– [Mn = 5820].](/image/article/2012/PY/c1py00325a/c1py00325a-f17.gif) | ||
| Fig. 17 UV-vis spectra of colloidal gold prepared by the use of fluoroalkyl end-capped oligomers (2.1 g dm−3) in CH2ClCH2Cl: (a) RF–(DOBAA)n–RF [RF = CF(CF3)OC3F7; Mn = 3710], (b) RF–(DOBAA)n–RF [RF = CF(CF3)OCF2CF(CF3)OC3F7; Mn = 6770], and (c) –(DOBAA)n– [Mn = 5820]. | ||
The modified cast PMMA film treated with these fluorinated oligomer/Au nanocomposites exhibited a similar plasmon absorption at around 540 nm, the same as for the 1,2-dichloroethane solution system (Fig. 18).106
![UV-vis spectra of gold nanoparticle-containing PMMA films treated with fluoroalkyl end-capped DOBAA oligomers [RF = CF(CF3)OC3F7].](/image/article/2012/PY/c1py00325a/c1py00325a-f18.gif) | ||
| Fig. 18 UV-vis spectra of gold nanoparticle-containing PMMA films treated with fluoroalkyl end-capped DOBAA oligomers [RF = CF(CF3)OC3F7]. | ||
XPS (X-ray photoelectron spectroscopy) analyses showed that gold nanoparticles should be well dispersed above the modified PMMA film (film thickness: 196 μm) surface as well as fluorinated DOBAA oligomers.106
As mentioned before, the shape of fluoroalkyl end-capped oligomeric aggregates is easily exchangeable under a variety of conditions (see Fig. 2).55 Therefore, it is of particular interest to prepare new fluoroalkyl end-capped oligomer/metal nanocomposites in which their structures are fixed from the developmental viewpoint of new fluorinated functional materials. Novel cross-linked fluorinated cooligomer/gold nanocomposites were prepared by the reduction of gold ions with poly(methylhydrosiloxane) (PMHS) and trioctylamine (TOA) in the presence of the corresponding cross-linked fluorinated cooligomeric nanoparticles in organic media as shown in Scheme 22.107
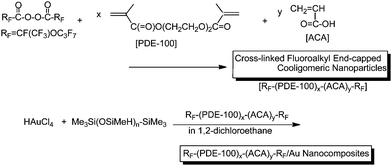 | ||
| Scheme 22 Preparation of novel cross-linked fluorinated cooligomer/gold nanocomposites. | ||
Interestingly, these fluorinated gold nanocomposite solutions can be easily isolated after simple centrifugal separation, and these nanocomposites thus obtained can be also redispersed into organic media to afford the same fluorinated nanocomposite solutions (Fig. 19).107
 | ||
| Fig. 19 RF–(PDE-100)x–(ACA)y–RF/gold nanocomposites in 1,2-dichloroethane solutions. | ||
The size of these composites increased effectively from 67 to 160 nm levels with increasing the amounts of trioctylamine. In particular, the plasmon absorption bands related to the gold nanocomposites were also dramatically red-shifted from around 530 nm to 900 nm levels under similar conditions (see Fig. 20).107
![Relationship between the concentration of TOA and the plasmon absorption bands related to the fluorinated Au nanocomposites in 1,2-dichloroethane solutions. Conditions: RF–(ACA)x–(PDE-100)y–RF [RF = CF(CF3)OC3F7] (50 mg), HAuCl4 (9.64 μmol) and poly(methylhydrosiloxane) (0.85 mmol).](/image/article/2012/PY/c1py00325a/c1py00325a-f20.gif) | ||
| Fig. 20 Relationship between the concentration of TOA and the plasmon absorption bands related to the fluorinated Au nanocomposites in 1,2-dichloroethane solutions. Conditions: RF–(ACA)x–(PDE-100)y–RF [RF = CF(CF3)OC3F7] (50 mg), HAuCl4 (9.64 μmol) and poly(methylhydrosiloxane) (0.85 mmol). | ||
Fluoroalkyl end-capped 2-acrylamido-2-methylpropanesulfonic acid (AMPS)–1-hydroxy-5-adamantylacrylate (Ad-HAc) cooligomers were applied to the preparation of novel fluorinated cooligomeric nanocomposite-encapsulated gold nanoparticles.108 These fluorinated gold nanocomposites were easily prepared by the reductions of gold ions with poly(methylhydrosiloxane) (PMHS) in the presence of the corresponding fluorinated nanoparticles and tri-n-octylamine (TOA) at room temperature (Scheme 23).108
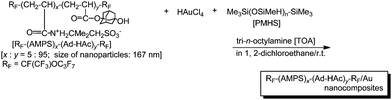 | ||
| Scheme 23 Preparation of fluorinated cooligomeric nanocomposite-encapsulated gold nanoparticles. | ||
These fluorinated gold nanoparticles were isolated as wine-red colored powders, and were found to exhibit a good dispersibility in a variety of traditional organic solvents to afford transparent wine-red colored solutions. UV-vis spectroscopy showed that these particles are nanometre size-controlled very fine nanoparticles (185–218 nm), which exhibit a plasmon absorption band around 530 nm. TEM images showed that these fluorinated cooligomeric nanocomposite-encapsulated gold nanoparticles can give linear arrays of these fluorinated nanoparticles with the increase of the feed amounts of TOA, and these fluorinated gold nanoparticles were able to exhibit the extremely red-shifted plasmon absorption band around 960 nm (see Fig. 21).108
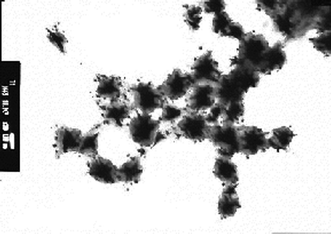 | ||
| Fig. 21 TEM (transmission electron microscopy) images of RF–(AMPS)x–(Ad-HAc)y–RF/Au nanocomposites in methanol. | ||
Fluoroalkyl end-capped N-(1,1-dimethyl-3-oxobutyl)acrylamide–acryloylmorpholine cooligomers exhibited the lower critical solution temperature (LCST) characteristic in aqueous solutions.109 The thermosensitive fluoroalkyl end-capped N-(1,1-dimethyl-3-oxobutyl)acrylamide–acryloylmorpholine cooligomer was applied to the autoreduction of gold ions to afford the fluorinated cooligomeric nanocomposite-encapsulated gold nanoparticles under very mild conditions (Scheme 24); although the corresponding non-fluorinated cooligomer was not able to afford the gold nanoparticles at all under similar conditions.110
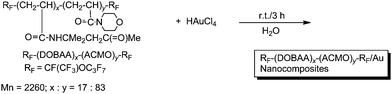 | ||
| Scheme 24 Preparation of fluoroalkyl end-capped DOBAA–ACMO cooligomeric nanocomposite-encapsulated gold nanoparticles. | ||
These fluorinated cooligomeric nanocomposites exhibited a plasmon absorption band around 520 nm related to the formation of gold nanoparticles in their aqueous solutions (Fig. 22).110 Interestingly, these fluorinated nanocomposite-encapsulated gold nanoparticles can exhibit the thermoswitchable sol–gel transition through the lower critical solution temperature (LCST)-triggered aggregation of the corresponding nanocomposites, and this sol–gel switching behavior is reversible during a heating and cooling cycle (Fig. 23).110
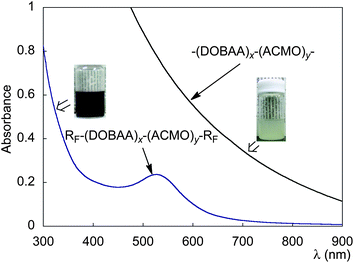 | ||
| Fig. 22 UV-vis spectra for aqueous solution of RF–(DOBAA)x–(ACMO)y–RF/Au nanocomposites at 20 °C, and aqueous solution containing HAuCl4 and –(DOBAA)x–(ACMO)y– cooligomer at 20 °C. | ||
![Temperature dependence of UV-vis spectra for aqueous solutions of RF–(DOBAA)x–(ACMO)y–RF [10 g dm−3]/Au nanocomposites and the photographs of their composite solutions.](/image/article/2012/PY/c1py00325a/c1py00325a-f23.gif) | ||
| Fig. 23 Temperature dependence of UV-vis spectra for aqueous solutions of RF–(DOBAA)x–(ACMO)y–RF [10 g dm−3]/Au nanocomposites and the photographs of their composite solutions. | ||
The LCST-triggered gel formation of RF–(DOBAA)x–(ACMO)y–RF/Au nanocomposites would be due to an intra-composite's hydrophilic–hydrophobic conformational change, which leads to hydrophobic interactions related to the oleophilic DOBAA segments in composites between RF–(DOBAA)x–(ACMO)y–RF/Au nanocomposites.110 Cooling the nanocomposite gels to below the LCST causes the architecture of RF–(DOBAA)x–(ACMO)y–RF/Au nanocomposites to the hydrophilic state, which affords the well-dispersed wine-red color nanocomposite sol solution (Fig. 24).110 Hitherto, there have been numerous reports for gold nanoparticle/hydrogel composites;111–116 however, to the best of our knowledge, our finding is the first example of the facile preparation of gold nanoparticles without any catalysts through fluorinated polysoaps possessing the LCST-triggered sol–gel switching characteristic.
![UV-vis spectra for aqueous solutions of RF–(DOBAA)x–(ACMO)y–RF [10 g dm−3]/Au nanocomposites after cooling from 50 to 20 °C.](/image/article/2012/PY/c1py00325a/c1py00325a-f24.gif) | ||
| Fig. 24 UV-vis spectra for aqueous solutions of RF–(DOBAA)x–(ACMO)y–RF [10 g dm−3]/Au nanocomposites after cooling from 50 to 20 °C. | ||
A variety of fluoroalkyl end-capped oligomer/silver nanocomposites were prepared by the reactions of silver ions with poly(methylhydrosiloxane) in the presence of the corresponding oligomers in organic media (Scheme 25).117UV-vis spectra of these fluorinated silver nanocomposites showed relatively broad plasmon absorptions at around 410 nm relative to the formation of silver nanoparticles (Fig. 25). However, narrow plasmon absorptions were observed by the addition of TOA (tri-n-octylamine) in these nanocomposites.117 These fluorinated silver nanocomposites were found to exhibit a good colloidal stability without their agglomeration in organic media. These nanocomposites were also applied to the surface modification of traditional organic polymers to exhibit not only a good oleophobicity imparted by fluorine but also high surface antibacterial activity related to the presence of silver nanoparticles on their surface.117
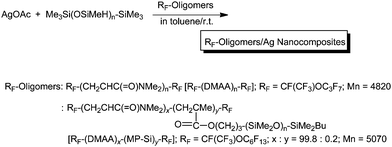 | ||
| Scheme 25 Preparation of fluoroalkyl end-capped oligomer/silver nanocomposites. | ||
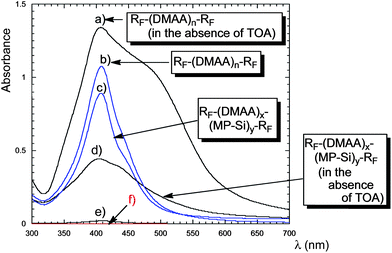 | ||
| Fig. 25 UV-vis spectra of colloidal silver particles prepared by the use of RF–(DMAA)n–RF and RF–(DMAA)x–(MP–Si)y–RF (4 g dm−3) in the presence of AgOAc (40 mol), poly(methylhydrosiloxane) (240 mol), and TOA (240 mol) in toluene, (e) in the absence of the oligomer and (f) in the absence of both oligomer and TOA. | ||
In a variety of metal nanoparticles, preparations of coppernanoparticles, whose typical characteristics are due to exhibiting a plasmon absorption peak near 570 nm, are relatively difficult because they are easily oxidized, although coppernanoparticles are very attractive materials from the high potential applicable viewpoint in a variety of practical fields.118–132 Therefore, the preparation of stable coppernanoparticles is very important, and in fact, many efforts have been directed so far towards their chemical synthesis.133Copper(II) chloride reacted with hydrazine hydrate in the presence of fluorinated oligomeric aggregates formed by fluoroalkyl end-capped N,N-dimethylacrylamide oligomer to afford coppernanoparticles (Scheme 26).134 These copper nanocomposites, protected by fluorinated oligomeric aggregates, exhibited a plasmon absorption in the visible region (λmax: 602 nm); however, their colloidal stability is relatively poor.134
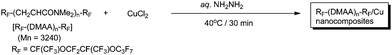 | ||
| Scheme 26 Preparation of fluoroalkyl end-capped N,N-dimethylacrylamide oligomer/copper nanocomposites. | ||
On the other hand, colloidal stable cross-linked fluoroalkyl end-capped cooligomer/copper nanocomposites were prepared by the reaction of copper(II) oxide with hydrazine in the presence of cross-linked fluoroalkyl end-capped cooligomers containing adamantyl segments.135 These fluorinated copper nanocomposites were stabilized by fluoroalkyl end-capped cooligomer containing benzotriazole segments, and these fluorinated copper nanocomposite powders were found to be stable for more than three months.135
Much attention has been paid to a practical application of zinc oxide nanoparticles in a variety of fields such as catalysts, gas sensors, semiconductors, varistors, piezoelectric devices, antibacterials and bactericides, field-emission displays and UV-shielding materials.136,137 It is of particular interest to develop new organic polymer/zinc oxide nanocomposites; however, we have some difficulties in isolating zinc oxide fine particles, because zinc oxide particles possess high surface energy, which might result in the agglomeration of particles when zinc oxide nanoparticles are dispersed in organic solvents and organic polymer matrices.136,137 A variety of fluoroalkyl end-capped oligomer/zinc oxide nanocomposites were prepared by the interaction of the corresponding oligomers with zinc oxide particles, which were obtained by the reaction of zinc acetate dihydrate with sodium hydroxide, in ethanol solutions at room temperature (Scheme 27).138 These fluorinated nanocomposites were applied to the surface modification of PMMA to exhibit not only a good surface active property imparted by fluorine but also unique characteristics such as surface electric conductivity around 10−11 S cm−1 level at room temperature and yellow-green luminescence under a UV lamp related to the presence of zinc oxide nanoparticles on the modified PMMA film surface (see Fig. 26).138
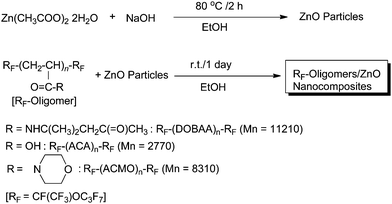 | ||
| Scheme 27 Preparation of fluoroalkyl end-capped oligomer/zinc oxide nanocomposites. | ||
 | ||
| Fig. 26 Photographs of the modified PMMA film treated with RF–(DOBAA)n–RF/ZnO nanocomposites (A) and the modified PMMA film treated with the RF–(DOBAA)n–RF oligomer (B). | ||
Similarly, novel fluoroalkyl end-capped oligomer/titanium dioxide nanocomposites were prepared by the hydrolysis of titanium isopropoxide in the presence of the fluoroalkyl end-capped N-(1,1-dimethyl-3-oxobutyl)acrylamide oligomer [RF–(DOBAA)n–RF], fluoroalkyl end-capped N,N-dimethylacrylamide oligomer [RF–(DMAA)n–RF], and fluoroalkyl end-capped acrylic acid oligomer [RF–(ACA)n–RF] in tetrahydrofuran under mild conditions.139 Among these fluorinated oligomers, the RF–(ACA)n–RF oligomer was more effective for the preparation of the corresponding oligomer/titanium dioxide nanocomposites.139 Fluoroalkyl end-capped oligomer/titanium dioxide nanocomposites were also applied to the surface modification of the common organic polymers such as poly(methyl methacrylate) to exhibit good oleophobic and hydrophilic characteristics on the surface.139
Fluoroalkyl end-capped vinyltrimethoxysilane oligomer caused the sol–gel reaction under alkaline conditions in the presence of titanium oxidenanoparticles in tetrahydrofuran to afford the corresponding fluorinated oligomer/titanium oxide nanocomposites [RF–(VM–SiO2)n–RF/TiO2] in excellent to moderate isolated yields (Scheme 28).140
 | ||
| Scheme 28 Preparation of fluoroalkyl end-capped vinyltrimethoxysilane oligomer/titanium oxide nanocomposites. | ||
These fluorinated composites thus obtained were nanometre size-controlled fine particles, and exhibited good dispersibility and stability in traditional organic solvents except for water. These fluorinated nanocomposites were applied to the surface modification of glass to exhibit not only a completely superhydrophobic characteristic (a water contact angle of 180°) with a non-wetting property against water droplets but also a good oleophobicity imparted by fluoroalkyl segments in the composites on their surface. Of particular interest, it was demonstrated that the wettability for water can be switched between superhydrophobicity and superhydrophilicity by alternation of ultraviolet (UV) irradiation and dark storage while keeping a good oleophobicity on the modified surface treated with the anatase-type titanium oxide composite.140
Hitherto, it is well-known that various transition-metal oxides, such as TiO2, ZnO, WO3, V2O5, and α-Fe2O3, exhibit photoinduced hydrophilicity as a result of UV irradiation.141–145 These metal oxide particles can exhibit UV-driven reversible switching behaviors between superhydrophobicity and superhydrophilicity. However, there have been no reports related to such switching behaviors with a good oleophobic characteristic, so far. In contrast, there have been some reports on the exploration of super-amphiphobic coating surfaces with fluorinated derivatives such as fluorinated monoalkyl phosphates,146,147 perfluoroalkyl methacrylic copolymer/silica composites,148 and fluorine-end-capped polyurethane.149 From this point of view, RF–(VM–SiO2)n–RF/TiO2 nanocomposites, especially, the anatase-type TiO2 nanocomposites are interesting fluorinated functional materials, because these nanocomposites can exhibit photoinduced hydrophilicity while keeping a good oleophobicity imparted by fluorine.
Calcium chloride reacted with sodium carbonate in the presence of a variety of self-assembled molecular aggregates formed by fluoroalkyl end-capped acrylic acid, 2-methacryloyloxyethanesulfonic acid, dimethylacrylamide, and acryloylmorpholine oligomers in aqueous solutions to afford the corresponding fluorinated oligomer/calcium carbonate composites in good isolated yields (Scheme 29).150 These fluorinated composites are nanometre size-controlled particles and well dispersed in aqueous and organic media.150
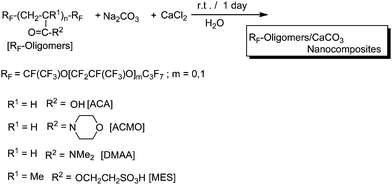 | ||
| Scheme 29 Preparation of fluoroalkyl end-capped oligomer/calcium carbonate nanocomposites. | ||
Cross-linked fluoroalkyl end-capped acrylic acid cooligomer containing poly(oxyethylene) units was also applied to the preparation of new cross-linked fluorinated calcium carbonate nanocomposites under similar conditions (see Scheme 30).150 The obtained cross-linked fluorinated calcium carbonate nanocomposites were found to have an extremely higher dispersibility in aqueous and organic media including fluorinated solvents, compared with that of the corresponding fluoroalkyl end-capped oligomeric nanocomposites. In particular, it was verified that these fluorinated calcium carbonate nanocomposites were applied to the dispersion of the above poly(methyl methacrylate) [PMMA] film surface. Field-emission SEM (FE-SEM) images of the cross-section of the modified PMMA films showed that calcium carbonate particles dispersed into these PMMA films could be arranged regularly above the modified film surface (see Fig. 27 and 28).
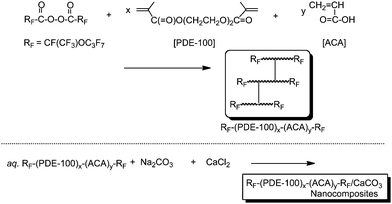 | ||
| Scheme 30 Preparation of cross-linked fluoroalkyl end-capped cooligomer/calcium carbonate nanocomposites. | ||
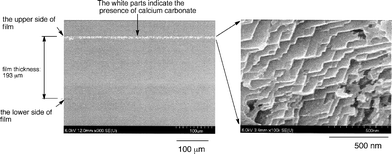 | ||
| Fig. 27 FE-SEM images of the cross-section of the modified PMMA films treated with RF–(PDE-100)x–(ACA)y–RF/CaCO3 nanocomposites. | ||
 | ||
| Fig. 28 FE-SEM images of the cross-section of the modified PMMA films treated with RF–(MES)n–RF/CaCO3 nanocomposites. | ||
Fluorinated oligomeric aggregates were able to provide suitable host moieties for the crystallization of calcium carbonate.150XRD analyses of the crystals formed in the cross-linked fluorinated aggregates show that calcite is mainly obtained. In contrast, interestingly, vaterite is mainly obtained in the case of the RF–(MES)n–RF oligomer [RF–(CH2CMeCO2CH2CH2SO3H)n–RF] nanocomposites.150
Self-assembled fluorinated molecular aggregates formed by fluoroalkyl end-capped oligomers were able to interact with nanodiamond powders to afford well-dispersed fluorinated oligomeric aggregate–diamond nanocomposites in organic media, and these fluorinated aggregate–diamond nanocomposites were applied to the surface modification of poly(methyl methacrylate) films.151
A variety of fluoroalkyl end-capped oligomers were applied to the preparation of nanometre-size controlled fluorinated oligomer/hydroxyapatite (HAp) composites, which exhibit a good dispersibility in water and traditional organic solvents.152,153 These fluoroalkyl end-capped oligomer/HAp nanocomposites were easily prepared by the reactions of disodium hydrogen phosphate and calcium chloride in the presence of self-assembled molecular aggregates formed by fluoroalkyl end-capped oligomers in aqueous solutions (Scheme 31).152,153
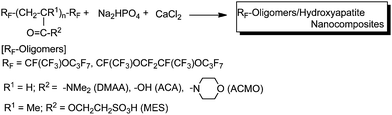 | ||
| Scheme 31 Preparation of nanometre-size controlled fluorinated oligomer/hydroxyapatite composites. | ||
These fluorinated oligomer/HAp nanocomposites were applied to the surface modification of glass and PVA [poly(vinyl alcohol)] to exhibit a good oleophobicity imparted by fluorine.153
New fluoroalkyl end-capped vinyltrimethoxysilane oligomer/HAp nanocomposites can be prepared by reaction of calcium nitrate tetrahydrate and phosphoric acid in the presence of the corresponding oligomer in good isolated yields under very mild conditions (Scheme 32).154 These fluorinated nanocomposites were also applied to the surface modification of glass and PMMA, and these modified glass and PMMA film surfaces exhibited good hydro- and oleo-phobic characteristics imparted by fluorine.154
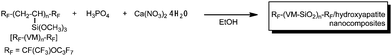 | ||
| Scheme 32 Preparation of fluoroalkyl end-capped vinyltrimethoxysilane oligomer/hydroxyapatite nanocomposites. | ||
HAp formation was newly observed on the modified PMMA film surface treated with RF–(VM–SiO2)n–RF/HAp nanocomposites by soaking this film into the simulated body fluid (SBF) (see Fig. 29).154 In this way, our new RF–(VM–SiO2)n–RF/HAp nanocomposites have high potential for new fluorinated functional materials through their unique properties imparted by both fluorine and HApnanoparticles, and are also expected to be applicable in a wide variety of fields including medical and biomaterials areas.
 | ||
| Fig. 29 SEM photographs of the modified PMMA film surface treated with RF–(VM–SiO2)n–RF/HAp nanocomposites before and after soaking in SBF at 37 °C for 7 days. | ||
7. Summary and outlook
The poor radical polymerizable characteristic of fluoroalkanoyl peroxide toward traditional radical polymerizable monomers enabled the preparation of fluoroalkyl end-capped oligomers and cross-linked fluoroalkyl end-capped oligomeric nanoparticles. Cross-linked fluorinated cooligomeric nanoparticles were applied to the encapsulation of magnetic nanoparticles and gold nanoparticles to afford the corresponding fluorinated nanoparticle/magnetite and/gold nanocomposites, respectively. These fluorinated nanocomposites were also applied to the surface modification of common organic polymers such as PMMA to exhibit not only the surface active characteristic imparted by fluoroalkyl groups but also unique properties related to the encapsulated guest molecules. Fluoroalkyl end-capped oligomers can form nanometre size-controlled molecular aggregates with the aggregation of end-capped fluoroalkyl groups in aqueous and organic media. These fluorinated molecular aggregates can afford suitable host moieties to interact with numerous guest molecules. Thus, a variety of fluorinated oligomer/guest molecule nanocomposites were developed from the interaction of fluorinated molecular aggregates with these guest molecules. In fact, fluorinated oligomer/silica nanocomposites and fluorinated oligomer/metal nanocomposites were prepared under mild conditions. Usual radical and living radical polymerization techniques are applicable for the preparation of fluorinated polymer/silica composites.42–47 These fluorinated polymer/silica composites thus obtained in general exhibit a clear weight loss characteristic corresponding to the contents of fluorinated polymers in the composites through the calcination process around 800 °C. This point is quite different from our present fluorinated oligomer/silica nanocomposites, and some fluorinated oligomers in the silica nanocomposite matrices can exhibit no weight loss behavior even after calcination at 800 °C. Fluorinated oligomers/gold, silver, copper, zinc oxide, titanium oxide, calcium carbonate, nanodiamond, and hydroxyapatite nanocomposites were found to exhibit not only a surface active characteristic imparted by fluoroalkyl groups but also unique properties related to these metal nanoparticles. Therefore, these fluorinated nanocomposites have high potential for novel fluorinated functional materials including their applications in a wide variety of fields.References
- M. Zanetti, S. Lomakin and G. Camino, Macromol. Mater. Eng., 2000, 279, 1–9 CrossRef CAS.
- P. M. Ajayan, L. S. Schadler and P. V. Braun, Nanocomposite Science and Technology, Wiley-VCH, Weinheim, 2003 Search PubMed.
- P. Gomez-Romero and C. Sanchez, Functional Hybrid Materials, Wiley-VCH, Weinheim, 2004 Search PubMed.
- G. He and Q. Pan, Macromol. Rapid Commun., 2004, 25, 1545–1548 CrossRef CAS.
- T. Basinska, S. Slomkowski, A. Dworak, I. Panchev and M. M. Chehimi, Colloid Polym. Sci., 2001, 279, 916–924 CAS.
- S. C. Yang, H. X. Ge, X. Q. Jiang and C. Z. Yang, Colloid Polym. Sci., 2000, 278, 285–292 CAS.
- T. Trimaille, C. Pichot, A. Elaissari, S. Briancon and T. Delair, Colloid Polym. Sci., 2003, 281, 1184–1190 CAS.
- N. Behan and C. Birkinshaw, Macromol. Rapid Commun., 2001, 22, 41–43 CrossRef CAS.
- K. Kauer, A. M. Imroz Ali and M. Sedlak, Colloid Polym. Sci., 2005, 283, 351–358 Search PubMed.
- C. Duan Vo, D. Kuckling, H.-J. P. Adler and M. Schonhoff, Colloid Polym. Sci., 2002, 280, 400–409 Search PubMed.
- L. M. Liz-Marzan, M. Giersig and P. Mulvaney, Langmuir, 1996, 12, 4329–4335 CrossRef CAS.
- M. Akashi, I. Kirikihira and N. Miyauchi, Angew. Makromol. Chem., 1985, 132, 81–89 CrossRef CAS.
- M. Akashi, T. Niikawa, T. Serizawa, T. Hayakawa and M. Baba, Bioconjugate Chem., 1998, 9, 50–53 CrossRef CAS.
- T. Hayakawa, M. Kawamura, M. Okamoto, M. Baba, T. Niikawa, S. Takehara, T. Serizawa and M. Akashi, J. Med. Virol., 1998, 56, 327–331 CrossRef CAS.
- M.-Q. Chen, T. Kaneko, M. Zhang, X.-Y. Liu, K. Wu and M. Akashi, Chem. Lett., 2003, 1138–1139 CrossRef CAS.
- T. Sanji, Y. Nakatsuka and H. Sakurai, Polym. J., 2005, 37, 1–6 CrossRef CAS.
- T. Serizawa, Y. Yanagisono, M. Ueno and M. Akashi, Polym. J., 2005, 37, 39–42 CrossRef CAS.
- T. Serizawa, S. Takehara and M. Akashi, Macromolecules, 2000, 33, 1759–1764 CrossRef CAS.
- Functional Hybrid Materials, ed. P. Gomez-Romero and C. Sanchez, Wiley-VCH, Weinheim, 2004 Search PubMed.
- P. Judeinstein and C. Sanchez, J. Mater. Chem., 1996, 6, 511–525 RSC.
- J. Wen and G. L. Wilkes, Chem. Mater., 1996, 8, 1667–1681 CrossRef CAS.
- C. J. Brinker and G. W. Scherer, Sol–Gel Science, Academic Press, Boston, 1990 Search PubMed.
- Z. Hua and K.-Y. Qiu, Polymer, 1997, 31, 521–526 Search PubMed.
- M. A. S. Pedroso, M. L. Dias, R. A. S. San Gil and C. G. Mothe, Colloid Polym. Sci., 2003, 281, 19–26 CAS.
- A. Bandyopadhyay, M. D. Sarkar and A. K. Bhowmick, J. Polym. Sci., Part B: Polym. Phys., 2005, 43, 2399–2412 CrossRef CAS.
- S. Li, Z. Zhou, H. Abernathy, M. Liu, W. Li, J. Ukai, K. Hase and M. Nakanishi, J. Mater. Chem., 2006, 16, 858–864 RSC.
- P. Hajii, L. David, J. F. Gerard, J. P. Pascault and G. Vigier, J. Polym. Sci., Part B: Polym. Phys., 1999, 37, 3172–3187 CrossRef.
- O. Prucker and J. Ruhe, Macromolecules, 1998, 31, 592–601 CrossRef CAS.
- B. De Boer, H. K. Simon, M. P. L. Werts, E. W. van Der Vegte and G. Hadziioannou, Macromolecules, 2000, 33, 349–356 CrossRef CAS.
- B. Zhao and W. J. Brittain, J. Am. Chem. Soc., 1999, 121, 3557–3558 CrossRef CAS.
- R. Jordan, A. Ulma, J. F. Kang, M. H. Rafailovich and J. Sokolov, J. Am. Chem. Soc., 1999, 121, 1016–1022 CrossRef CAS.
- S.-W. Zhang, S.-X. Zhou, Y.-M. Weng and L.-M. Wu, Langmuir, 2005, 21, 2124–2128 CrossRef CAS.
- R. Ranjan and W. J. Brittain, Macromol. Rapid Commun., 2008, 29, 1104–1110 CrossRef CAS.
- Y. Li and B. C. Benicewicz, Macromolecules, 2008, 41, 7986–7992 CrossRef CAS.
- C.-H. Liu and C.-Y. Pan, Polymer, 2007, 48, 3679–3685 CrossRef CAS.
- D. H. Nguyen and P. Vana, Polym. Adv. Technol., 2006, 17, 625–633 CrossRef CAS.
- C. Bartholome, E. Beyou, E. Bourgeat-Lami, P. Chumont and N. Zydowicz, Macromolecules, 2003, 36, 7946–7952 CrossRef CAS.
- M. Husseman, E. E. Maalmstrom, M. McNamara, M. Mate, D. Mecerreyes, D. G. Benoit, J. L. Hedrick, P. Mansky, E. Huang, T. P. Russell and C. J. Hawker, Macromolecules, 1999, 32, 1424–1431 CrossRef CAS.
- K. Ueno, A. Inaba, M. Kondoh and M. Watanabe, Langmuir, 2008, 24, 5253–5259 CrossRef CAS.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS.
- S.-W. Zhang, S.-X. Zhou, Y.-M. Weng and L.-M. Wu, Langmuir, 2005, 21, 2124–2128 CrossRef CAS.
- Y.-L. Liu and S.-H. Li, Macromol. Rapid Commun., 2004, 25, 1392–1395 CrossRef CAS.
- G. Schmid, Nanoparticles, Wiley-VCH, Weinheim, 2004 Search PubMed.
- L. Matejka and J. Plestil, Macromol. Symp., 1997, 122, 191–196 CrossRef CAS.
- N. Juangvanich and K. A. Maurite, J. Appl. Polym. Sci., 1998, 67, 1799 CrossRef CAS.
- A. M. Granville and W. J. Brittain, Macromol. Rapid Commun., 2004, 25, 1298–1302 CrossRef CAS.
- L. Andruzzi, A. Hexemer, X. Li, C. K. Ober, E. J. Krmaer, G. Galli, E. Chiellini and D. A. Fischer, Langmuir, 2004, 20, 10498–10506 CrossRef CAS.
- H. Sawada, Chem. Rev., 1996, 96, 1779–1808 CrossRef CAS.
- H. Sawada, J. Fluorine Chem., 2000, 105, 219–220 CrossRef CAS.
- H. Sawada, Prog. Polym. Sci., 2007, 32, 509–533 CrossRef CAS.
- H. Sawada, Polym. J., 2007, 39, 637–650 CrossRef CAS.
- H. Sawada, N. Itoh, T. Kawase, M. Mitani, H. Nakajima, M. Nishida and Y. Moriya, Langmuir, 1994, 10, 994–995 CrossRef CAS.
- J. Nakagawa, K. Kamogawa, H. Sakai, T. Kawase, H. Sawada, J. Manosroi, A. Manosroi and M. Abe, Langmuir, 1998, 14, 2055–2060 CrossRef CAS.
- J. Nakagawa, K. Kamogawa, N. Momozawa, H. Sakai, T. Kawase, H. Sawada, Y. Sano and M. Abe, Langmuir, 1998, 14, 2061–2067 CrossRef CAS.
- H. Sawada, K. Ikeno and T. Kawase, Macromolecules, 2002, 35, 4306–4313 CrossRef CAS.
- H. Sawada, T. Narumi, A. Kajiwara, K. Ueno and K. Hamazaki, Colloid Polym. Sci., 2006, 284, 551–555 CAS.
- B. Ameduri and B. Boutevin, Well-Architectured Fluoropolymers: Synthesis, Properties and Applications, Elsevier, Amsterdam, 2004, pp. 231–348 Search PubMed.
- J.-F. Berret, D. Calvet, A. Collet and M. Viguier, Curr. Opin. Colloid Interface Sci., 2003, 8, 296–306 CrossRef CAS.
- T. Imae, Curr. Opin. Colloid Interface Sci., 2003, 8, 307–314 CrossRef CAS.
- T. Imae, H. Tabuchi, K. Funayama, A. Sato, T. Nakamura and N. Amaya, Colloids Surf., A, 2000, 167, 73–81 CrossRef CAS.
- P. Lebreton, B. Ameduri, B. Boutevin and J. M. Corpart, Macromol. Chem. Phys., 2002, 203, 522–537 CrossRef CAS.
- M. Destarac, D. Charmot, S. Zard and X. Franck, WO 00/75207 A1, 2000.
- M. J. Monteiro, M. M. Adamy, B. J. Leeuwen, A. M. Van Herk and M. Destarac, Macromolecules, 2005, 38, 1538–1541 CrossRef CAS.
- A. E. Feiring, E. R. Wonchoba, F. Davdson, V. Percec and B. Barboiu, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 3313–3335 CrossRef CAS.
- M. Destarac, K. Matyjaszewski, E. Silverman, B. Ameduri and B. Boutevin, Macromolecules, 2000, 33, 4613–4615 CrossRef CAS.
- Z. Shi and S. Holdcroft, Macromolecules, 2005, 38, 4193–4201 CrossRef CAS.
- K. Jankova and S. Hvilsted, J. Fluorine Chem., 2005, 126, 241–250 CrossRef CAS.
- H. Sawada, Y.-F. Gong, Y. Minoshima, T. Matsumoto, M. Nakayama, M. Kosugi and T. Migita, J. Chem. Soc., Chem. Commun., 1992, 537–538 RSC.
- H. Sawada, Y. Minoshima and H. Nakajima, J. Fluorine Chem., 1992, 65, 169–173 CrossRef.
- D. M. Young and W. N. Stoops, US Pat., 2792423, 1957.
- O. H. Bullitt, US Pat., 2559630, 1951.
- H. Sawada, J. Fluorine Chem., 2003, 121, 111–130 CrossRef CAS.
- H. Sawada, K. Tanba, N. Itoh, C. Hosoi, M. Oue, M. Baba, T. Kawase, M. Mitani and H. Nakajima, J. Fluorine Chem., 1996, 77, 51–64 CrossRef CAS.
- H. Yoshioka, T. Narumi and H. Sawada, J. Oleo Sci., 2007, 56, 377–383 CrossRef CAS.
- M. Mugisawa, K. Ueno, K. Hamazaki and H. Sawada, Macromol. Rapid Commun., 2007, 28, 733–739 CrossRef CAS.
- H. Sawada, T. Kijima and M. Mugisawa, Polym. J., 2010, 42, 494–500 CrossRef CAS.
- M. Mugisawa, R. Kasai and H. Sawada, Langmuir, 2009, 25, 415–421 CrossRef CAS.
- M. Seno, M. Hasegawa, T. Hirano and T. Sato, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5864–5871 CrossRef CAS.
- H. Sawada, M. Suzuki and M. Mugisawa, Polym. Adv. Technol., 2006, 17, 66–69 CrossRef CAS.
- H. Sawada and M. Nakayama, J. Chem. Soc., Chem. Commun., 1991, 677–678 RSC.
- H. Yoshioka, M. Suzuki, M. Mugisawa, N. Naitoh and H. Sawada, J. Colloid Interface Sci., 2007, 308, 4–10 CrossRef CAS.
- H. Sawada, J. Iidzuka, T. Maekawa, R. Takahashi, T. Kawase, K. Oharu, H. Nakagawa and K. Ohira, J. Colloid Interface Sci., 2003, 263, 1–3 CrossRef CAS.
- M. Mugisawa, K. Ohnishi and H. Sawada, Langmuir, 2007, 23, 5848–5851 CrossRef CAS.
- M. D. C. Topp, P. J. Dijksta, H. Talsma and J. Feijin, Macromolecules, 1997, 30, 8518–8520 CrossRef CAS.
- H.-H. Lin and Y.-L. Cheng, Macromolecules, 2001, 34, 3710–3715 CrossRef CAS.
- H. Sawada, T. Suzuki, H. Takashima and K. Takishita, Colloid Polym. Sci., 2008, 286, 1569–1574 CAS.
- H. Sawada, Y. Goto and T. Narumi, Bull. Chem. Soc. Jpn., 2010, 83, 82–91 CrossRef CAS.
- H. Sawada, T. Narumi, M. Kiyohara and M. Baba, J. Fluorine Chem., 2007, 128, 1416–1420 CrossRef CAS.
- B. Ameduri, Chem. Rev., 2009, 109, 6632–6682 CrossRef CAS.
- B. Ameduri and B. Boutevin, J. Fluorine Chem., 2000, 104, 53–62 CrossRef CAS.
- Y.-C. Chen, C.-C. Tsai and Y.-D. Lee, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 1789–1807 CrossRef CAS.
- H. Sawada, T. Narumi, S. Kodama, M. Kamijo, R. Ebara, M. Sugiya and Y. Iwasaki, Colloid Polym. Sci., 2007, 285, 977–983 CAS.
- H. Sawada, T. Tashima and S. Kodama, Polym. Adv. Technol., 2008, 19, 739–747 CrossRef CAS.
- H. Sawada, T. Tashima, H. Kakehi, Y. Nishiyama, M. Kikuchi, M. Miura, Y. Sato and N. Isu, Polym. J., 2010, 42, 167–171 CrossRef CAS.
- H. Sawada, H. Kakehi, T. Tashima, Y. Nishiyama, M. Miura and N. Isu, J. Appl. Polym. Sci., 2009, 112, 3482–3487 CrossRef CAS.
- H. Sawada, T. Tashima, Y. Nishiyama, M. Kikuchi, G. Kostov, Y. Goto and B. Ameduri, Macromolecules, 2011, 44, 1114–1124 CrossRef CAS.
- H. Sawada, M. Kikuchi and M. Nishida, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1070–1078 CrossRef CAS.
- H. Sawada, X. Liu, Y. Goto, M. Kikuchi, T. Tashima and M. Nishida, J. Colloid Interface Sci., 2011, 356, 8–15 CrossRef CAS.
- H. Sawada, Y. Matsuki, Y. Goto, S. Kodama, M. Sugiya and Y. Nishiyama, Bull. Chem. Soc. Jpn., 2010, 83, 75–81 CrossRef CAS.
- Y. Sun and Y. Xia, Science, 2002, 298, 2176–2179 CrossRef CAS.
- M. A. Van Dijk, M. Lippitz and M. Orrit, Acc. Chem. Res., 2005, 38, 594–601 CrossRef CAS.
- Colloids and Colloid Assemblies, ed. F. Caruso, Wile-VCH, Weinheim, 2004 Search PubMed.
- S. Forster and M. Antonietti, Adv. Mater., 1998, 10, 195–217 CrossRef.
- C. W. Chen, T. Serizawa and M. Akashi, Chem. Mater., 2002, 14, 2232–2239 CrossRef CAS.
- S. Pothukuchi, Y. Li and C. P. Wong, J. Appl. Polym. Sci., 2004, 93, 1531–1538 CrossRef CAS.
- H. Sawada, A. Sasaki, K. Sasazawa, T. Kawase, K. Ueno and K. Hamazaki, Colloid Polym. Sci., 2005, 283, 583–586 CAS.
- H. Sawada, A. Sasaki and K. Sasazawa, Colloids Surf., A, 2009, 337, 57–60 CrossRef CAS.
- M. Mugisawa and H. Sawada, Langmuir, 2008, 24, 9215–9218 CrossRef CAS.
- H. Sawada, K. Takahashi, M. Mugisawa, T. Oya and S. Ogino, Langmuir, 2007, 23, 11947–11950 CrossRef CAS.
- H. Sawada and K. Takahashi, J. Colloid Interface Sci., 2010, 351, 166–170 CrossRef CAS.
- J.-H. Kim and T. R. Lee, Drug Dev. Res., 2006, 67, 61–69 CrossRef CAS.
- X. Jiang, D. Xiong, Y. An, P. Zheng, W. Zhang and L. Shi, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 2812–2819 CrossRef CAS.
- X. Zhao, X. Ding, Z. Deng, Z. Zheng, Y. Peng and X. Long, Macromol. Rapid Commun., 2005, 26, 1784–1787 CrossRef CAS.
- V. Pardo-Yissar, R. Gabai, A. N. Shipway, T. Bourenko and I. Willner, Adv. Mater., 2001, 13, 1320–1323 CrossRef CAS.
- C. Wang, N. T. Flynn and R. Langer, Adv. Mater., 2004, 16, 1074–1079 CrossRef CAS.
- B. Jing, X. Chen, X. Wang, Y. Zhao and H. Qiu, ChemPhysChem, 2009, 9, 249–252 CrossRef.
- H. Sawada, A. Sasaki, K. Sasazawa, K. Toriba, H. Kakehi, M. Miura and N. Isu, Polym. Adv. Technol., 2008, 19, 419–424 CrossRef CAS.
- X. Wang, W. Liu, F. Yan, Z. Zhang and B. Xu, Chem. Lett., 2004, 196–197 CrossRef CAS.
- H.-T. Zhu, Y.-S. Lin and Y.-S. Yin, J. Colloid Interface Sci., 2004, 277, 100–103 CrossRef CAS.
- S. Kapoor and T. Mukherjee, Chem. Phys. Lett., 2003, 370, 83–87 CrossRef CAS.
- A. N. Pestryakov, V. P. Petranovskii, A. Kryazhov, O. Ozhereliev, N. Pfander and A. Knop-Gericke, Chem. Phys. Lett., 2004, 385, 173–176 CrossRef CAS.
- I. Lisiecki, F. Billoudet and M. P. Pileni, J. Mol. Liq., 1997, 72, 251–261 CrossRef CAS.
- A. G. Nasibulin, P. P. Ahonen, O. Richard and E. I. Kauppinen, J. Aerosol Sci., 2000, 31, S552–S553 CrossRef.
- S. Illy-Cherrey, O. Tillement, J. M. Dubois, F. Massicot, Y. Fort, J. Ghanbaja and S. Begin-Colin, Mater. Sci. Eng., A, 2002, 338, 70–75 CrossRef.
- J. Hambrock, R. Becker, A. Birkner, J. Weiss and R. A. Fischer, Chem. Commun., 2002, 68–69 RSC.
- X. Somg, S. Sun, W. Zhang and Z. Yin, J. Colloid Interface Sci., 2004, 273, 463–469 CrossRef.
- M. P. Pileni and I. Lisiecki, Colloids Surf., A, 1993, 80, 63–68 CrossRef CAS.
- S. Kapoor, D. K. Palit and T. Mukherjee, Chem. Phys. Lett., 2002, 355, 383–387 CrossRef CAS.
- S.-Y. Xie, Z.-J. Ma, C.-F. Wang, S.-C. Lin, Z.-Y. Jiang, R.-B. Huang and L.-S. Zheng, J. Solid State Chem., 2004, 177, 3743–3747 CrossRef CAS.
- L. Qi, J. Ma and J. Shen, J. Colloid Interface Sci., 1997, 186, 498–500 CrossRef CAS.
- I. Lisiecki and M. P. Pileni, J. Am. Chem. Soc., 1993, 115, 3887–3896 CrossRef CAS.
- H. Wang, Y. Huang, Z. Tan and X. Hu, Anal. Chim. Acta, 2004, 526, 13–17 CrossRef CAS.
- A. Nandi, M. D. Gupita and A. A. K. Banthia, Colloids Surf., A, 2002, 197, 119–124 CrossRef CAS.
- H. Sawada, R. Furukuwa, K. Sasazawa, K. Toriba, K. Ueno and K. Hamazaki, J. Appl. Polym. Sci., 2006, 100, 1328–1334 CrossRef CAS.
- H. Sawada, R. Furukuwa, K. Sasazawa, M. Mugisawa and K. Ohnishi, Eur. Polym. J., 2007, 43, 3258–3263 CrossRef CAS.
- T. K. Gupita, J. Am. Ceram. Soc., 1990, 73, 1817–1840 CrossRef.
- D. Wang and C. Somng, J. Phys. Chem. B, 2005, 109, 12697–12700 CrossRef CAS.
- K. Sasazawa, Y. Hirayama and H. Sawada, Polym. Int., 2008, 58, 177–182 CrossRef.
- H. Sawada, E. Sawada, H. Kakehi, T. Kariya, M. Mugisawa, Y. Chounan, M. Miura and N. Isu, Polym. Compos., 2009, 30, 1848–1853 CrossRef CAS.
- E. Sawada, H. Kakehi, Y. Chounan, M. Miura, Y. Sato, N. Isu and H. Sawada, Composites, Part B, 2010, 41, 498–502 CrossRef.
- R. Wang, K. Hashimoto, A. Fujishima, M. Chikuni, E. Kojima, A. Kitamura, M. Shimohigoshi and T. Watanabe, Adv. Mater., 1998, 10, 135–138 CrossRef CAS.
- X. Feng, L. Feng, M. Jin, J. Zhai, L. Jiang and D. Zhu, J. Am. Chem. Soc., 2004, 126, 62–63 CrossRef CAS.
- H. S. Lim, D. Kwak, D. Y. Lee, S. G. Lee and K. Cho, J. Am. Chem. Soc., 2007, 129, 4128–4129 CrossRef CAS.
- B. Yan, J. Tao, C. Pamg, Z. Zheng, Z. Shen, C. H. A. Huan and T. Yu, Langmuir, 2008, 24, 10569–10571 CrossRef CAS.
- S. Wang, X. Feng, J. Yao and L. Jiang, Angew. Chem., Int. Ed., 2006, 45, 1264–1267 CrossRef CAS.
- K. Tsujii, T. Yamamoto, T. Onda and S. Shibuichi, Angew. Chem., Int. Ed. Engl., 1997, 36, 1011–1012 CrossRef CAS.
- S. Shibuichi, T. Yamamoto, T. Onda and K. Tsujii, J. Colloid Interface Sci., 1998, 208, 287–294 CrossRef CAS.
- C.-T. Hsieh, F.-L. Wu and W.-Y. Chen, Surf. Coat. Technol., 2009, 203, 3377–3384 CrossRef CAS.
- Q. Xie, J. Xu, L. Feng, W. Tang, X. Luo and C. C. Han, Adv. Mater., 2004, 16, 302–305 CrossRef CAS.
- H. Sawada, Y. Shikauchi, H. Kakehi, Y. Katoh and M. Miura, Colloid Polym. Sci., 2007, 285, 499–506 CAS.
- H. Sawada, J. Kurachi, H. Takahashi, K. Ueno and K. Hamazaki, Polym. Adv. Technol., 2005, 16, 651–654 CrossRef CAS.
- H. Sawada, H. Takashima, K. Iwaki, R. Furukuwa and K. Takishita, Macromol. Mater. Eng., 2007, 292, 403–406 CrossRef CAS.
- H. Takashima, K. Iwaki, R. Furukuwa, K. Takishita and H. Sawada, J. Colloid Interface Sci., 2008, 320, 436–444 CrossRef CAS.
- H. Takashima, K. Iwaki, K. Takishita and H. Sawada, Polym. Adv. Technol., 2009, 20, 887–891 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |