Comparison of DNA damage responses following equimutagenic doses of UVA and UVB: A less effective cell cycle arrest with UVA may render UVA-induced pyrimidine dimers more mutagenic than UVB-induced ones†
Thomas M.
Rünger
*,
Benyamin
Farahvash
,
Zsofia
Hatvani
and
Adam
Rees
Boston University School of Medicine, Department of Dermatology, 609 Albany Street, Boston, MA 02118, USA. E-mail: truenger@bu.edu; Fax: 001 617 638 5515; Tel: 001 617 638 5551
First published on 18th October 2011
Abstract
Mechanisms of UVA-mutagenesis remain a matter of debate. Earlier described higher rates of mutation formation per pyrimidine dimer with UVA than with UVB and other evidence suggested that a non-pyrimidine dimer-type of DNA damage contributes more to UVA- than to UVB-mutagenesis. However, more recently published data on the spectra of UVA-induced mutations in primary human skin cells and in mice suggest that pyrimidine dimers are the most common type of DNA damage-inducing mutations not only with UVB, but also with UVA. As this rebuts a prominent role of non-dimer type of DNA damage in UVA-mutagenesis, we hypothesized that the higher mutation rate at UVA-induced pyrimidine dimers, as compared to UVB-induced ones, is caused by differences in the way UVA- and UVB-exposed cells process DNA damage. Therefore, we here compared cell cycle regulation, DNA repair, and apoptosis in primary human fibroblasts following UVB- and UVA-irradiation, using the same physiologic and roughly equimutagenic doses (100–300 J m−2 UVB, 100–300 kJ m−2 UVA) we have used previously for mutagenesis experiments with the same type of cells. ELISAs for the detection of pyrimidine dimers confirmed that much fewer dimers were formed with these doses of UVA, as compared to UVB. We found that cell cycle arrests (intra-S, G1/S, G2/M), mediated at least in part by activation of p53 and p95, are much more prominent and long-lasting with UVB than with UVA. In contrast, no prominent differences were found between UVA and UVB for other anti-mutagenic cellular responses (DNA repair, apoptosis). Our data suggest that less effective anti-mutagenic cellular responses, in particular different and shorter-lived cell cycle arrests, render pyrimidine dimers induced by UVA more mutagenic than pyrimidine dimers induced by UVB.
Introduction
In addition to shortwave ultraviolet light (UVB, 280–315 nm), longwave ultraviolet light (UVA, 315–400 nm) has recently also been classified as a class I carcinogen by the WHO International Agency for Research on Cancer Monograph Working Group.1 However, the mechanisms by which UVA induces mutations and with that skin cancer remain a matter of debate.It is well established that solar ultraviolet light produces a variety of different types of DNA damage that in turn may result in mutation formation and ultimately photocarcinogenesis.2DNA damage profiles are dependent on wavelength. UVB generates DNA photoproducts (cis–syn cyclobutane pyrimidine dimers (CPDs), pyrimidine (6–4) pyrimidone photoproducts (6-4PPs), and Dewar valence isomers (DewPPs)) through direct absorption by DNA. While UVA is less energetic than UVB, it is also far more abundant than UVB and constitutes the majority of solar-available UV. While UVA is much less absorbed by DNA than UVB, it still generates pyrimidine dimers.2–11
Similar to UVB-induced mutations, mutations induced by UVA have also been shown to be predominantely C → T transitions, both in vitro and in vivo.11–14 As C → T transitions are mostly formed at sites of pyrimidine dimers, this indicates that pyrimidine dimers are the most common pre-mutagenic DNA lesions not only in UVB-, but also in UVA-mutagenesis.
In the past, findings of a higher yield of mutations per pyrimidine dimer with UVA, as compared to UVB,15 and of a second peak of skin cancer formation in the UVA2 range (340–400 nm) without a peak of pyrimidine dimer formation 16,17 were interpreted to support a contribution of a non-pyrimidine dimer-type DNA damage to UVA mutagenesis, including e.g. oxidative DNA base damage or DNA double strand breaks. However, the above mentioned predominance of C → T transitions with UVA is not consistent with that explanation, and we have proposed an alternative explanation:11,12
The vast majority of UV-induced DNA lesions are not translated into a mutation, because cells are equipped with a variety of anti-mutagenic mechanisms that prevent mutation formation at sites of DNA damage or limit survival of severely damaged cells. These antimutagenic cellular DNA damage responses include DNA repair (some of which is inducible18,19), cell cycle arrest, and apoptosis. We hypothesize that a UVA-induced pyrimidine dimer may be more mutagenic than a UVB-induced pyrimidine dimer, because UVA induced antimutagenic cellular responses are not as effective as those induced by UVB. And indeed, p53, a central regulator of antimutagenic cellular DNA damage responses, was found to be much less activated by UVA, as compared to UVB, when comparing equimutagenic doses.12
In order to further substantiate this hypothesis, we are describing here in much further detail the cellular DNA damage responses to equimutagenic doses of UVA and UVB, both by function (cell cycle arrest, DNA repair, and apoptosis) and by related DNA damage signaling. Toward that goal we have used the same model system we used for our studies with the hprt-mutagenesis assay12 (primary cultured human fibroblasts) and the same range of UVA and UVB doses. This avoids the problem that investigations from different laboratories can often not be directly compared because of the use of different model systems (mouse vs. human; transformed cellsvs. primary cells; cultured cellsvs. in vivo studies), because of different emission spectra of UVB and UVA lamps, and because of poorly standardized dosimetry. The use of primary, non-transformed cells assures that DNA damage responses are not altered, as is often the case in transformed cell lines, e.g. through mutation of p53. In addition, human cells are better suited for mutagenesis studies than e.g. rodent cells, as the latter are characterized by a reduced efficiency of DNA repair, which is an important DNA damage response following UV-exposure. In addition, the UVB- and UVA-doses used here can easily be reached by exposure to natural sunlight and are therefore clinically relevant. Despite all these advantages of our model system, it is nevertheless not ideal, because in humans, exposure to ultraviolet light is not known to induce fibroblast-derived malignancies and because cultured cells grow in an artificial environment that only poorly represents the in vivo situation.
Methods
A solar simulator (LH 153, Kratos Analytical, Ramsey, NJ) was used for UVB irradiations. The tissue culture lid removes contaminating UVC and only slightly reduces irradiance of the shorter wavelengths of the UVB range. The resulting emission spectrum was published by Werninghaus et al.21 The UVA emitted by this source at the maximum UVB doses used was less than 2.5 kJ m−2, consistent with previous reports that solar simulators often emit insufficient longwave UVA to adequately simulate natural sunlight.22,23
Radiometric measurements were performed for each experiment. For dosimetry, an IL-1700 Research Radiometer (International Light, MA) was used, equipped with a UVB-sensor (SED 240) or a UVA-sensor (SEF 015). A 137Cs gamma irradiator was used to expose cells to 10 Gy of gamma radiation.
Table 1 lists the doses of UVA and UVB used here and compares them to the range of UVA- and UVB-doses we had used previously in our mutagenesis experiments with the same type of cells and identical UV sources and dosimetry.12 It demonstrates that the UVA and UVB conditions used here induce mutations at rates that are in the same order of magnitude (roughly equimutagenic). These doses of UVA and UVB also have similar effects on cell growth/cell yields, as previously reported.12
| Doses used to study UV-induced damage responses | Mutation frequencies (from Kappes et al.12) | |
|---|---|---|
| UV dose | UV dose | mutation frequency (number of mutants/106 colony forming cells) |
| UVA | ||
| 100 kJ m−2 | 150 kJ m−2 | 5.9 |
| 200 kJ m−2 | 200 kJ m−2 | 70.4 |
| 300 kJ m−2 | 300 kJ m−2 | 136.4 |
| UVB | ||
| 100 J m−2 | 70 J m−2 | 8.7 |
| 200 J m−2 | 200 J m−2 | 20.4 |
| 300 J m−2 | 400 J m−2 | 62.3 |
| 600 J m−2 | 97.7 | |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1000) was then detected by sequential treatment with a biotinylated goat anti-mouse IgG antibody (1:10,000; Invitrogen, Carlsbad, CA) and streptavidin-peroxidase (Invitrogen). Finally, the absorbance of colored products derived from O-phenylene diamine was measured at 492 nm. All samples were measured in quadruplets. The unpaired Student t-test was used to test for differences.
:1000) was then detected by sequential treatment with a biotinylated goat anti-mouse IgG antibody (1:10,000; Invitrogen, Carlsbad, CA) and streptavidin-peroxidase (Invitrogen). Finally, the absorbance of colored products derived from O-phenylene diamine was measured at 492 nm. All samples were measured in quadruplets. The unpaired Student t-test was used to test for differences.
0.25 μg of treated or untreated plasmid was transfected into exponentially growing cells in 35 mm tissue culture dishes at 50% confluence, using Lipofectamine Plus (Gibco/Invitrogen) and the manufacturer's protocol. Triplicate samples were used for each cell line and each dose of UV. After 2 days, luciferase activity was determined using the luciferase reagent kit and luminometer (Promega). In order to compensate for variations of transfection rates, plasmid replication or condition of the cells used, triplicate parallel reference samples of unirradiated plasmid were used in every experiment. The relative luciferase activity with the UV-irradiated samples, which reflects repair of DNA photoproducts on the plasmids by the host cells (host cell reactivation), was calculated in percent of the parallel control samples transfected with unirradiated plasmid. While the read-out of this assay depends on the transcription of the repaired reporter gene, defects in global-genome repair, as e.g. in cells from patients with xeroderma pigmentosum complementation group C, are also detectable with this assay.28,31 This indicates that this assay is not limited to the measurement of transcription-coupled repair, and does not distinguish between global genome and transcription-coupled repair. The unpaired Student t-test was used to test for differences.
Here, we used this assay to investigate the effects of UVA and UVB on DNA repair capacity. For that, cells were pre-irradiated with different doses of UVA (50, 100, or 200 kJ m−2) or UVB (50, 100, or 200 J m−2) 24 h before transfection of the damaged plasmids for the repair assay. This permits investigation of UVA- and UVB- effects on DNA repair capacity separate from UV-induced toxicity, which is much higher for UVA than for UVB when comparing doses that generate the same amount of pyrimidine dimers.
:1000; mouse monoclonal; Cell Signaling, Danvers, MA), p95 (NBS1) phosphorylated at serine 343 (1:1000; rabbit polyclonal; Cell Signaling), XPC (1:500; mouse monoclonal; AbCam, Cambridge, MA), or actin as loading control (1:5000; HRP conjugate; Santa Cruz, Santa Cruz, CA).
:2000).
Results
Formation of DNA damage with UVA and UVB
Formation of CPDs and 6–4PPs is readily detectable with the range of UVB doses used for this study (Fig. 1, left panel). In contrast, the range of UVA doses, which generated mutation rates in the same order of magnitude as the UVB doses (Table 1, and 12), induced only a slight, statistically non-significant increase in CPDs and no increase in 6–4PPs (Fig. 2, right panel). While the ELISA used for this measurement is a rather crude assay to measure formation of pyrimidine dimers, it nevertheless confirms that UVA generates much less pyrimidine dimers than equimutagenic doses of UVB, and with that supports the premise that UVA produces more mutations per dimer than UVB (see discussion in introduction).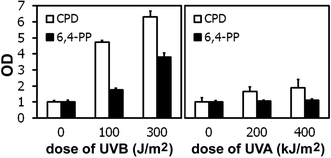 | ||
| Fig. 1 UVB induces more cyclobutane pyrimidine dimers (CPDs) and 6–4 photoproducts (6,4-PPs) than UVA, when using roughly equimutagenic doses. Shown are means of quadruple samples ± standard deviation. | ||
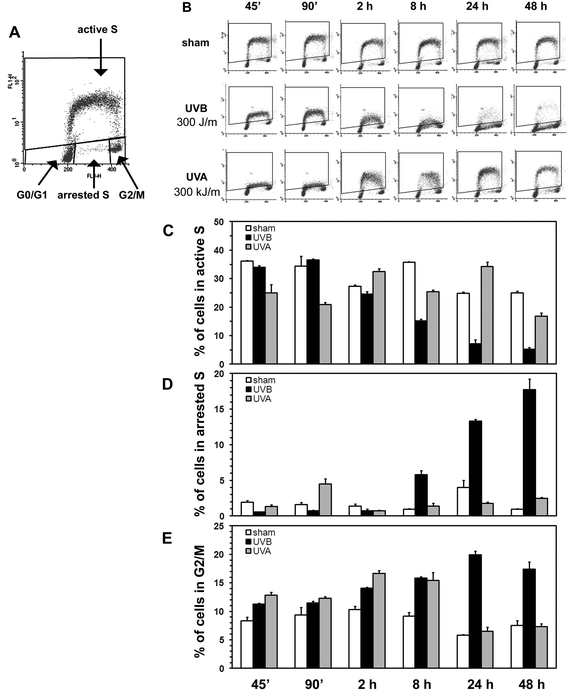 | ||
| Fig. 2 Bivariate BrdU/PI flow-cytometric analysis of cell cycle progression in non-synchronized cells reveals profound differences in cell cycle profiles following irradiation with roughly equimutagenic doses of UVB and UVA. Panel A depicts the attribution of cells to the different phase of the cells cycle. The cell cycle profiles at various time points after irradiation with sham, UVB (300 J m−2), or UVA (300 kJ m−2) are shown in panel B. The numerical analysis of the percentage of cells in the active S, in the arrested S, and in the G2/M phase are shown in panels C, D, and E respectively (averages of duplicate samples ± standard deviation). | ||
Effects of UVB and UVA on DNA replication and cell cycling
A detailed analysis of cell cycle changes in response to 300 J m−2 UVB and 300 kJ m−2 UVA reveals profound differences in how DNA synthesis and cell cycling of fibroblasts are affected by UVB and UVA (Fig. 2). An immediate slowing of DNA replication is observed both with UVB and UVA, as seen by the lowering of the S-phase arch (less incorporation of BrdU) at the 45 min time point (Fig. 2, panel B). DNA replication does not recover for at least 48 h after UVB-irradiation, with a decreasing fraction of cells in active S phase from 2 h to 48 h (Fig. 2, panels B and C) and an increasing fraction of cells being completely arrested in S-phase starting 8 h after irradiation (Fig. 2, panels B and D). After UVA-irradiation, the slowing of replication results in an early, but very transient arrest in active S phase for a small fraction of cells only at the 90 min time point, but rapid recovery of DNA replication already 2 h after irradiation (Fig. 2, panels B and D).UVB-irradiated cells also demonstrate a long-lasting G2/M arrest (2–48 h), whereas a mild G2/M arrest with UVA is observed only at the 2 and 8 h time points (Fig.2, panels B and E).
A G1/S arrest was not observed with non-synchronized fibroblasts (Fig. 2, analysis of cells in G1/G0 not shown). In order to study the failure of UVB- or UVA-irradiated cells to enter S-phase (G1/S-arrest), we synchronized cells by serum starvation and irradiated them with UVB or with UVA just prior to release from growth arrest by re-feeding with complete medium (10% calf serum). By 24 h after irradiations and release from serum starvation, sham- and low-dose (100 J m−2) UVB-irradiated fibroblasts readily entered S-phase, whereas the intermediate dose of UVB (200 J m−2) significantly reduced the fraction of cells entering S-phase by that time (partial S-phase arrest; Fig. 3 A). The highest dose of UVB (300 J m−2) completely blocked S-phase entry (complete S-phase arrest; Fig. 3 A). In contrast, none of the UVA doses affected the fraction of cells with S-phase entry, indicating a complete lack of a G1/S-arrest (Fig. 3 A).
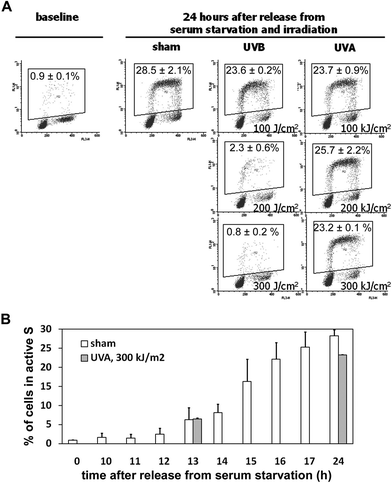 | ||
| Fig. 3 Dose-dependent block of S-phase entry only after UVB-, but not after UVA-irradiation. Cells were synchronized by serum starvation and released from serum starvation immediately after irradiation. Cell cycle profiles show efficient S-phase entry 24 h after sham, low-dose UVB (100 J m−2), and all doses of UVA (100, 200, and 300 kJ m−2; panel A; shown are representative examples of duplicates, the numbers are averages of the percentage of cells in active S-phase ± standard deviation). The intermediate dose of UVB (200 J m−2) delayed and reduced S-phase entry, and the highest dose of UVB (300 J m−2) blocked S-phase entry completely. Panel B describes the hour-by-hour S-phase entry of sham-irradiated cells and that S-phase entry after release from serum starvation is first detectable after 13 h. This was not delayed after irradiation with the highest UVA-dose, as the S-phase entry was also observed at the same time and in an almost identical fraction of cells. | ||
In order to determine whether UVA delays S-phase entry temporarily, which may not be detectable anymore 24 h after release from serum starvation, we observed S-phase entry of sham-irradiated fibroblasts hour-by-hour and determined the number of cells in active S-phase after UVA-irradiation exactly at the time point at which sham-irradiated cells started entering S-phase (13 h after release from serum starvation; Fig. 3 B). At that time point, the number of cells in active S-phase was almost identical after sham- and after UVA-irradiation. This indicates that UVA does not delay S-phase entry at all, not even by one hour (Fig. 3 B).
The cell cycle data (Fig. 2 and 3) demonstrate that the responses of fibroblasts to equimutagenic doses of UVA and UVB are not only quantitatively different (more pronounced cell cycle arrests with UVB than with UVA), but also qualitatively different (e.g. an early and very transient intra-S-arrest only with UVA, whereas the intra-S-arrest with UVB is observed later and persists much longer; prominent G1/S-arrest with higher doses of UVB, but no G1/S-arrest with UVA).
The rapid slowing of DNA replication after UVB with the sustained intra-S arrest may be mediated by activation of p95, because phosphorylation of p95 can be detected as early as 15 min after UVB irradiation (Fig. 4 A). The rapid, but short-lived slowing of DNA replication with brief intra-S-arrest after UVA cannot be due to activation of p95, as phosphorylation of p95 was not detected after UVA (Fig. 4 A). It may be due to (temporary) replication stops at pyrimidine dimers. However, if this were the case, it should be more prominent with UVB, because UVB generates more pyrimidine dimers (as shown in Fig. 1). Brief replication stops at UVA-induced DNA lesions other than pyrimidine dimers, including oxidative base modifications or single-strand breaks, may therefore account for the brief and transient slowing of DNA replication early after UVA irradiation.
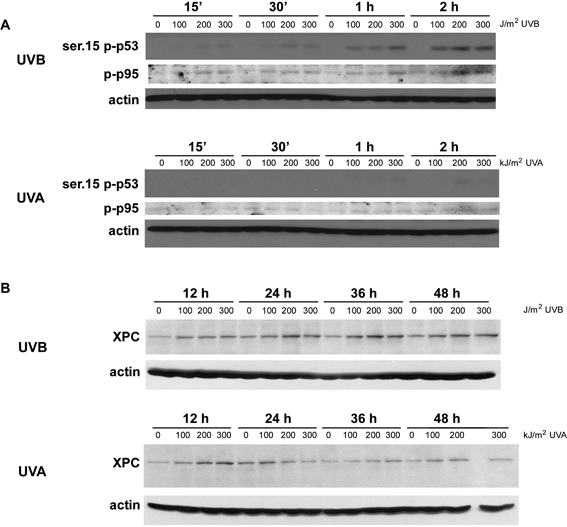 | ||
| Fig. 4 Dose-dependent activation of p53 and p95 (NBS1) early after UVB, but not after UVA (panel A). Consistent with much shorter-lived and less prominent activation of p53 at the early time points with UVA, as shown in panel A, and as reported previously for later time points,12 the induction of XPC (113 kD), which is known to be p53-dependent,33,34 is less prominent and shorter-lived after UVA irradiation, as compared to UVB-irradiation (panel B). | ||
Activation of ATM, an upstream regulator of p95, was detected with both, UVA and UVB, both at an early and at a later time point, 30 min and 4 h after irradiation (Fig. 5). However, this was only seen in a subset of cells and therefore appears to be cell cycle dependent. This is in contrast to ionizing radiation, with activates ATM in all cells (Fig. 5). The difference in p95 activation between UVA and UVB (no p95 activation with UVA, prominent activation with UVB) cannot be attributed to differences in phosphorylation of ATM at serine 1981, because UVA and UVB both appear to activate ATM equally. Other upstream kinases, e.g. ATR, or phosphorylation sites of ATM other than serine 1981 may be differentially activated by UVA and UVB and account for the difference in p95 activation.
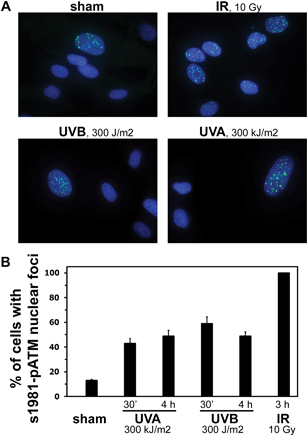 | ||
| Fig. 5 No difference in the activation of ATM (phosphorylation at serine 1981) by UVB (300 J m−2) or UVA (300 kJ m−2). Shown are representative examples of immunostainings for serine 1981–ATM nuclear foci (green; counterstaining with DAPI, blue, to mark nuclei; panel A) and the numerical analysis of the percent of cells with nuclear foci of activated ATM early (30 min) and later (4 h) after irradiation. One hundred cells in three different areas of the slide were counted; shown are averages ± standard deviation. Activation of ATM was only seen in a subset of cells, both after UVB and UVA, whereas ionizing radiation activated ATM in all cells. | ||
Activation of the G1/S-checkpoint is commonly thought to be mediated by p21via p53. Consistent with the activation of the G1/S checkpoint with UVB and the lack of it with UVA, we already reported a much weaker and less long lasting activation of p53 with UVA, as compared to UVB (2–48 h after irradiation12). Here, we add to this data by describing that p53 activation is already detectable one hour after irradiation with UVB, and confirm the lack of p53 activation with UVA (Fig. 4 A).
Effects of UVB and UVA on DNA repair
Nucleotide excision repair is the principal DNA repair pathway for the removal of pyrimidine dimers. It can be upregulated through activation of p53.33–37 P53 targets of the NER pathway are XPC, p48, and GADD45. Consistent with the stronger and longer lasting p53 activation by UVB, as compared to UVA, we also found a less prominent and shorter-lived induction of the XPC protein with UVA (Fig. 4 B). In order to investigate the functional consequences of this p53-mediated induction of XPC, we measured DNA repair capacity using a host cell reactivation assay 24 h after pre-irradiation of cells with sham, UVB, or UVA. Pre-irradiation of fibroblasts with 200 J m−2 UVB or 200 kJ m−2 UVA significantly reduced the repair capacity, as compared to sham irradiated cells (Fig. 6 A). Pre-irradiation with lower doses of UVB (Fig. 5 B) or lower doses of UVA (Fig. 5 C), did neither reduce nor increase DNA repair capacity of such pretreated cells. It therefore appears that a possible DNA repair capacity-increasing effect of XPC induction is offset by damaging effects of the UV exposures on the cells and their DNA repair machinery, with no net-increase in DNA repair capacity, and that these effects are very similar for the equimutagenic doses of UVB and UVA. Induction of XPC and with that adaptive increases in DNA repair capacity to UVA and UVB may be more important in cellular responses to repeated UV-exposures. These were not studied here.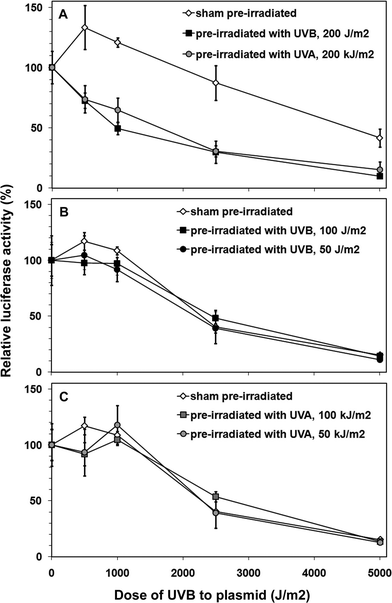 | ||
| Fig. 6 No difference in how UVA and UVB affect DNA repair capacity of fibroblasts. Cells were irradiated with intermediate doses of UVA (200 kJ m−2) or UVB (200 J m−2; panel A), with low doses of UVB (50 or 100 J m−2; panel B), or with low doses of UVA (50 or 100 kJ m−2; panel C), and DNA repair capacity was determined 24 h later using a host cell reactivation assay with plasmids containing increasing amounts of pyrimidine dimers generated by increasing doses of UVB to the plasmid prior to transfection. The repair of the DNA damage on the plasmids by the host cells' DNA repair machinery (host cell reactivation) was measured 24 h after transfection (= 48 h after pretreatment of host cells with UVA or UVB) by measuring expression of the plasmid-encoded luciferase. Shown are averages of triplicates samples ± standard deviation. Pre-irradiation with the higher doses of UVA and UVB (panel A) reduced DNA repair capacity of the host cells, whereas pre-irradiation with the lower doses of UVB (panel B) or UVA (panel C) did not affect DNA repair capacity. | ||
Effects of UVB and UVA on apoptosis
In order to assess the percentage of cells undergoing apoptosis, flow cytometry was used to measure the fraction of cells with sub-G1 DNA content. Only UVB, and only at the 48 h time point induced apoptosis in a very small fraction of cells (less than 4%; Fig. 7). The cell cycle changes observed after UVA and UVB (Fig. 2 and 3) are therefore observed in the absence of a significant induction of cell death. In addition, the prominent reductions in cell yield after UVB- or UVA-irradiation (see growth curves published by us earlier 12) are therefore mostly the result of cell cycle arrests, and hardly the result of induced cell death. Consequently, the large difference of mutation formation at UVB- and UVA-induced pyrimidine dimers cannot be explained by differences in induction of apoptosis.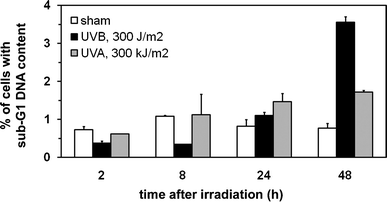 | ||
| Fig. 7 Both UVA and UVB induce apoptosis only in a small fraction of cells irradiated with the maximum doses used in this study. The fraction of cells with sub-G1 DNA content (apoptotic cells) was determined at several time points after UVA (300 kJ m−2) or UVB (300 J m−2) irradiation. Shown are averages of duplicate samples ± standard deviation. | ||
Discussion
UV-induced cellular responses to DNA damage that reduce the formation of mutations at sites of DNA damage include activation of cell cycle checkpoints and upregulation of DNA repair. In addition, apoptosis reduces the survival of damaged cells and with that also reduces formation/survival of UV-induced mutations. Using a range of UVB- and UVA-doses, which we have previously shown to generate similar rates of mutations, we demonstrate here in extensive time-course experiments that the most prominent difference in activation of DNA damage responses between UVB and UVA is in the arrest of the cell cycle at various checkpoints. Unlike UVB, the reaction to UVA is characterized by mostly a lack of cell cycle checkpoint activation (no G1/S-arrest, no sustained intra S-arrest or G2/M-arrest) and a much shorter-lived slowing of DNA replication. As replication of damaged and unrepaired DNA is the most important circumstance of mutation formation, these differences between UVB and UVA are likely to profoundly affect the rate by which UVB- and UVA-induced pyrimidine dimers are translated into mutations. The much weaker activation of p53 and p95 by UVA with subsequent lack of cell cycle checkpoint activation results in replication of DNA with pyrimidine dimers and formation of C → T transitions. In contrast, activation of cell cycle checkpoints through p53 and p95 by UVB will prevent formation of C → T transition mutations at most sites of pyrimidine dimers.Using an experimental design very similar to ours (similar doses of UVA and UVB; primary human fibroblasts), de Laat et al.26 also described suppression of DNA replication following both UVA and UVB. Very similar to our results, this effect was also more long-lasting with UVB than with UVA. However, they did not specifically analyze the fraction of cells arrested in S-phase, where we found an early, but short-lived intra-S arrest after UVA, and a late, but persistent and more profound intra-S arrest after UVB. Very similar to our results, they also did not see dramatic differences in the percentage of cells in G0/G1 when using non-synchronized cells. They did not examine S-phase entry in synchronized cells, with which we here describe a clear G1/S arrest after UVB (but not after UVA). A more prominent and longer-lasting suppression of DNA replication by UVB, as compared to UVA, was also described in vivo in epidermal cells of irradiated hairless mice.38 Using transformed human or mouse embryonal fibroblasts, Girard et al.39 also described slowing of DNA replication/BrdU incorporation following UVA-irradiation, again very similar to our results. We hypothesize that in our experiments this slowing is not due to replication stops at pyrimidine dimers, because it is more prominent after UVA, as compared to UVB, while our UVA doses produce less pyrimidine dimers than our UVB doses. Instead, we hypothesize that the slowing of DNA replication may be due to brief replication stops at sites of non-pyrimidine dimer types of DNA damage, including e.g. oxidative base modifications and single-strand breaks. In line with this interpretation, Girard et al.39 describe a partial rescue of UVA-inhibited DNA replication by N-acetyl-L-cystein (NAC), a scavenger of reactive oxygen species, confirming that oxidative DNA damage is indeed at least in part responsible for the immediate slowing of DNA replication after UVA exposure. Such slowing of replication is different from a complete block of replication, and may be due to recruitment of translesional DNA polymerases for bypassing oxidative DNA damage, including e.g.8-oxo-guanine.40 Given the lack of p53- and p95-activation early after UVA-irradiation, we conclude from our data that the early slowing of DNA replication after UVA is not due to activation of cell cycle checkpoints. Again very much in line with this interpretation, Girard et al.39 describe that in their transformed cells, the slowing of DNA replication after UVA is independent of the ATM/ATR/Chk1/Chk2 pathways, which are upstream regulators of p53 and p95. Using the same model system (primary human fibroblasts) and the same UVA sources as in this report, we recently published evidence that UVA does not generate DNA double strand breaks.41 This excludes the possibility that DNA double strand breaks cause the observed slowing of DNA replication after UVA exposure. Finally, given that UVA has been suggested to generate more pyrimidine dimers than oxidative DNA damage,3,42 mechanisms other than impaired replication at UVA-induced DNA lesions still need to be considered for the observed slowing of replication with UVA.
Our data support the hypothesis that UVA-induced dimers are more mutagenic than UVB-induced dimers because of less protective, anti-mutagenic cellular responses. This, however, only applies to exposure to pure UVA. For example, following exposure to natural solar radiation, which contains both UVA and UVB, the UVB-induced DNA damage responses will also protect against mutation formation at the additional UVA-induced dimers. Nevertheless, life in modern times does involve situations of exposure to pure UVA, including e.g. exposure to sunlight filtered through window glass, sun-exposure with sunscreens that effectively filter UVB, but do not (or less effectively) filter UVA, or UVA-phototherapy. In those circumstances, the mutagenic and with that the carcinogenic potential of UVA-induced pyrimidine dimers, in the absence of UVB-induced protective responses, may be of particular clinical significance.
References
- E. El Ghissassi, R. Baan, K. Straif, Y. Grosse, B. Secretan, V. Bouvard, L. Benbrahim-Talla, N. Guha, C. Freeman and L. Galichet, V. Cogliano, and on behalf of the WHO International Agency for Research on Cancer Monograph Working Group, A review of human carcinogens - part D: radiation, Lancet Oncol., 2009, 10, 751–752 CrossRef.
- J. Cadet, E. Sage and T. Douki, Ultraviolet radiation-mediated damage to cellular DNA, Mutat. Res., Fundam. Mol. Mech. Mutagen., 2005, 571, 3–17 CrossRef CAS.
- C. Kielbassa, L. Roza and B. Epe, Wavelength dependence of oxidative DNA damage induced by UV and visible light, Carcinogenesis, 1997, 18, 811–816 CrossRef CAS.
- S. E. Freeman, R. W. Gange, J. C. Sutherland, E. A. Matzinger and B. M. Sutherland, Production of pyrimidine dimers in DNA of human skin exposed in situ to UVA radiation, J. Invest. Dermatol., 1987, 88, 430–433 CAS.
- T. Matsunaga, K. Hieda and O. Nikaido, Wavelength dependent formation of thymine dimers and (6–4) photoproducts in DNA by monochromatic ultraviolet light ranging from 150 to 365 nm, Photochem. Photobiol., 1991, 54, 403–410 CrossRef CAS.
- R. D. Ley and A. Fourtanier, UVA1-induced edema and pyrimidine dimers in murine skin, Photochem. Photobiol., 2000, 72, 485–487 CrossRef CAS.
- A. R. Young, C. S. Potten, O. Nikaido, P. G. Parsons, J. Boenders, J. M. Ramsden and C. A. Chadwick, Human melanocytes and keratinocytes exposed to UVB or UVA in vivo show comparable levels of thymine dimers, J. Invest. Dermatol., 1998, 111, 936–940 CrossRef CAS.
- Z. Kuluncsics, D. Perdiz, E. Brulay, B. Muel and E. Sage, Wavelengths dependence of ultraviolet-induced DNA damage distribution: involvement of direct or indirect mechanisms and possible artefacts, J. Photochem. Photobiol., B, 1999, 49, 71–80 CrossRef CAS.
- T. Douki, D. Perdiz, P. Grof, Z. Kuluncsics, E. Moustacchi, J. Cadet and E. Sage, Oxidation of guanine in cellular DNA by solar UV radiation: biological role, Photochem. Photobiol., 1999, 70, 184–190 CrossRef CAS.
- S. Courdavault, C. Baudouin, M. Charveron, A. Favier, J. Cadet and T. Douki, Larger yield of cyclobutane dimers than 8-oxo-7,8-dihydroguanine in the DNA of UVA-irradiated human skin cells, Mutat. Res., 2004, 556, 135–142 CrossRef CAS.
- T. M. Rünger and U. P. Kappes, Mechanisms of mutation formation with long-wave ultraviolet light (UVA), Photodermatol., Photoimmunol. Photomed., 2008, 24, 2–10 CrossRef.
- U. P. Kappes, D. Luo, M. Potter, K. Schulmeister and T. M. Rünger, Short- and long-wave ultraviolet light (UVB and UVA) induce similar mutations in human skin cells, J. Invest. Dermatol., 2006, 126, 667–675 CrossRef CAS.
- C. Robert, H. Mueller, A. Benoit, L. Dubertret, A. Sarasin and A. Stary, Cell survival and shuttle vector mutagenesis induced by ultraviolet A and ultraviolet B radiation in a human cell line, J. Invest. Dermatol., 1996, 106, 721–728 CAS.
- H. Ikehata, K. Kawai, J. Komura, K. Sakatsume, L. Wang, M. Imai, S. Higashi, O. Nikaido, K. Yamamoto, K. Hieda, M. Watanabe, H. Kasai and T. Ono, UVA1 genotoxicity is meditated not by oxidative damage but by cyclobutane pyrimidine dimers in normal mouse skin, J. Invest. Dermatol., 2008, 128, 2289–2296 CrossRef CAS.
- I. C. Enninga, R. T. L. Groenendijk, A. R. Filon, A. A. van Zeeland and J. W. I. M. Simons, The wavelength dependence of U.V.-induced pyrimidine dimer formation, cell killing and mutation induction in human diploid skin fibroblasts, Carcinogenesis, 1986, 7, 1829–1836 CrossRef CAS.
- F. R. de Gruijl, H. J. Sterenborg, P. D. Forbes, R. E. Davies, C. Cole, G. Kelfkens, W. H. van, H. Slaper, J. C. van der Leun, F. R. de Gruijl, H. J. Sterenborg, P. D. Forbes, R. E. Davies, C. Cole, G. Kelfkens, H. van Weelden, H. Slaper and J. C. van der Leun, Wavelength dependence of skin cancer induction by ultraviolet irradiation of albino hairless mice, Cancer Res., 1993, 53, 53–60 CAS.
- F. R. de Gruijl, Photocarcinogenesis: UVA vs. UVB, Methods Enzymol., 2000, 319, 359–366 CAS.
- P. C. Hanawalt, D. J. Crowley, J. M. Ford, A. K. Ganesan, D. R. Lloyd, T. Nouspikel, C. A. Smith, G. Spivak and S. Tornaletti, Regulation of nucleotide excision repair in bacteria and mammalian cells, Cold Spring Harbor Symp. Quant. Biol., 2000, 65, 183–191 CAS.
- S. Bhana and D. R. Lloyd, The role of p53 in DNA damage-mediated cytotoxicity overrides its ability to regulate nucleotide excision repair in human fibroblasts, Mutagenesis, 2008, 23, 43–50 CrossRef CAS.
- B. M. Stanulis-Praeger and B. A. Gilchrest, Effect of donor age and prior sun exposure on growth inhibition of cultured human dermal fibroblasts by all trans-retinoic acid, J. Cell. Physiol., 1989, 139, 116–124 CrossRef CAS.
- K. Werninghaus, R. M. Handjani and B. A. Gilchrest, Protective effect of alpha-tocopherol in carrier liposomes on ultraviolet-mediated human epidermal cell damage in vitro, Photodermatol., Photoimmunol. Photomed., 1991, 8, 236–242 CAS.
- A. Stary and A. Sarasin, Ultraviolet A- and singlet oxygen-induced mutation spectra,, Methods Enzymol., 2000, 319, 153–165 CAS.
- R. M. Sayre, C. Cole, W. Billhimer, J. Stanfield and R. D. Ley, Spectral comparison of solar simulators and sunlight, Photodermatol., Photoimmunol. Photomed., 1990, 7, 159–165 CAS.
- K. M. Thoms, C. Kuschal, E. Oetjen, T. Mori, N. Kobayashi, P. Laspe, L. Boeckmann, M. P. Schon and S. Emmert, Cyclosporin A, but not everolimus, inhibits DNA repair mediated by calcineurin: implications for tumorigenesis under immunosuppression, Exp. Dermatol., 2011, 20, 232–236 CrossRef CAS.
- T. Mori, M. Nakane, T. Hattori, T. Matsunaga, M. Ihara, O. Nikaido, T. Mori, M. Nakane, T. Hattori, T. Matsunaga, M. Ihara and O. Nikaido, Simultaneous establishment of monoclonal antibodies specific for either cyclobutane pyrimidine dimer or (6–4)photoproduct from the same mouse immunized with ultraviolet-irradiated DNA, Photochem. Photobiol., 1991, 54, 225–232 CrossRef CAS.
- A. de Laat, M. van Tilburg, J. C. von der Leun, W. A. van Vloten and F. R. de Gruijl, Cell cycle kinetics following UVA irradiation in comparison to UVB and UVC irradiation, Photochem. Photobiol., 1996, 63, 492–497 CrossRef CAS.
- R. M. Snapka, Bromodeoxyuridine photodamage in studies of UVA damage and the cell cycle, DNA Repair, 2009, 8, 3 CrossRef CAS.
- T. M. Rünger, B. Epe and K. Möller, Repair of ultraviolet B and singlet oxygen-induced DNA damage in xeroderma pigmentosum cells, J. Invest. Dermatol., 1995, 104, 68–73 Search PubMed.
- S. Emmert, H. Slor, D. B. Busch, S. Batko, R. B. Albert, D. Coleman, S. G. Khan, B. Abu-Libdeh, J. J. DiGiovanna, B. B. Cunningham, M. M. Lee, J. Crollick, H. Inui, T. Ueda, M. Hedayati, L. Grossman, T. Shahlavi, J. E. Cleaver and K. H. Kraemer, Relationship of neurologic degeneration to genotype in three xeroderma pigmentosum group G patients, J. Invest. Dermatol., 2002, 118, 972–982 CrossRef CAS.
- T. M. Rünger, B. Epe and K. Möller, Processing of directly and indirectly ultraviolet-induced DNA damage in human cells, Recent Results Cancer Res., 1995, 139, 31–42 Search PubMed.
- S. Emmert, N. Kobayashi, S. G. Khan and K. H. Kraemer, The xeroderma pigmentosum group C gene leads to selective repair of cyclobutane pyrimidine dimers rather than 6-4 photoproducts, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 2151–2156 CrossRef CAS.
- J. Dunn, M. Potter, A. Rees and T. M. Rünger, Activation of the Fanconi anemia/BRCA pathway and recombination repair in the cellular response to solar UV, Cancer Res., 2006, 66, 11140–11147 CrossRef CAS.
- J. M. Ford, Regulation of DNA damage recognition and nucleotide excision repair: another role for p53, Mutat. Res., Fundam. Mol. Mech. Mutagen., 2005, 577, 195–202 CrossRef CAS.
- S. Adimoolam and J. M. Ford, p53 and DNA damage-inducible expression of the xeroderma pigmentosum group C gene, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 12985–12990 CrossRef CAS.
- M. L. Smith and Y. R. Seo, p53 regulation of DNA excision repair pathways, Mutagenesis, 2002, 17, 149–156 CrossRef CAS.
- L. Latonen and M. Laiho, Cellular UV damage responses - functions of tumor suppressor p53, Biochim. Biophys. Acta, 2005, 1755, 71–89 CAS.
- D. Decraene, P. Agostinis, A. Pupe, P. de Haes and M. Garmyn, Acute response of human skin to solar radiation: regulation and function of the p53 protein, J. Photochem. Photobiol., B, 2001, 63, 78–83 CrossRef CAS B - Biology.
- L. A. de, E. D. Kroon and F. R. de Gruijl, Cell cycle effects and concomitant p53 expression in hairless murine skin after longwave UVA (365 nm) irradiation: a comparison with UVB irradiation, Photochem. Photobiol., 1997, 65, 730–735 CrossRef.
- P. M. Girard, M. Pozzebon, F. Delacote, T. Douki, V. Smirnova and E. Sage, Inhibition of S-phase progression triggered by UVA-induced ROS does not require a functional DNA damage checkpoint response in mammalian cells, DNA Repair, 2008, 7, 1500–1516 CrossRef CAS.
- G. Maga, G. Villani, E. Crespan, U. Wimmer, E. Ferrari, B. Bertocci and U. Hubscher, 8-oxo-guanine bypass by human DNA polymerases in the presence of auxiliary proteins, Nature, 2007, 447, 606–608 CrossRef CAS.
- J. L. Rizzo, J. Dunn, A. Rees and T. M. Rünger, No formation of DNA double-strand breaks and no activation of recombination repair with UVA, J. Invest. Dermatol., 2011, 131, 1139–1148 CrossRef CAS.
- T. Douki, A. Reynaud-Angelin, J. Cadet and E. Sage, Bipyrimidine photoproducts rather than oxidative lesions are the main type of DNA damage involved in the genotoxic effect of solar UVA radiation, Biochemistry, 2003, 42, 9221–9226 CrossRef CAS.
Footnote |
| † Contribution to the themed issue on the biology of UVA. |
| This journal is © The Royal Society of Chemistry and Owner Societies 2012 |