Constructing star polymersvia modular ligation strategies
Ozcan
Altintas
a,
Andrew P.
Vogt
a,
Christopher
Barner-Kowollik
*a and
Umit
Tunca
*b
aPreparative Macromolecular Chemistry, Institut für Technische Chemie und Polymerchemie, Karlsruhe Institute of Technology (KIT), Engesserstr. 18, 76128, Karlsruhe, Germany. E-mail: christopher.barner-kowollik@kit.edu; Web: www.macroarc.de Fax: +49 721 608-45740; Tel: +49 721 608-45741
bDepartment of Chemistry, Istanbul Technical University, Maslak, Istanbul, 34469, Turkey. E-mail: tuncau@itu.edu.tr; Web: www.kimya.itu.edu.tr/polimer Fax: +90 212 285 63 86; Tel: +90 212 285 60 48
First published on 10th August 2011
Abstract
Branched polymers result in a more compact structure in comparison to linear polymers of identical molecular weight, due to their high segment density which affects the crystalline, mechanical, and viscoelastic properties of the polymer. Star polymers constitute the simplest form of branched macromolecules where all of the chains—or arm segments—of one macromolecule are linked to a centre defined as the core. Over recent years, modular ligation reactions—some of which adhere to click criteria—have enabled the synthesis of a variety of star polymersvia efficient polymer–polymerconjugations. While the modified Huisgen [3 + 2] dipolar copper catalyzed azide and alkynecycloaddition (CuAAC) has been widely employed for macromolecular star synthesis, Diels–Alder and hetero Diels–Alder reactions offer alternative pathways which allow for similarly efficient macromolecular conjugations. Moreover, combinations of these protocols afford the synthesis of more complex star polymer structures which previously had not been achievable.
 Ozcan Altintas | Ozcan Altintas obtained his BSc in Chemistry from Istanbul Technical University (ITU), Turkey in June 2004. In February 2006, he received his Master degree in Polymer Science and Technology at the ITU. Since July 2009, he has been performing his PhD research working on self-assembly of single polymer chains under the supervision of Christopher Barner-Kowollik at the Karlsruhe Institute of Technology in Karlsruhe, Germany. His main research interests include controlled/living polymerizations, modular ligation reactions, supramolecular chemistry and conducting polymers. |
 Andrew P. Vogt | Andrew P. Vogt received his PhD in Chemistry from Southern Methodist University in Dallas, TX, under the direction of Prof. Brent S. Sumerlin. His dissertation research focused on combining controlled radical polymerization (CRP) and click chemistry techniques for the creation of well-defined polymeric materials. Dr Vogt then undertook a postdoctoral research fellow position in the Centre for Nanoscale Science and Technology in the School of Chemistry and Physics at Flinders University in Adelaide, Australia. Currently, Dr Vogt is a postdoctoral research fellow under the advisement of Prof. Christopher Barner-Kowollik at the Karlsruhe Institute of Technology in Karlsruhe, Germany. |
 Christopher Barner-Kowollik | Christopher Barner-Kowollik completed a PhD in Physical Chemistry at the University of Göttingen, before joining the Centre for Advanced Macromolecular Design at the University of New South Wales, where he led a research team as Full Professor, after holding ranks from research associate to associate professor. He is currently Full Professor at the Karlsruhe Institute of Technology. His main research interests range from the synthesis of complex macromolecular architectures, their applications and material properties, the development of polymer and surface conjugation as well as polymerization protocols, mass spectrometry on polymer systems coupled with chromatographic techniques to polymer reaction kinetics. |
 Umit Tunca | Umit Tunca completed his PhD under the supervision of Prof. Yagci in 1990. Since 1998, he has been a professor at Chemistry Department of Istanbul Technical University, Turkey. He has published over 80 papers and 1 book chapter. His research interests are mainly living radical polymerizations and modular ligation reactions. His awards include Monbusho (Japan) (1987) and Alexander von Humboldt (Germany) (1990) research fellowships, as well as the TUBITAK (The Scientific and Technological Research Council of Turkey) Young Investigator Award (1997). |
1. Introduction
Polymer properties are mainly influenced by the chemical composition and topology of the constituting macromolecules. Therefore, the synthesis of complex macromolecular architectures to control the polymer properties is a key field of study in polymer science.The degree of branching of polymers is a useful structural variable that can be exploited in the presence of various functionalities. Star polymer architectures can modify the polymers' physical and processing characteristics by varying its melt, solution, and solid-state properties.1,2 It has been shown that branching results in a more compact structure in comparison to linear polymers of identical molecular weight, due to their high segment density, which affects the crystalline, mechanical, and viscoelastic properties of the polymer.
A branched polymer structure is described as a nonlinear polymer with more than one backbone chain radiating from branch points (junction points such as atoms or functional groups from which more than two polymer chains emanate).2–6 Star polymers constitute the simplest form of branched macromolecules in which all the polymer chains (or arm segments) of one macromolecule are linked to a central core.
A key aim of polymer chemistry is to prepare polymers with well-defined properties via the precise control of molecular weight, polydispersity, composition, and topology. Highly successful synthetic strategies to enable control over both structure and functionality include living anionic and cationic polymerization processes.3–6
In the last decade, based on significant advances in living radical polymerization (LRP) techniques7 such as atom transfer radical polymerization (ATRP), nitroxide mediated radical polymerization (NMP), and reversible addition fragmentation chain transfer (RAFT) polymerization, the synthesis of polymers with complex architectures and predetermined chemical composition was significantly simplified and has received increased attention due to the exceptional variety of applicable monomers and tolerance to experimental conditions in comparison with living ionic polymerization techniques.8
The click chemistry philosophy—introduced by Sharpless and coworkers in 2001—comprises chemical reactions which display high stereo- and regio-selectivity, high yields, simple recovery of the main product, mild reaction conditions, compatibility in a wide range of solvents and a tolerance to a wide range of functional groups.9 This conjugation chemistry concept has been adapted to a vast variety of applications10 and has possibly led to a ‘paradigm shift’11 toward the modular construction approach within polymer chemistry. This ‘paradigm shift’ is due in part to the applicability of the click chemistry concept to prepare macromolecular structures that would have otherwise not been achievable by such simple quantitative methods, and has opened up a particularly rapid and efficient synthetic route to functional materials with predefined polymeric architectures.11 Recently, Barner-Kowollik et al. refined the requirements that a click reaction should fulfill within synthetic polymer chemistry in adaptation of the original definitions by Sharpless and co-workers.12 These refined requirements include the use of equimolar amounts of the polymeric building blocks, a simple large-scale purification process, high yields, reasonable time scales, and mild reaction conditions.12 It is important to note that the term ‘click chemistry’ has been employed in the literature in an inflationary fashion and reactions have been included under the click definition, which definitely do not fall under its strict criteria. Thus, we will adopt a cautious approach herein when denoting a reaction as the click-type.
2. Modular ligation reactions that can fulfill the click criteria
The most prominent modular ligation reactions widely adaptable to polymer chemistry include the Cu(I) catalyzed azide–alkynecycloaddition (CuAAC), Diels–Alder, hetero Diels–Alder, thiol–ene, thiol–yne, and the nitroxide radical coupling (NRC) reactions (Scheme 1).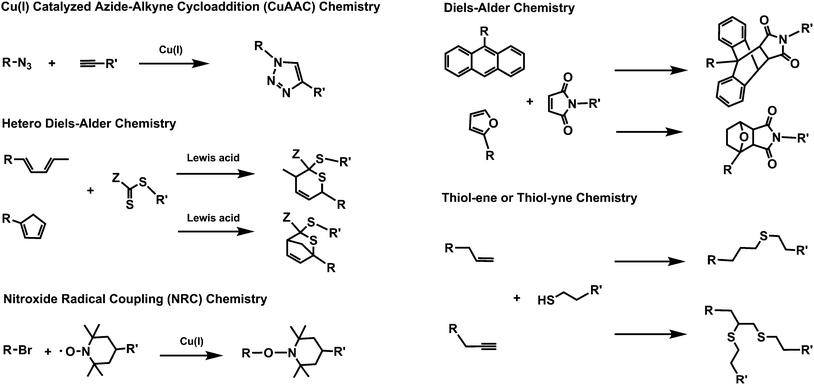 | ||
| Scheme 1 Schematic representation of modular ligation reactions, which can adhere to click criteria, that have been used for the preparation of complex macromolecular architectures. | ||
2.1 Cu(I) catalyzed azide–alkynecycloaddition (CuAAC)
The CuAAC reaction is a modified Huisgen 1,3-dipolar [3 + 2] cycloaddition, where the presence of a coppercatalyst affords a quantitative 1,4 triazole product which proceeds at non-demanding reaction conditions.13 Here, the [3 + 2] notation describes the number of atoms in the two components involved in the 1,3-dipolar cycloaddition. Huisgen type reactions are exergonic processes, which combine two (e.g.azide and alkyne) unsaturated reactants and produce a racemic mixture of 1,4- and 1,5-disubstituted-1,2,3-triazoles, whereas the CuAAC reaction is highly regioselective and yields only a 1,4-disubstituted-1,2,3-triazole. The CuAAC reaction was first adapted14 to the field of polymer chemistry due of its often quantitative yields, mild reaction condition, tolerance to a wide range of functional groups, and harmony with the LRP techniques.10 In many circumstances, the CuAAC process fulfills the click criteria. Among the LRP techniques utilized in conjunction with the CuAAC, ATRP shares a number of important features with the CuAAC reaction including robustness, tolerance to many functional groups, and the same catalytic system. Most significantly, however, ATRP and the CuAAC constitute a beneficial combined/tandem synthetic route due in part to ATRP offering well-defined polymer chains with chlorine or bromine end-groups which may consequently be converted efficiently to an azide using a classic substitution reaction.102.2 Diels–Alder and hetero Diels–Alder reactions
The Diels–Alder [4 + 2] cycloaddition involves the reaction of a conjugated diene (4π) with a dieneophile (2π) to yield a 6-membered ring, where the [4 + 2] denotes the number of π-electrons that are involved in the cycloaddition process.15 The most encountered diene/dienophile functionalities extensively employed in synthetic polymer chemistry are anthracene/maleimide, butadiene or cyclopentadiene/electron deficient dithioesters, and furan/maleimide. The anthracene–maleimidecycloaddition reactions are conducted at high temperatures, commonly 125 °C. Additionally, furan must be used as a protecting agent for the maleimide to avoid a possible radical polymerization during the preparation of maleimide end-functionalized polymeric precursors when used in conjunction with LRP techniques.16 A more attractive cycloaddition occurs with a diene and dithioester in a hetero Diels–Alder (HDA) process,17 which is carried out at relatively lower temperatures of 50 °C for butadiene or ambient temperature for cyclopentadiene.18 Compared to the anthracene–maleimide Diels–Alder cycloaddition, the dithioester end-functionality may be introduced into the polymer backbone via the RAFT process. The obtained adducts are not thermally stable over 80 °C for cyclopentadiene/dithioester and over 120 °C for furan/maleimide, limiting the applicability of the resulting polymers at high temperatures.19 However, the same instability conversely enables the design of macromolecular structures with reversible thermoresponsive properties.20,99 Just as with the CuAAC process, some of the above noted (H)DA systems fulfill the polymer ligation click criteria.212.3 Thiol–ene and thiol–yne reactions
Thiol–ene or thiol–yne radical addition reactions are based on the free radical reaction between a thiol and an alkene or an alkyne by using a thermal or photochemical process resulting in thioether products with high degree of anti-Markovnikov selectivity.22,23 Additionally, it has been shown that the thiol–ene reaction can be catalyzed by employing a primary amine,24 which is advantageous when used in conjunction with a RAFT precursor because the primary amine both aminolyzes the RAFT agent and catalyzes the thiolene-Michael type reaction. Commonly, the thiolene-Michael addition is used to conjugate thiols to an acrylate or a maleimide, yielding thioether–ester or thioether–imide functionalized adducts, respectively.25 In a thiol–yne reaction the alkyne reacts first with a thiol to result in a vinyl sulfide followed by a subsequent reaction of the vinyl sulfide with another thiol to yield the 1,2-disubstituted thioether adduct.22,23 However, the thiol–ene and thiol–yne reactions have some limitations for polymer–polymerconjugation due to disulfide coupling, which has resulted in research exhibiting a higher efficiency for thiol–ene reactions ligating small organic molecules to polymers.26 Importantly, there exists significant doubt whether the radical version of thiol–ene reaction can be classified as a click reaction, due to the occurrence of radical–radical coupling reactions during the ligation process.262.4 Nitroxide radical coupling (NRC) reaction
The NRC reaction proceeds between a halide- and 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO)-terminated polymers in the presence of a CuBr/ligand complex at elevated temperatures based on an ATRP type mechanism.27 Huang and coworkers first applied the NRC reaction for the preparation of linear triblock terpolymers together with the CuAAC reaction.27 Later, the NRC process was carried out—according to the authors—under single electron transfer living radical polymerization (SET-LRP) conditions.28 In the SET process, Cu(0) (equimolar) transfers an electron to a halogen-terminated polymer to provide a macroradical and Cu(I) at ambient temperature via a SET-LRP mechanism, in which the macroradical is then efficiently trapped by a TEMPO-terminated polymer. The modular nature of the NRC reaction was proven in the preparation of well-defined polymers with different topologies such as block, star, and graft copolymers.29Although several reviews have described the widespread use of modular ligation reactions throughout the preparation of polymers with different topologies, there has been no distinct critical collation focusing only on the preparation of star polymers employing modular ligation reactions.10,22,23 Therefore, the aim of the current compilation is to review the various conjugation strategies employed to generate star polymers.
3. Syntheses of star polymers in general
Star polymers may be classified into two categories based on the chemical composition of the arm species, being a homoarm (regular) star polymer or miktoarm (heteroarm) star polymer (Fig. 1).4–6Homoarm star polymers consist of a symmetric structure comprising radiating arms with similar molecular weight and identical chemical composition. Within this classification, star block copolymers may be defined as homoarm star polymers, in which each is constituted of identical copolymers (diblock, triblock, etc.). In contrast, a miktoarm star polymer contains two or more arm species with different chemical compositions and/or molecular weights.30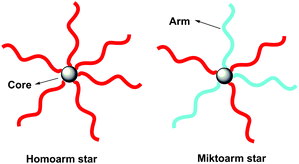 | ||
| Fig. 1 Schematic representation of star polymer structures. | ||
Star polymers are usually synthesized via one of three common methodologies, i.e. core-first, arm-first, and coupling-onto.31 These three synthetic methods differ from each other based on the sequence employed for the formation of the core and the arms (Fig. 2). In the “core-first” approach (divergent), multifunctional initiators are used to grow arms vialiving polymerization techniques. The “arm-first” approach (convergent) is carried out by linking telechelic preformed linear arm precursors commonly using a divinyl compound to form the final star polymer with a highly cross-linked core. In the “coupling-onto” methodology, linear arm precursors with reactive functional end-groups are attached via ligation reactions onto a multifunctional core to create a star polymer. Although these methodologies provide a powerful tool to produce star polymers, a number of challenges remain regarding the structural homogeneity, purity, and molecular weight distribution of star molecules. Star–star coupling is the main reason for broad molecular weight distribution in the “core-first” method due to the high number of initiating sites with propensity for radical–radical recombination; therefore, “core-first” star polymerizations are usually limited to low monomer conversion (<20%) to avoid star–star radical coupling. Moreover, in the “arm-first” approach, poor structural homogeneity and broad molecular weight distribution arises from the result of a random distribution of arms per molecule during the cross-linking reaction of the core. The main problem encountered in the “coupling-onto” methodology is commonly inefficient coupling reactions between the polymer chains and the multifunctional core. Fortunately, the developing modular ligation strategies have emerged as a powerful tool to solve the problem of inefficient coupling.
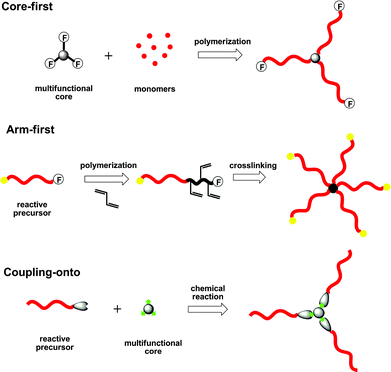 | ||
| Fig. 2 General synthetic methodologies for the preparation of star polymers. | ||
3.1 Homoarm star polymers and star block copolymers
Matyjaszewski and Gao first utilized the CuAAC reaction for the preparation of A3 and A4 type homoarm star polymersvia the coupling-onto methodology.32 In their study, ATRP generated polystyrene (PS) (Mn,GPC = 1400–18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100 g mol−1, Mw/Mn = 1.03) with a terminal azidegroup was attached to a tri- and tetra-alkyne functionalized core with 90 and 83% efficiency, respectively. All of the CuAAC reactions for star formation were complete within 3 h at ambient temperature. These authors also indicated that the presence of a small amount of Cu(0) enhanced the coupling efficiency and the highest product yield was obtained when the molar ratio of azide/alkynegroups was close to unity. However, the efficiency of the CuAAC reaction decreased with increasing molecular weight of the arm precursors, which was most likely due to higher steric congestion and propensity for the reactive end group to become suffocated within the polymeric coil as the molecular weight increases. Nevertheless, the CuAAC (coupling-onto) strategy has proven to be a versatile method for the synthesis of numerous star polymers with high yield. In a similar study by Altintas et al., a variety of 3-arm star polymers have been prepared using the CuAAC reaction via coupling onto, and the coupling efficiencies of these polymers were found to be similar in comparison to the previous work of Matyjaszewski.33 The effectiveness of modular ligation chemistry in the synthesis of star polymers using the CuAAC (via the coupling onto method) was again demonstrated with a series of 3-arm star polymers prepared by reacting a trialkyne containing core with azide-terminated PS (PS–N3) (Mn,GPC = 3500 g mol−1, Mw/Mn = 1.10), azide-terminated-poly(tert-butyl acrylate) (PtBA–N3) (Mn,GPC = 6700 g mol−1, Mw/Mn = 1.29), and azide-terminated poly(ethylene glycol) (PEG–N3) (Mn,GPC = 2600 g mol−1, Mw/Mn = 1.07). The application of the CuAAC to the A3 star formation was found to be reasonably efficient, as 87% of the PS chains reacted to give a PS3 star. A similar efficiency of the CuAAC reaction was also observed during the generation of PEG3 and PtBA3 star polymers, which had formation efficiencies of 82% and 85%, respectively.
100 g mol−1, Mw/Mn = 1.03) with a terminal azidegroup was attached to a tri- and tetra-alkyne functionalized core with 90 and 83% efficiency, respectively. All of the CuAAC reactions for star formation were complete within 3 h at ambient temperature. These authors also indicated that the presence of a small amount of Cu(0) enhanced the coupling efficiency and the highest product yield was obtained when the molar ratio of azide/alkynegroups was close to unity. However, the efficiency of the CuAAC reaction decreased with increasing molecular weight of the arm precursors, which was most likely due to higher steric congestion and propensity for the reactive end group to become suffocated within the polymeric coil as the molecular weight increases. Nevertheless, the CuAAC (coupling-onto) strategy has proven to be a versatile method for the synthesis of numerous star polymers with high yield. In a similar study by Altintas et al., a variety of 3-arm star polymers have been prepared using the CuAAC reaction via coupling onto, and the coupling efficiencies of these polymers were found to be similar in comparison to the previous work of Matyjaszewski.33 The effectiveness of modular ligation chemistry in the synthesis of star polymers using the CuAAC (via the coupling onto method) was again demonstrated with a series of 3-arm star polymers prepared by reacting a trialkyne containing core with azide-terminated PS (PS–N3) (Mn,GPC = 3500 g mol−1, Mw/Mn = 1.10), azide-terminated-poly(tert-butyl acrylate) (PtBA–N3) (Mn,GPC = 6700 g mol−1, Mw/Mn = 1.29), and azide-terminated poly(ethylene glycol) (PEG–N3) (Mn,GPC = 2600 g mol−1, Mw/Mn = 1.07). The application of the CuAAC to the A3 star formation was found to be reasonably efficient, as 87% of the PS chains reacted to give a PS3 star. A similar efficiency of the CuAAC reaction was also observed during the generation of PEG3 and PtBA3 star polymers, which had formation efficiencies of 82% and 85%, respectively.
While the CuAAC reaction has received substantial attention in synthetic polymer chemistry, the Diels–Alder reaction between anthracene and maleimide derivatives has recently been utilized for the synthesis of graft copolymers34 and block copolymers.35 By employing a Diels–Alder conjugation reaction strategy, 3-arm star polymers have been obtained by linking the linear precursors to a trifunctional core.36 In this approach, furan-protected maleimide-functionalized ATRPinitiators were used to synthesize poly(methyl methacrylate) (PMMA–MI) (Mn,GPC = 3200 g mol−1, Mw/Mn = 1.19) and PtBA–MI (Mn,GPC = 3500 g mol−1, Mw/Mn = 1.16); additionally, a furan-protected maleimide was linked to the PEG–MI (Mn,GPC = 3200 g mol−1, Mw/Mn = 1.06) through an esterification reaction to produce three polymers with furan-protected maleimide terminal groups. The resulting furan–maleimide terminated polymers were subsequently conjugated to a trifunctional core with three anthracene moieties to yield PEG3, PMMA3, and PtBA3 star polymers. The relatively high efficiencies of 82, 89, and 93% obtained for the PEG3, PMMA3, and PtBA3 3-arm star polymers, respectively, indicate that the Diels–Alder reaction of furan-protected maleimides with anthracene derivatives is a robust and efficient method for the synthesis of star type macromolecular architectures.
Barner-Kowollik, Stenzel, and colleagues also reported the synthesis of star polymers using the hetero Diels–Alder (HDA) reaction (coupling-onto) for the conjugation of linear polymer chains generated by RAFT onto a diene core (Fig. 3).37 The polymeric arms of the star were composed of PS using benzyl (diethoxyphosphoryl) dithioformate as the chain transfer agent (Mn,GPC = 3600 g mol−1, Mw/Mn = 1.15). The electron-withdrawing nature of the diethoxyphosphorylgroup activates the thiocarbonyl thio end group on the PS as a reactive hetero-dienophile, which was employed in a HDA reaction with a multi-diene core to yield 3- and 4-arm PS stars. The efficiencies calculated from the result of peak splitting by GPC analysis indicated that 86 and 82% were achieved for respective PS3 and PS4 star polymers.
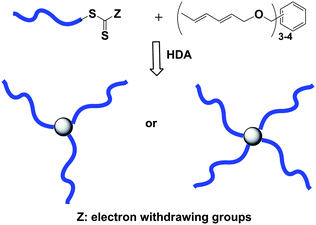 | ||
| Fig. 3 Preparation of PS3 or PS4 star polymersvia a HDA cycloaddition sequence. | ||
Recently, Hoyle and Lowe et al. reported the convergent synthesis of a 3-arm star polymervia a thiol–ene radical reaction.38 In their study, a linear homopolymer of poly(N,N-diethylacrylamide) (PDEAm) (Mn,GPC = 2900 g mol−1, Mw/Mn = 1.18) was synthesized initially by RAFT using 1-cyano-1-methylethyldithiobenzoate as the chain transfer agent, followed by reduction of the dithioester which yielded thiol-terminated polymers. Subsequently, addition of the polymer to a trimethylolpropane triacrylate was carried out in situ to yield the desired 3-arm PDEAm star polymer. The authors stressed that the coupling reaction was accelerated by using dimethylphenylphosphine (DMPP) as a catalyst. The efficiency for the thiol–ene reaction to generate the A3 star formation was not provided.
The synthesis of star block copolymersvia a modular ligation strategy was reported by Matyjaszewski and coworkers,39 in which they accomplished the synthesis of a 3-arm star block copolymer(AB)3 by sequential core-first and CuAAC (coupling-onto) methods. In this strategy, 3-arm star PS (PS–Br)3 with highly functionalized bromine chain-ends was first synthesized by ATRP using the ‘core-first’ method (Fig. 4). After complete conversion of the bromo-terminated polymers into azido-terminated polymers by nucleophilic substitution with sodium azide, the product (PS–N3)3 (Mn,GPC = 3300 g mol−1, Mw/Mn = 1.07) was further conjugated with alkyne-terminated PEG (PEG–alkyne) (Mn,GPC = 2100 g mol−1, Mw/Mn = 1.05) yielding 3-arm star block copolymers (PS-b-PEG)3via the CuAAC (‘coupling-onto’) method with high coupling efficiencies. The weight fraction of the (PS-b-PEG)3 star block copolymer was 85% in the final conjugated product between (PS–N3)3 and PEG–alkyne.
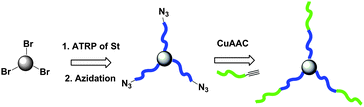 | ||
| Fig. 4 Synthesis of 3-arm star block copolymer(PS-b-PEG)3 by a sequential core-first and CuAAC reaction sequence. | ||
A CuAAC-Diels–Alder tandem reaction strategy for the synthesis of star polymers was first explored for the preparation of 3-arm star block copolymers, (PS-b-PMMA)3 and (PS-b-PEG)3 using a one-pot technique.40 The linear precursors, PS with α-anthracene-ω-azide (anth–PS–N3) (Mn,GPC = 5200 g mol−1; Mw/Mn = 1.12) and PMMA–MI (Mn,GPC = 2500 g mol−1; Mw/Mn = 1.19), were generated using suitable initiators for ATRP of St and MMA. Moreover, a PEG–MI (Mn,GPC = 3200 g mol−1, Mw/Mn = 1.03) was obtained from an esterification reaction of PEG–OH with 3-acetyl-N-(2-hydroxyethyl)-7-oxabicyclo[2.2.1]hept-5-ene-2-carboxamide. Finally, PS/PMMA or PS/PEG blocks were linked efficiently with a trialkyne functional linking agent in the presence of a CuBr–PMDETA ligand complex at 120 °C in DMF for 48 h to yield 3-arm star block copolymers. The efficiencies for both star block formations were calculated to be 82–88% via peak splitting of traces from gel permeation chromatography (GPC) analysis of the reaction mixture. The observed efficiencies are comparable to the efficiency obtained for star block copolymers synthesized by employing only the CuAAC reaction. The use of a one-pot technique for star block copolymer formation clearly demonstrates the orthogonality of the Diels–Alder and the CuAAC reactions.
Barner-Kowollik, Stenzel, and coworkers applied the HDA and the CuAAC combination for the production of star block copolymers containing PS and polycaprolactone (PCL) building blocks.41 In the first step of a ligation sequence, an α-diene-ω-alkyne-terminated PCL yielded a PS-b-PCL in the HDA reaction with a RAFT generated PS (Mn,GPC = 2900 g mol−1, Mw/Mn = 1.14). The block copolymer was subsequently reacted with a triazide core via the CuAAC to yield a star block copolymer. In a second sequence, an α-diene-ω-alkyne-terminated PCL (Mn,GPC = 2400 g mol−1, Mw/Mn = 1.39) was first ligated with a trialkyne core to result in a PCL star polymer through the CuAAC which was followed by a HDA ligation reaction between the PCL star polymer and RAFT-generated PS to achieve the star block copolymer. The authors indicated that the CuAAC reaction proceeded to quantitative conversion, whereas the HDA proceeded to 94% and 81% conversions, respectively, for the two sequences described above. A collation of all homoarm star and star block copolymers prepared via combining core-first and (coupling-onto) reactions employing various sequences are provided in Table 1.
| Core | Arm | Sequence of reactions | Type of star and star block copolymerref. |
|---|---|---|---|
| Cyclodextrin–(N3)7 | PCL–alkyne | ROP, CuAAC | Cyclodextrin–(PCL)742 |
| Diol-dibromide | PS-b-PCL and PCL-b-PS | PS (ATRP), PCL (ROP), CuAAC, PCL (ROP), PS (ATRP) | (PS-b-PCL)2–(PS-b-PCL)243 |
| Dendrimer bromide | P(iBorA) and PS | P(iBorA) (ATRP), PS (ATRP), CuAAC44a, P(iBorA) (ATRP), PS (RAFT), HDA44b | (P(iBorA)-b-PS)12 44 |
| Trialkyne | PONB–N3 | PONB (ROMP), azidation, CuAAC | (PONB)3 45 |
| Trialkyne | POSS–N3 | CuAAC | (POSS)3 46 |
| Trialkyne | PS–N3 | PS (ATRP), CuAAC | (PS)3 47 |
| Triacrylate | PnBA and PDEAm | PnBA (RAFT), PDEAm (RAFT) and thiol–ene | (PnBA)3 and (PDEAm)348 |
| Trialcohol | PLA and PEG–N3 | PLA (ROP), esterification, CuAAC | (PLA-b-PEG)3 49 |
| POSS–(N3)8 | Alkyne–PS–Br | CuAAC and PS (ATRP) | POSS–(PS)850 |
| Trinitroxide | PS–Br | PS (ATRP), NRC | (PS)3 51 |
| Cyclotriphosphazene–(OH)6 | PCL–thiophene | PCL (ROP), azidation, CuAAC | (PCL–thiophene)652 |
| (PiB–N3)3 | PEG–alkyne | CuAAC | (PiB-b-PEG)3 53 |
| (Glucose–Br)5 | PS and POSS–alkyne | PS (ATRP), CuAAC. | (Glucose-PS-b-POSS)5 54 |
| Trialkyne | Isotactic polypropylene–N3 | Isotactic polypropylene (CP) and CuAAC | (iPP)3 55 |
| Fullerene–(crown ether–N3)6 | PEG–alkyne, PS–alkyne or PNIPAM–alkyne | PS (RAFT), PNIPAM (RAFT), PS (ATRP), CuAAC | Fullerene–(crown ether–PEG)6, fullerene–(crown ether–PS)6, fullerene–(crown ether–PNIPAM)656 |
| Azide–dibromide | CA–PS and PS2–HW | PS (ATRP), CuAAC, H-Bonds | PS3 57 |
3.2 Miktoarm star polymers
A 3-miktoarm star terpolymer (ABC type) was first achieved utilizing the core-first methodology and CuAAC reaction.58 In this study, a trifunctional miktoinitiator containing bromine, TEMPO, and alkyne moieties was synthesized and successively used as an initiator in the ATRP of MMA (Mn,GPC = 6200 g mol−1, Mw/Mn = 1.2) and NMP of St. Finally, the CuAAC reaction was accomplished between the alkyne mid-functional copolymer (PMMA–PS–alkyne) and PtBA–N3 (Mn,GPC = 6000 g mol−1, Mw/Mn = 1.28) or PEG–N3 (Mn,GPC = 2000 g mol−1, Mw/Mn = 1.07) to yield PMMA–PS–PtBA or PMMA–PS–PEG miktoarm star terpolymers, respectively (Fig. 5).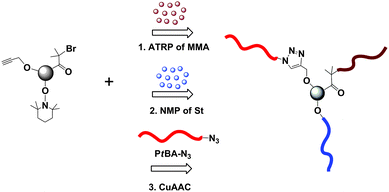
Similarly, Monteiro and coworkers demonstrated the synthesis of AB2 miktoarm star polymers (PS, Mn,GPC = 5000 g mol−1, Mw/Mn = 1.09, PtBA, Mn,GPC = 6900 g mol−1, Mw/Mn = 1.08, and PMA, Mn,GPC = 5600 g mol−1, Mw/Mn = 1.06) with low polydispersity and high yields using the CuAAC reaction strategy within short reaction times of 60 min.59 In their study, the authors added an excess amount of a trialkyne core into the reaction medium containing one arm via a syringe pump to ensure exclusive monosubstitution. Subsequently, the solution was added to another solution of the second arm through a syringe pump. It should be noted that this ligation reaction requires careful control with respect to the rate of core addition and it proceeds at a relatively high temperature (80 °C).
The Diels–Alder reaction (coupling-onto) has also been applied to create a 3-miktoarm star terpolymer.60 A linear PEG–MI (Mn,GPC = 740 g mol−1, Mw/Mn = 1.07) was initially reacted via a Diels–Alder reaction with an initiator containing anthracene, bromine and TEMPO functionalities. Secondly, PS and PtBA arms were grown from this macroinitiator viaNMP and ATRP, resulting in a final PEG–PS–PtBA miktoarm star terpolymer. The Diels–Alder reaction efficiency was obtained by monitoring the UV spectra of the Diels–Alder adduct between 300 and 400 nm and found to be quantitative.
In addition to these efficient linking reactions, Huang and coworkers applied a different reaction type for the synthesis of 3-miktoarm star terpolymers.61 In this strategy, ATRP generated PtBA–N3 (Mn,GPC = 2600–3000 g mol−1, Mw/Mn = 1.07–1.13), PEG–TEMPO (Mn,GPC = 3600 g mol−1, Mw/Mn = 1.20) or PCL–TEMPO (Mn,GPC = 6200 g mol−1; Mw/Mn = 1.13) and ATRP generated alkyne–PS–(Br) (Mn,GPC = 3000–6300 g mol−1; Mw/Mn = 1.04–1.03) were conjugated in a one-pot fashion employing a combination of the CuAAC and NRC reactions catalyzed by a Cu/CuBr/PMDETA complex in DMF at 70 °C for 24 h yielding the corresponding miktoarm star terpolymers (PtBA–PS–PEG and PtBA–PS–PCL). The formation efficiencies of miktoarm star terpolymers were found to be in the range of 90–91% calculated from 1H NMR spectra. The CuAAC reaction in a combination with ROP, NMP, and ATRP has afforded miktoarm star polymers with more complex structures, such as 4-miktoarm star quarterpolymers (ABCD type).62 In this approach, a PCL-b-PS copolymer with an azidegroup located in the center was synthesized in a one-pot, two-step reaction; the first step involved the growth of PS and PCL from a bifunctional initiator using ROP and NMP techniques, respectively. The second step involved a transformation of a central reactive bromine into an azidegroup. A second block copolymer was prepared by coupling a carboxylic acid-terminated PEG onto an alkyne functionalized ATRPinitiator. The PEG–macroinitiator was used to initiate ATRP of MMA or tBA to form PEG-b-PMMA or PEG-b-PtBA with an alkyne at the junction point. To obtain the desired ABCD 4-miktoarm star quarterpolymers, the azide-functionalized PCL-b-PS copolymer was reacted with either the alkyne-containing PEG-b-PMMA or PEG-b-PtBA in the presence of CuBr and PMDETA to yield PCL–PS–PEG–PMMA and PCL–PS–PEG–PtBA miktoarm star quarterpolymers via the CuAAC reaction. Similarly, the group of Pan prepared a 4-arm star quarterpolymer with PS, PEG, PCL, and PMA arms.63 Another 4-arm star quarterpolymer example (PS–polyisoprene–PtBA–PEG) comes from Huang and coworkers employing a similar strategy of coupling two block copolymers together from their junction points through the CuAAC reaction.64 Many other miktoarm star polymersvia modular ligation reactions reported in literature are summarized in Table 2.
| A arm | B arm | C or D arm | Sequence of reactions | Structure of miktoarm star polymerref |
|---|---|---|---|---|
| PMMA–N3 (ATRP), PtBA–N3 (ATRP), PEG–N3 (anionic ROP) | PCL (ROP) | PS (NMP) | One-pot: NMP, ROP, and CuAAC | PS–PCL–PMMA, or PS–PCL–PEG or PS–PCL–PtBA65 |
| PEG–N3 (anionic ROP) | PS (ATRP) | PCL (ROP) | CuAAC, ATRP, ROP | PEG–PS–PCL66 |
| PS–N3 (RAFT) | PMMA (ATRP) | — | RAFT, CuAAC, ATRP | (PS)3–PMMA67 |
| PEG–N3 (anionic ROP) | PCL–alkyne (ROP) | Poly (2-ethoxy-2-oxo-1,3,2-dioxaphospholane) (ROP) | ROP, CuAAC, ROP | PEG–PCL–polyphosphoester68 |
| Poly(vinyl acetate)-b-polyacrylonitrile | PEG–N3 | — | CMRP, NMRC, CuAAC | (Poly(vinyl acetate)-b-polyacrylonitrile)2-PEG69 |
| PS (NMP) | PMMA (ATRP) | PNBONI–N3 (ROMP) | ROMP, NMP, ATRP, CuAAC | PS–PMMA–PNBONI70 |
| PS–alkyne (RAFT) | PNIPAM (RAFT) | PDMAEMA–N3 (ATRP) | RAFT, RAFT, ATRP, CuAAC | PS–PNIPAM–PDMAEMA71 |
| PEG–Br (–alkyne) (anionic ROP) | PNIPAM (ATRP) | PDEMA–N3 (ATRP) | ROP, ATRP, ATRP, CuAAC | PEG–PNIPAM–PDEMA72 |
| PEG–Br (–N3) (anionic ROP) | PNIPAM (ATRP) | PLL–alkyne (ROP) | ROP, ATRP, ROP, CuAAC | PEG–PNIPAM–PLL73 |
| PS–N3 (ATRP) | PCL (ROP) | PNIPAM–N3 (ATRP) | ATRP, ATRP, ROP, CuAAC | PS–PCL–PNIPAM74 |
| PEG–N3 (Anionic ROP) (PS–N3 (ATRP)) | PCL (ROP) | PDMA (ATRP) | One-pot: ROP, ATRP, CuAAC | PEG–PCL–PDMA or PS–PCL–PDMA75 |
| PEG–alkyne | PNIPAM (ATRP) | PtBMA–alkyne (ATRP) | CuAAC, ATRP, CuAAC | PEG–PNIPAM–PtBMA76 |
| PEG–alkyne (anionic ROP) | PtBMA (ATRP) | PDEAm–alkyne (ATRP) | CuAAC, ATRP, CuAAC, PtBMA hydrolyzed to PMMA | PEG–PtBMA–PDEAm and PEG–PMAA–PDEAm77 |
| PEG–COOH | PMMA (ATRP) | POSS–N3 | Esterification, ATRP, CuAAC | PEG–PMMA–POSS46 |
| PtBA–maleimide (–TEMPO) (ATRP) | PCL–(anth.) (–N,N-dimethylaniline) (ROP) | PS (NMP) (C arm) PMMA (FRP) (D arm) | ATRP, ROP, Diels–Alder, NMP, FRP | PtBA–PCL–PS–PMMA78 |
| PCL (ROP) | PEG–alkyne | — | ROP, esterification (ibuprofen), CuAAC | (PCL)2–(PEG)279 |
| PDMAEMA–N3 (ATRP) | PS-N3 (ATRP) | — | ATRP, CuAAC | (PDMAEMA)2–PS47 |
| PNIPAM (ATRP) | PDEAm–alkyne (ATRP) | — | ATRP, CuAAC | (PDEAm)7–cyclodextrin–(PNIPAM)1480 |
| PLL–(alkyne)2 | PLGA–N3 | — | CuAAC | PLL–(PLGA)281 |
| PNIPAM–(N3)2 | PLL-alkyne | — | ATRP, ROP, CuAAC | PNIPAM–(PLL)282 |
| PEG–N3 (anionic ROP) | PS–N3 (LAP) | PCL (ROP) | LAP, CuAAC, CuAAC, ROP. | PEG–PS–PCL83 |
| PCL from β-cyclodextrin (ROP) | PEG–alkyne | — | ROP, esterification (ibuprofen), CuAAC | (PCL)14–(PEG)784 |
| PEG–N3 (anionic ROP) | PCL (ROP) | — | CuAAC, ROP | (PEG)2–PCL85 |
| PiBOA-alkyne (ATRP) | PS–N3 (ATRP) | — | ATRP, ATRP, NMRC, CuAAC | (PiBOA)2–PS86 |
| PS2HW | CA–PMMA (ATRP) | — | ATRP, CuAAC, H-bonds | (PS)2–PMMA57 |
| Polyethylene–alkyne | PS (ATRP) | — | CuAAC, ATRP | (PE)2–(PS)287 |
| PS–OH(–N3) (ATRP) | PCL (ROP) | PEG–alkyne (anionic ROP) | ATRP, Thiol–ene, ROP, CuAAC | PS–PCL–PEG88 |
3.3 Multiarm star polymers with cross-linked cores
Multiarm star polymers containing a cross-linked core have mainly been produced utilizing an arm-first methodology followed by either a single or combinatorial ligation reaction process. A less common way of incorporating a linkage reaction is achieved by modifying the precursor linear polymer arm followed by the arm-first methodology to generate a final multiarm star polymer structure.As a first example of the above concept, a linear anthracene-terminated PS macroinitiator (anth–PS–Br) (Mn,GPC = 5700 g mol−1, Mw/Mn = 1.08) was prepared by ATRP of St with low monomer conversion to ensure a high degree of bromine chain end-functionality. Next, the anth–PS–Br macroinitiator and divinyl benzene (DVB), as the cross-linker, were reacted with CuBr/PMDETA at 110 °C to yield a poly(DVB)(PS–anth)m multiarm star polymer with DVB as the cross-linked core and an anthracene periphery.89 It was demonstrated that soluble star polymers could be obtained when a suitable molar ratio of DVB to macroinitiator (i.e. 15) was employed. The DVB conversion was followed by GC analysis and the polymerization was stopped after 10 h at 95% conversion. The weight average arm number (f) of the poly(DVB)(PS–anth)m multiarm star polymer was calculated to be close to 33. Subsequently, the obtained poly(DVB)(PS–anth)m multiarm star polymer was reacted with maleimide terminated-PMMA (PMMA–MI) or -PtBA (PtBA–MI) to yield the resulting multiarm star block copolymersvia the Diels–Alder reaction. The Diels–Alder reaction efficiencies were calculated to be 96 and 88% for poly(DVB)(PS–anth)m–(PMMA)n and poly(DVB)(PS–anth)m–(PtBA)k multiarm star block copolymers, respectively, which were obtained by observing the decrease in absorbance of the anthracenechromophore at 368 nm with UV spectroscopy.
The first application of the CuAAC reaction strategy for the synthesis of multiarm star copolymers using the arm-first methodology was reported by Qiao and Wiltshire.90 A pendant alkyne-functional multiarm star polymer (f = 38) obtained from the ROP of a bis-lactone monomer initiated by a hydroxyl end-functionalized block copolymer was ligated with a linear azide-terminated PS (Mn,GPC = 10400 g mol−1, Mw/Mn = 1.02) to generate a multiarm star copolymer with brush-like arms. A similar synthetic approach using the CuAAC reaction strategy and the arm-first approach for multiarm star block copolymers (and mixed-block) was carried out by Durmaz et al.91 A linear α-silyl protected alkyne-terminated PS (α-silyl-alkyne–PS–Br) was prepared by ATRP of St and further employed as the macroinitiator in the ATRP of DVB to successfully generate a multiarm star homopolymer with alkyne periphery (f = 27) at 110 °C for 9 h. Next, a linear azide-terminated-PEG (PEG–N3) (Mn,GPC = 450 g mol−1, Mw/Mn = 1.05) and PtBA–N3 (Mn,GPC = 4050 g mol−1, Mw/Mn = 1.10) were efficiently conjugated with the multiarm star polymer described previously to yield multiarm star block copolymers, poly(DVB)–(PS)n–(PEG)k and poly(DVB)–(PS)n–(PtBA)m or a multiarm star mixed-block copolymer, poly(DVB)–(PS)n–(PEG)k–(PtBA)m catalyzed by the CuBr/PMDETA complex in DMF at ambient temperature for 24 h.
The tandem ligation strategy (CuAAC and Diels–Alder) of coupling three blocks to create a linear triblock terpolymer was further employed for the preparation of multiarm star polymers with triblock terpolymer arms (Fig. 6).92 The poly(DVB)–(PS–alkyne)m multiarm star polymer (f = 23–25) was obtained by ATRP of DVB in the presence of an α-silyl-alkyne–PS–Br as the linear macroinitiator and CuBr/PMDETA as the catalyst complex at 110 °C. Next, this poly(DVB)–(PS–alkyne)m multiarm star polymer was sequentially linked with a linear ATRP-generated PMMA containing anthracene–azide terminal groups (anthracene–PMMA–N3) (Mn,GPC = 4100 g mol−1, Mw/Mn = 1.23) and ATRP-generated PtBA–MI (Mn,GPC = 4100 g mol−1, Mw/Mn = 1.14) or PEG–MI (Mn,GPC = 550 g mol−1, Mw/Mn = 1.09) precursors to yield corresponding poly(DVB)–(PS)m–(PMMA)n–(PtBA)k and poly(DVB)–(PS)m–(PMMA)n–(PEG)p multiarm star triblock terpolymers. The Diels–Alder reaction efficiencies were as high as 95–99% for the formation of the described multiarm star triblock terpolymers.
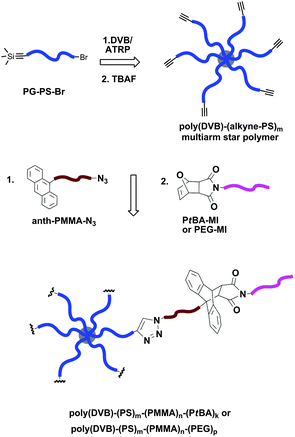 | ||
| Fig. 6 Synthesis of multiarm star polymer with triblock terpolymer arms using a sequential CuAAC and Diels–Alder reaction sequence. | ||
Another example that demonstrated the preparation of multi-miktoarm star block copolymers employed the arm-first and sequential tandem ligation reactions involving the CuAAC and Diels–Alder reactions.93 A PS multiarmed star with both alkyne and anthracene moieties at the periphery, (alkyne–PS)n–polyDVB–(PS–anthracene)n, was obtained viaATRP of DVB concurrently initiated with linear α-silyl protected alkyne- and α-anthracene-terminated PS macroinitiators, followed by sequential CuAAC and Diels–Alder reactions with PtBA–N3 and α-maleimide terminated PMMA (PMMA–MI) respectively, resulting in the formation of the target multi-miktoarm star block copolymer(PtBA)m–(PS)n–polyDVB–(PS)n–(PMMA)k.
Qiao and coworkers reported the synthesis of multiarm star polymers (f = 26–40) comprised entirely of amino acidsviaROP of the amino acidN-carboxyanhydride in a one-pot arm-first technique.94 These water-soluble multiarm star polymers have selectively degradable cores and hierarchical functionalities at the periphery, the arms, and the core. The alkynegroups within the core of multiarm star polymers were further reacted with an azidopyrenyl compound via the CuAAC reaction.
A variety of multiarm star polymers modified by using the modular ligation reactions are collected in Table 3.
| Cross-linker and inner arm | Periphery | Reactions sequence | Structure, fref |
|---|---|---|---|
| DVB (ATRP), PS–Br (ATRP) | Cyclic-PS | ATRP, CuAAC, ATRP, ATRP | Poly(DVB)–PS-cyclic-PS, f = 17–7795 |
| 1,4-Butanediol dimethacrylate (BDMA), and methacryloyl-PCL–alkyne | Dendritic-L-Lysine–N3 | ROP, CuAAC, ATRP of BDMA | Poly(BDMA)–PCL–dendritic L-lysine96 |
6,6′-(Ethane-1,2-diylbis(oxy)) bis(3-vinylbenzaldehyde) (EVBA), C12H25S(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S)S–PS–COOH S)S–PS–COOH |
— | RAFT, non-aldol reaction between aldehyde and PEG–ONH2 | Poly(EVBA)–(PS–PEG), f = 1497 |
| DVB and silyl-alkyne–PS–Br (ATRP) silyl-alkyne–PtBA–Br (ATRP) | POSS–N3 | ATRP, CuAAC | Poly(DVB)–PS–POSS and poly(DVB)–PtBA–POSS, f = 2798 |
| Furan–maleimide adduct diacrylate, PMMA–Br (ATRP) | — | ATRP, Retro Diels–Alder. | Poly(furan-maleimide)–PMMA99 |
| DVB, anth–PS–Br (ATRP) | (PMMA)2n–MI (2-arm) (ATRP), (PMMA)4n–MI (4-arm) (ATRP) | ATRP, Diels–Alder | Poly(DVB)–(PS)m–(PMMA)2n and poly(DVB)–(PS)m–(PMMA)4n, f = 28100 |
| 4,4′-Bioxepanyl-7,7′-dione (BOD) (ROP), PCL–(PMA-g-silyl alkyne)–Br (ATRP) | PEG–N3, PtBA–N3 (ATRP) or Naphthyl–N3, anthryl–N3, pyrenyl–N3, saccharide–N3 | ROP, ATRP, ROP, CuAAC | Poly(BOD)–PCL–(PMA-g-PEG); poly(BOD)–PCL–(PMA-g-PtBA), Poly(BOD)–PCL–(PMA-g-naphthyl) etc., f = 25101 |
4. Conclusions
Living anionic polymerization techniques have frequently been employed for the preparation of star polymers. However, in recent years, LRPs have become important techniques not only for the synthesis of linear polymers but also for branched architectures, due to a wide variety of applicable monomers and great tolerance to experimental conditions in comparison to living ionic polymerization techniques. As there is no perfect all-encompassing polymerization process, all living polymerization techniques have their own potential limitations, particularly during the synthesis of increasingly complex macromolecular structures. The click chemistry concept has brought the modular construction approach to preparative polymer chemistry and afforded the preparation of new macromolecular structures that would have otherwise been unachievable. Modular ligation strategies and LRP techniques are compatible partners because LRPs affords well-defined polymers with functional groups suitable to further conjugations. This partnership is most pronounced in the syntheses of complex macromolecular structures, such as miktoarm star polymers and multi-arm star polymers with crosslinked cores. CuAAC and Diels–Alder reactions are the most effective processes for the preparation of homoarm star polymers, with efficiencies ranging from 85 to 95% depending on the molecular weight of the polymer precursor to be coupled. The NRC reaction has some limitations due to the incompatibility with LRP techniques via the incorporation of alkoxyamines into the polymer backbone. However, for star formation, the NRC ligation reaction demonstrates similar yields to the CuAAC and DA sequences described above. It is highly possible that radical thiol–ene reactions are effective only for conjugation of polymers with low molecular weights, underpinned by only one single example regarding 3-arm star formation employing the radical thiol–ene reaction reported by the Hoyle group.38 It appears that radical thiol–ene reactions do not fulfill the basic requirements of a click reaction.26Further, tandem modular ligation chemistries afford polymers with diverse topology and composition that cannot be prepared by using only one conjugation technique. Tandem ligation reactions can reduce the reaction time and the number of required steps. To date, only a few highly efficient reaction combinations, the CuAAC–Diels–Alder (or HDA) and the CuAAC–NRC reactions, have been extensively adapted for tandem reaction strategies for star polymer formation. In the future, different reaction combinations other than the CuAAC–Diels–Alder and the CuAAC–NRC may emerge for polymerconjugation leading to various polymeric star architectures.
Abbreviations
| Anth | anthracene |
| ATRP | Atom Transfer Radical Polymerization |
| BDMA | 1,4-butanediol dimethacrylate |
| BOD | 4,4′-bioxepanyl-7,7′-dione |
| CA | Cyanuric Acid |
| CMRP | Cobalt-mediated radical polymerization |
| CP | Coordination Polymerization |
| CRP | Controlled Radical Polymerization |
| CuAAC | Cu(I) Catalyzed Azide–AlkyneCycloaddition |
| DEAm | N,N-diethylacrylamide |
| DMPP | dimethylphenylphosphine |
| DVB | divinylbenzene |
| ε-CL | epsilon-caprolactone |
| EVBA | 6,6′-(ethane-1,2-diylbis(oxy)) bis(3-vinylbenz aldehyde) |
| FRP | Free Radical Polymerization |
| GPC | Gel Permeation Chromatography |
| HDA | Hetero Diels–Alder |
| HW | Hamilton Wedge |
| iBoA | isobornyl Acrylate |
| LAP | Living Anionic Polymerization |
| LRP | Living Radical Polymerization |
| MI | maleimide |
| MMA | methyl methacrylate |
| nBA | n-butylacrylate |
| NIPAM | N-isopropylacrylamide |
| NMP | Nitroxide Mediated Radical Polymerization |
| NMRC | Nitroxide Mediated Radical Coupling |
| NRC | Nitroxide Radical Coupling |
| ONB | exo-N-butyl-7-oxabicyclo[2.2.1]-hept-5-ene-2,3-dicarboximide |
| PCL | polycaprolactone |
| PDEAm | poly(N,N-diethylacrylamide) |
| PDEMA | poly(2-(diethylamino)ethyl methacrylate) |
| PDMAEMA | poly(2-(dimethyl-amino)ethyl-methacrylate) |
| PEG | poly(ethylene glycol) |
| PiB | polyisobutylene |
| PiBoA | poly(isobornyl acrylate) |
| PLA | poly(L-lactide) |
| PLGA | poly(L-glutamic acid) |
| PLL | poly(L-lycine) |
| PMA | poly(methyl acrylate) |
| PMDETA | N,N,N′,N′′,N′′-pentamethyldiethylenetriamine |
| PMMA | poly(methyl methacrylate) |
| PnBA | poly(n-butyl acrylate) |
| PNBONI | poly(N-butyl oxanorbornene imide) |
| PNIPAM | poly(N-isopropylacrylamide) |
| PONB | poly(exo-N-butyl-7-oxabicyclo[2.2.1]-hept-5-ene-2,3-dicarboximide) |
| POSS | Polyhedral Oligomeric Silsesquioxane |
| PP | polypropylene |
| PS | polystyrene |
| PtBA | poly(tert-butyl acrylate) |
| PtBMA | poly(tert-butyl methacrylate) |
| RAFT | Reversible Addition Fragmentation Chain Transfer Polymerization |
| ROMP | Ring Opening MetathesisPolymerization |
| ROP | Ring Opening Polymerization |
| SET-LRP | Single Electron Transfer Living Radical Polymerization |
| St | styrene |
| tBA | tert-butyl acrylate |
| tBMA | tert-butyl methylacrylate |
| TEMPO | 2,2,6,6-tetramethylpiperidine-1-oxyl |
Acknowledgements
C.B.-K. acknowledges support from the Karlsruhe Institute of Technology (KIT) in the context of Excellence Initiative for leading German universities and O.A. acknowledges the Islamic Development Bank (IDB) for a PhD scholarship.References and notes
- J. Roovers, in Star and Hyperbranched Polymers, ed. M. K. Mishra and S. Kobayashi, Marcel Dekker, New York, 1998, 285 Search PubMed.
- (a) A. Hult, M. Johansson and E. Malmstrom, Adv. Polym. Sci., 1999, 143, 1 CAS; (b) W. Burchard, Adv. Polym. Sci., 1999, 143, 113 CAS.
- J. P. Kennedy and S. Jacob, Acc. Chem. Res., 1998, 31, 835 CrossRef CAS.
- N. Hadjichristidis, M. Pitsikalis, S. Pispas and H. Iatrou, Chem. Rev., 2001, 101, 3747 CrossRef CAS.
- N. Hadjichristidis, H. Iatrou, M. Pitsikalis and J. Mays, Prog. Polym. Sci., 2006, 31, 1068 CrossRef CAS.
- A. Hirao, M. Hayashi, S. Loykulnant and K. Sugiyama, Prog. Polym. Sci., 2005, 30, 111 CrossRef CAS.
- (a) J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559 CrossRef CAS; (b) C. Barner-Kowollik and S. Perrier, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5715 CrossRef CAS; (c) M. K. Georges, R. P. N. Veregin, P. M. Kazmaier and G. K. Hamer, Macromolecules, 1993, 26, 2987 CrossRef CAS; (d) C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661 CrossRef CAS; (e) M. Kato, M. Kamigaito, M. Sawamoto and T. Higashimura, Macromolecules, 1995, 28, 1721 CrossRef CAS; (f) M. Ouchi, T. Terashima and M. Sawamoto, Chem. Rev., 2009, 109, 4963 CrossRef CAS; (g) J. S. Wang and K. Matyjaszewski, Macromolecules, 1995, 28, 7901 CrossRef CAS; (h) F. Di Lena and K. Matyjaszewski, Prog. Polym. Sci., 2010, 35, 959 CrossRef CAS; (i) V. Percec and B. Barboiu, Macromolecules, 1995, 28, 7970 CrossRef CAS; (j) B. M. Rosen and V. Percec, Chem. Rev., 2009, 109, 5069 CrossRef CAS; (k) K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921 CrossRef CAS; (l) L. Barner, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, Macromol. Rapid Commun., 2007, 28, 539 CrossRef CAS; (m) Handbook of RAFT polymerization, ed. C. Barner-Kowollik, Wiley-VCH, Weinheim, Germany, 2008 Search PubMed.
- Y. Yagci and M. A. Tasdelen, Prog. Polym. Sci., 2006, 31, 1133 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS.
- (a) B. S. Sumerlin and A. P. Vogt, Macromolecules, 2010, 43, 1 CrossRef CAS; (b) R. K. Iha, K. L. Wooley, A. M. Nystrom, D. J. Burke, M. J. Kade and C. J. Hawker, Chem. Rev., 2009, 109, 5620 CrossRef CAS; (c) K. Khanna, S. Varshney and A. Kakkar, Polym. Chem., 2010, 1, 1171 RSC; (d) U. Mansfeld, C. Pietsch, R. Hoogenboom, C. R. Becer and U. S. Schubert, Polym. Chem., 2010, 1, 1560 RSC; (e) O. Altintas and U. Tunca, Chem.–Asian J., 2011, 6, DOI:10.1002/asia.201100138; (f) P. L. Golas and K. Matyjaszewski, Chem. Soc. Rev., 2010, 39, 1338 RSC; (g) P. Lecomte, R. Riva, C. Jerome and R. Jerome, Macromol. Rapid Commun., 2008, 29, 982 CrossRef CAS; (h) C. R. Becer, R. Hoogenboom and U. S. Schubert, Angew. Chem., Int. Ed., 2009, 48, 4900 CrossRef CAS.
- C. Barner-Kowollik and A. J. Inglis, Macromol. Chem. Phys., 2009, 210, 987 CrossRef CAS.
- C. Barner-Kowollik, F. E. Du Prez, P. Espeel, C. J. Hawker, T. Junkers, H. Schlaad and W. Van Camp, Angew. Chem., Int. Ed., 2011, 50, 60 CrossRef CAS.
- (a) V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596 CrossRef CAS; (b) C. W. Tornoe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057 CrossRef CAS.
- J. A. Opsteen and J. C. M. van Hest, Chem. Commun., 2005, 57 RSC.
- (a) O. Diels and K. Alder, Ber. Dtsch. Chem. Ges. B, 1929, 62, 554 CrossRef; (b) H. Kwart and K. King, Chem. Rev., 1968, 68, 415 CrossRef CAS.
- G. Hizal, U. Tunca and A. Sanyal, J. Polym. Sci., Part A: Polym. Chem., 2011, 49 DOI:10.1002/pola.24835.
- A. J. Inglis and C. Barner-Kowollik, Macromol. Rapid Commun., 2010, 31, 1247 CrossRef CAS.
- A. J. Inglis, T. Paulöhrl and C. Barner-Kowollik, Macromolecules, 2010, 43, 33 CrossRef CAS.
- M. L. Szalai, D. V. McGrath, D. R. Wheeler, T. Zifer and J. R. McElhanon, Macromolecules, 2007, 40, 818 CrossRef CAS.
- (a) A. J. Inglis, L. Nebhani, O. Altintas, F. G. Schmidt and C. Barner-Kowollik, Macromolecules, 2010, 43, 5515 CrossRef CAS; (b) N. Aumsuwan and M. W. Urban, Polymer, 2009, 50, 33 CrossRef CAS.
- (a) H. Durmaz, B. Colakoglu, U. Tunca and G. Hizal, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 1667 CrossRef CAS; (b) A. J. Inglis, S. Sinnwell, M. H. Stenzel and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2009, 48, 2411 CAS.
- (a) M. J. Kade, D. J. Burke and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 743 CrossRef CAS; (b) A. B. Lowe, Polym. Chem., 2010, 1, 17 RSC.
- C. E. Hoyle, A. B. Lowe and C. N Bowman, Chem. Soc. Rev., 2010, 39, 1355 RSC.
- X.-P. Qiu and F. M. Winnik, Macromol. Rapid Commun., 2006, 27, 1648 CrossRef CAS.
- (a) A. H. Soeriyadi, G.-Z. Li, S. Slavin, M. W. Jones, C. M. Amos, C. R. Becer, M. R. Whittaker, D. M. Haddleton, C. Boyera and T. P. Davis, Polym. Chem., 2011, 2, 815 RSC; (b) G.-Z. Li, R. K. Randev, A. H. Soeriyadi, G. Rees, C. Boyer, Z. Tong, T. P. Davis, C. R. Becer and D. M. Haddleton, Polym. Chem., 2010, 1, 1196 RSC; (c) J. W. Chan, C. E. Hoyle, A. B. Lowe and M. Bowman, Macromolecules, 2010, 43, 6381 CrossRef CAS.
- S. P. S. Koo, M. M. Stamenovic, R. A. Prasath, A. J. Inglis, F. E. Du Prez, C. Barner-Kowollik, W. van Camp and T. Junkers, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1699 CrossRef CAS.
- W. Lin, Q. Fu, Y. Zhang and J. Huang, Macromolecules, 2008, 41, 4127 CrossRef CAS.
- V. Percec, T. Guliashvili, J. S. Ladislaw, A. Wistrand, A. Stjerndahl, M. J. Sienkowska, M. J. Monteiro and S. Sahoo, J. Am. Chem. Soc., 2006, 128, 14156 CrossRef CAS.
- H. Durmaz, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1962 CrossRef CAS.
- N. Hadjichristidis, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 857 CrossRef CAS.
- (a) H. F. Gao and K. Matyjaszewski, Prog. Polym. Sci., 2009, 34, 317 CrossRef CAS; (b) A. Blencowe, J. F. Tan, T. K. Goh and G. G. Qiao, Polymer, 2009, 50, 5 CrossRef CAS; (c) E. H. H. Wong, T. Junkers and C. Barner-Kowollik, Polym. Chem., 2011, 2, 1008 RSC.
- H. F. Gao and K. Matyjaszewski, Macromolecules, 2006, 39, 4960 CrossRef CAS.
- O. Altintas, B. Yankul, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 6458 CrossRef CAS.
- B. Gacal, H. Durmaz, M. A. Tasdelen, G. Hizal, U. Tunca, Y. Yagci and A. L. Demirel, Macromolecules, 2006, 39, 5330 CrossRef CAS.
- H. Durmaz, A. Dag, O. Altintas, T. Erdogan, G. Hizal and U. Tunca, Macromolecules, 2007, 40, 191 CrossRef CAS.
- A. Dag, H. Durmaz, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 302 CrossRef CAS.
- A. J. Inglis, S. Sinnwell, T. P. Davis, C. Barner-Kowollik and M. H. Stenzel, Macromolecules, 2008, 41, 4120 CrossRef CAS.
- J. W. Chan, B. Yu, C. E. Hoyle and A. B. Lowe, Chem. Commun., 2008, 4959 RSC.
- H. Gao, K. Min and K. Matyjaszewski, Macromol. Chem. Phys., 2007, 208, 1370 CrossRef CAS.
- H. Durmaz, A. Dag, A. Hizal, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7091 CrossRef CAS.
- S. Sebastian, A. J. Inglis, M. H. Stenzel and C. Barner-Kowollik, Macromol. Rapid Commun., 2008, 29, 1090 CrossRef CAS.
- R. Hoogenboom, B. C. Moore and U. S. Schubert, Chem. Commun., 2006, 4010 RSC.
- L.-P. Yang, X.-H. Dong and C.-Y. Pan, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7757 CrossRef CAS.
- (a) M. Lammens, D. Fournier, M. W. M. Fijten, R. Hoogenboom and F. Du Prez, Macromol. Rapid Commun., 2009, 30, 2049 CrossRef CAS; (b) S. Sinnwell, M. Lammens, M. H. Stenzel, F. E. Du Prez and C. Barner-Kowollik, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 2207 CrossRef CAS.
- A. Dag, H. Durmaz, O. Sirkecioglu, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 2344 CrossRef CAS.
- E. Gungor, C. Bilir, H. Durmaz, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5947 CrossRef CAS.
- W. Zhang, W. Zhang, Z. Zhang, J. Zhu, Q. Pan and X. Zhu, Polym. Bull., 2009, 63, 467 CrossRef CAS.
- J. W. Chan, B. Yu, C. E. Hoyle and A. B. Lowe, Polymer, 2009, 50, 3158 CrossRef CAS.
- M. J. Stanford and A. P. Dove, Macromolecules, 2009, 42, 141 CrossRef CAS.
- Z. Ge, D. Wang, Y. Zhou, H. Liu and S. Liu, Macromolecules, 2009, 42, 2903 CrossRef CAS.
- J. Kulis, C. A. Bell, A. S. Micallef, Z. Jia and M. J. Monteiro, Macromolecules, 2009, 42, 8218 CrossRef CAS.
- M. Gorur, F. Yilmaz, A. Kilic, A. Demirci, Y. Ozdemir, A. Kosemen and S. E. San, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 3668 CrossRef CAS.
- M. Rother, H. Barqawi, D. Pfefferkorn, J. Kressler and W. H. Binder, Macromol. Chem. Phys., 2010, 211, 204 CrossRef CAS.
- W. Zhang and A. H. E. Mueller, Macromolecules, 2010, 43, 3148 CrossRef CAS.
- H. Huang, H. Niu and J.-Y. Dong, Macromolecules, 2010, 43, 8331 CrossRef CAS.
- A. J. Inglis, P. Pierrat, T. Muller, S. Brase and C. Barner-Kowollik, Soft Matter, 2010, 6, 82 RSC.
- O. Altintas, U. Tunca and C. Barner-Kowollik, Polym. Chem., 2011, 2, 1146 RSC.
- O. Altintas, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 5699 CrossRef CAS.
- M. R. Whittaker, C. N. Urbani and M. J. Monteiro, J. Am. Chem. Soc., 2006, 128, 11360 CrossRef CAS.
- H. Durmaz, F. Karatas, U. Tunca and G. Hizal, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 499 CrossRef CAS.
- Q. Fu, G. Wang, W. Lin and J. Huang, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 986 CrossRef CAS.
- O. Altintas, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 1218 CrossRef CAS.
- L. Yang, H. Zhou, G. Shi, Y. Wang and C.-Y. Pan, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6641 CrossRef CAS.
- G. Wang, X. Luo, C. Liu and J. Huang, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2154 CrossRef CAS.
- O. Altintas, B. Yankul, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 3588 CrossRef CAS.
- G. Deng, D. Ma and Z. Xu, Eur. Polym. J., 2007, 43, 1179 CrossRef CAS.
- J. Zhu, X. Zhu, E. T. Kang and K. G. Neoh, Polymer, 2007, 48, 6992 CrossRef CAS.
- Y.-Y. Yuan, Y.-C. Wang, J.-Z. Du and J. Wang, Macromolecules, 2008, 41, 8620 CrossRef CAS.
- C. Detrembleur, A. Debuigne, O. Altintas, M. Conradi, E. H. H. Wong, C. Jerome, C. Barner-Kowollik and T. Junkers, Polym. Chem., 2011 Search PubMed , in press.
- A. Gozgen, A. Dag, H. Durmaz, O. Sirkecioglu, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 497 CrossRef CAS.
- J. Li, W.-D. He, S.-C. Han, X.-L. Sun, L.-Y. Li and B.-Y. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 786 CrossRef CAS.
- Y. Zhang, H. Liu, J. Hu, C. Li and S. Liu, Macromol. Rapid Commun., 2009, 30, 941 CrossRef CAS.
- J. Li, W.-D. He, N. He, S.-C. Han, X.-L. Sun, L.-Y. Li and B.-Y. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 1450 CrossRef CAS.
- Y. Zhang, H. Liu, H. Dong, C. Li and S. Liu, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 1636 CrossRef CAS.
- Y. Zhang, C. Li and S. Liu, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3066 CrossRef CAS.
- C. Li, Z. Ge, H. Liu and S. Liu, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 4001 CrossRef CAS.
- H. Liu, C. Li, H. Liu and S. Liu, Langmuir, 2009, 25, 4724 CrossRef CAS.
- O. Altintas, G. Hizal and U. Tunca, Des. Monomers Polym., 2009, 12, 83 Search PubMed.
- P.-F. Gou, W.-P. Zhu, N. Xu and Z.-Q. Shen, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 6962 CrossRef CAS.
- Z. Ge, J. Xu, J. Hu, Y. Zhang and S. Liu, Soft Matter, 2009, 5, 3932 RSC.
- Z. Ge and S. Liu, Macromol. Rapid Commun., 2009, 30, 1523 CrossRef CAS.
- L.-Y. Li, W.-D. He, J. Li, B.-Y. Zhang, T.-T. Pan, X.-L. Sun and Z.-L. Ding, Biomacromolecules, 2010, 11, 1882 CrossRef CAS.
- K. Khanna, S. Varshney and A. Kakkar, Macromolecules, 2010, 43, 5688 CrossRef CAS.
- P.-F. Gou, W.-P. Zhu and Z.-Q. Shen, Biomacromolecules, 2010, 11, 934 CrossRef CAS.
- G. M. Soliman, R. Sharma, A. O. Choi, S. K. Varshney, F. M. Winnik, A. K. Kakkar and D. Maysinger, Biomaterials, 2010, 31, 8382 CrossRef CAS.
- E. H. H. Wong, M. H. Stenzel, T. Junkers and C. Barner-Kowollik, Macromolecules, 2010, 43, 3785 CrossRef CAS.
- R. Liu, Z. Li, D. Yuan, C. Meng, Q. Wu and F. Zhu, Polymer, 2011, 52, 356 CrossRef CAS.
- B. Iskin, G. Yilmaz and Y. Yagci, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 2417 CrossRef CAS.
- A. Dag, H. Durmaz, U. Tunca and G. Hizal, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 178 CrossRef CAS.
- J. T. Wiltshire and G. G. Qiao, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 1485 CrossRef CAS.
- H. Durmaz, A. Dag, E. Erdogan, A. L. Demirel, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 99 CrossRef CAS.
- H. Durmaz, A. Dag, D. Gursoy, A. L. Demirel, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1557 CrossRef CAS.
- A. Dag, H. Durmaz, V. Kirmizi, G. Hizal and U. Tunca, Polym. Chem., 2010, 1, 621 RSC.
- A. Sulistio, A. Widjaya, A. Blencowe, X. Zhangb and G. Qiao, Chem. Commun., 2011, 47, 1151 RSC.
- Y. Peng, H. Liu, X. Zhang, S. Liu and Y. Li, Macromolecules, 2009, 42, 6457 CrossRef CAS.
- I. Javakhishvili and S. Hvilsted, Polym. Chem., 2010, 1, 1650 RSC.
- Z.-M. Wu, H. Liang, J. Lu and W.-L. Deng, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 3323 CrossRef CAS.
- E. Gungor, C. Bilir, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 4835 CrossRef CAS.
- J. A. Syrett, G. Mantovani, W. R. S. Barton, D. Pricec and D. M. Haddleton, Polym. Chem., 2010, 1, 102 RSC.
- H. Durmaz, A. Dag, C. Onen, O. Gok, A. Sanyal, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 4842 CrossRef CAS.
- J. M. Ren, J. T. Wiltshire, A. Blencowe and G. G. Qiao, Macromolecules, 2011, 44, 3189 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |