Iron, oxidative stress and the example of solar ultraviolet A radiation†
Asma
Aroun‡
,
Julia Li
Zhong‡§
,
Rex M.
Tyrrell
and
Charareh
Pourzand
*
Department of Pharmacy and Pharmacology, University of Bath, Bath, BA2 7AY, United Kingdom. E-mail: prscap@bath.ac.uk; Fax: (+44)1225386114; Tel: (+44)1225383590
First published on 11th October 2011
Abstract
Iron has outstanding biological importance as it is required for a wide variety of essential cellular processes and, as such, is a vital nutrient. The element holds this central position by virtue of its facile redox chemistry and the high affinity of both redox states (iron II and iron III) for oxygen. These same properties also render iron toxic when its redox-active chelatable ‘labile’ form exceeds the normal binding capacity of the cell. Indeed, in contrast to iron bound to proteins, the intracellular labile iron (LI) can be potentially toxic especially in the presence of reactive oxygen species (ROS), as it can lead to catalytic formation of oxygen-derived free radicals such as hydroxyl radical that ultimately overwhelm the cellular antioxidant defense mechanisms and lead to cell damage. While intracellular iron homeostasis and body iron balance are tightly regulated to minimise the presence of potentially toxic LI, under conditions of oxidative stress and certain pathologies, iron homeostasis is severely altered. This alteration manifests itself in several ways, one of which is an increase in the intracellular level of potentially harmful LI. For example acute exposure of skin cells to ultraviolet A (UVA, 320–400 nm), the oxidising component of sunlight provokes an immediate increase in the available pool of intracellular LI that appears to play a key role in the increased susceptibility of skin cells to UVA-mediated oxidative membrane damage and necrotic cell death. The main purpose of this overview is to bring together some of the new findings related to intracellular LI distribution and trafficking under physiological and patho-physiological conditions as well as to discuss mechanisms and consequences of oxidant-induced alterations in the intracellular pool of LI, as exemplified by UVA radiation.
1 Introduction
1.1 Iron and oxidative stress
Iron is the second most abundant metal, after aluminium, and the fourth most abundant element in the earth's crust (5%). Living organisms from bacteria to mammals use iron for vital biological processes, as part of, or as a cofactor of proteins and enzymes.1Iron plays a key role in mammalian cells' growth, respiration and replication. Many iron-containing proteins catalyze key reactions involved in energy metabolism (cytochromes, mitochondrial aconitase, iron-sulfur proteins of the electron transport chain), respiration (e.g.hemoglobin), and DNA synthesis (i.e.ribonucleotide reductase) and it is well known that iron depletion leads to G1/S cell cycle arrest and apoptosis.2 Additionally, iron-containing proteins are required for the metabolism of collagen, tyrosine and catecholamines.3Iron is a transition metal that can exist in two stable configurations: electron donor ferrous (Fe2+) and electron acceptor ferric (Fe3+). The easy access to two oxidation states allows iron to act as a catalyst in mammalian cellular pathways that involve redox mechanisms, however this same property makes iron toxic when its redox-active chelatable ‘labile’ form exceeds the normal binding capacity of the organism.4 Indeed in contrast to iron bound to proteins, the intracellular labile iron (LI) can undergo redox cycling between its most stable oxidation states Fe+2/Fe+3 and react with ROS such as superoxide anion (O2˙−) and hydrogen peroxide (H2O2) giving rise to hydroxyl radical (OH˙) via the Fenton reaction (eqn (2)) or via superoxide-driven Fenton chemistry (eqn (3)).5 Such reactive ROS are capable of interacting with most biomolecules, depending on the site of bound iron, including sugars, lipids, proteins, and nucleic acids. These interactions promote various harmful processes in cells including lipid peroxidation, protein oxidation, DNA/RNA oxidation and DNA lesions and ultimately overwhelm the cellular antioxidant defense mechanisms and lead to cell damage and death.
| O˙−2 + Fe3+ → Fe2+ + O2 | (1) |
| H2O2 + Fe2+ → OH˙ + OH− + Fe3+ (Fenton reaction) | (2) |
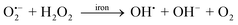 | (3) |
1.2 UVA and oxidative stress
At the cellular level, the interaction of solar ultraviolet (UV) radiation with biological material changes as a function of wavelength and requires the absorption of the radiation by biomolecules. The UVB (290–320 nm) region of sunlight overlaps with the DNA absorption spectrum and, as a result, the direct absorption of UVB by cellular DNA causes DNA photodamage and mutagenesis.6 In contrast, the UVA (320–400 nm) region of sunlight is weakly absorbed by most biomolecules but is predominantly oxidative in nature, generating ROS via photochemical interactions with intracellular chromophores.7 The ROS generated by UVA are certainly involved in cytotoxicity, as it has been shown that UVA inactivation of mammalian cells is strongly oxygen-dependent.8 Furthermore there is evidence from in vitro studies that UVA irradiation of macromolecules can cause the generation of H2O2 and O2˙− and that iron-catalyzed reduction of H2O2 by O2˙− can further generate the highly reactive OH˙.9,10 Studies in prokaryotic and eukaryotic cells indicate that ROS may also be generated in vivo by UVA irradiation.7,9,11UVA may also trigger the formation of ROS long after UVA exposure via non-photosensitised mechanisms. This may occur by activation of enzymatic systems such as NADPH oxidase that generates O2˙−.12 Based on such considerations, the UVA component of sunlight is now considered as a generator of intracellular oxidative stress.The uncontrolled production of ROS by UVA is undesirable and thus a number of protective strategies are adopted by cells to prevent their formation. Mammalian cells possess several antioxidant enzymes including superoxide dismutase (SOD), catalase and glutathione peroxidase (GPx). In addition, tissues contain many antioxidant molecules such as the endogenous compound glutathione (GSH), which is a major contributor to cellular reducing equivalents and compounds derived from the diet such as vitamins (e.g.alpha-tocopherol, ascorbate), carotenoids and flavonoids. It has been shown that GSH plays a critical role in cellular defence against the lethal action of both UVA and UVB radiations.13 All these antioxidant enzymes and molecules would be expected to be important in cellular defence against UVA-induced oxidative stress. However the release of LI in skin cells that occurs immediately after UVA irradiation14 provokes the excess production of highly reactive ROS such as OH˙ which can overwhelm the antioxidant capacity of the cells leading to cell damage and possible death (see sections 1.3 and 2.3).
1.3 UVA, iron and oxidative stress
Under normal conditions, iron levels in cells and body are under extremely tight control and there is little opportunity for iron-catalysed free radical generating reactions to occur. However upon exposure to oxidising agents such as UVA, the intracellular iron status changes dramatically as UVA leads to both LI and free heme releases in cells.14,15 Although heme itself is not a source of LI, it is a substrate for heme-catabolising enzyme heme oxygenase (HO) which could release the heme iron and expand the pool of potentially harmful cytosolic LI.16 The dual role of UVA as ROS generator and LI enhancer classifies this radiation as a potent oxidising component of sunlight. Indeed UVA-mediated generation of ROS not only induces direct oxidative damage to skin constituents but also leads to an immediate increase in cytosolic labile iron pool (LIP), which in turn exacerbates the oxidative damage already occurring in the skin cells. Both singlet oxygen (1O2) and H2O2, the most important ROS species generated by UVA, exert their biological damage in skin cells predominantly viairon-catalysed oxidative reactions.17–20 For example it has been shown that physiologically relevant doses of UVA induce lipid peroxidation in membranes of human primary fibroblasts and keratinocytesvia pathways involving iron, H2O2 and 1O2.17,21,22 In fact, lipid peroxidation, which is considered as the prominent marker of UVA-induced photo-oxidative damage in skin, involves iron as the major contributor for initiation and propagation of peroxidative damage.17,21–23 This is because iron ‘at’ or ‘near’ strategic targets such as cell membranes, can undergo redox cycling by reacting sequentially with one electron reductants and oxidants, thereby generating toxic oxidants such as OH˙ and lipid derived alkoxyl and peroxyl radicals and can elicit biological damage.5,24Lipid peroxidation results in the destruction of membrane function and structure, disturbed membrane fluidity and ultimately loss of membrane integrity and cell lysis.25The UVA-induced expansion of intracellular LIP ultimately overwhelms the cellular antioxidant defense mechanisms and leads to cell damage.26 Depending on the severity of the UVA dose applied, the LI-mediated oxidative damage may lead to cell death by apoptosis or necrosis.27,28 Alternatively the damage may be processed in such way that the carcinogenesis process is initiated or promoted.26 The injurious consequences of iron-catalysed damage exerted by UVA have been shown to play a key role in skin photoaging and the promotion of skin cancer.23,26,28,29
The understanding of the mechanisms underlying the disturbance of iron homeostasis by UVA appears therefore crucial to understand how UVA interacts with cells and tissues. Here we first provide an overview of iron distribution in cells and body and then introduce the iron homeostasis machinery composed of a complex network of transporters, storage molecules and regulators that coordinately govern iron absorption, iron recycling, and the mobilisation of stored iron in the organism. While these regulatory networks minimise the pool of potentially toxic LIP in cells and tissues, we describe how iron homeostasis is altered under oxidative and pathological conditions. Using this knowledge, we then discuss the mechanisms and consequences of oxidant-induced alterations in the intracellular pool of LI, as exemplified by UVA radiation.
2 Iron absorption, storage, transport and distribution
2.1 The body iron pool
Iron is absorbed from the mammalian gastrointestinal tract by two protein-mediated mechanisms, one absorbing iron as Fe2+ and the other iron as heme.30 Primates have evolved not to be able to excrete iron, and therefore body iron levels are totally controlled by the absorption process.31 The total amount of iron in an average human body is about 4–5 g, the majority (i.e. 75%) of which is incorporated into the heme complex and can be found in proteins such as hemoglobin and myoglobin.32 Only a very small amount of the total body iron is present as an essential component of a very large number of heme (e.g.cytochromes, catalase, oxidases and peroxidases) and non-heme proteins and enzymes (e.g.ribonucleotide reductase and iron-sulfur proteins). All of these may account for no more than 10% of the total body iron pool. The remaining 15% can be found principally in the cytoplasm in the form of the iron storage protein ferritin (Ft) but also in lysosomes as hemosiderin.Storage iron is found in the liver, mainly in the hepatocytes, but it is also found in macrophages in the liver, bone marrow, spleen and muscles, where it is readily available as a reserve in case of blood loss.33,34 Due to epithelial shedding in the gastrointestinal tract and the skin and because of blood loss in women, there is an average daily loss of 1–2 mg. This loss is usually compensated for by the absorption of iron through the diet which contains approximately 10–20 mg of iron of which 1–2 mg is absorbed under normal circumstances. This absorption is increased several-fold when iron levels are low (e.g. in anaemia and hypoxia) and decreased when the iron stores are replete (e.g. in iron-overload conditions and inflammation).31,35
2.2 The labile iron pool
All the iron-complexing molecules leave body fluids and cells with an extremely low concentration of free transit iron. Nevertheless there is now strong evidence for the existence of a transit pool of catalytically active iron complexes which is distinct from intracellular iron associated with proteins and is known as the labile iron pool (LIP). Iron belonging to this pool is considered to be in steady-state equilibrium, loosely bound to low-molecular-weight compounds, accessible to permeant chelators and metabolically and catalytically reactive.36 The LIP has been detected both in the systemic circulation and inside cells and is likely that all daily uptake of the dietary iron (1–2 mg) will pass via the LIP stage before it is sequestered by target proteins.37The LIP is associated with important functions: (a) physiologically, as readily available source of iron for incorporation into proteins; (b) pharmacologically, as target for chelators or metal scavengers; and (c) toxicologically, as vehicle for promoting the formation of free radicals.38
2.3 Iron homeostasis
To minimise damage caused by LI, both plasma and intracellular LIP are tightly regulated by means of a complex network of transporters, storage molecules and regulators that coordinately govern iron absorption, iron recycling, and the mobilisation of stored iron. These regulatory networks maintain an adequate level of iron for vital cell, tissue and organ functions, while also minimising the pool of potentially toxic LIP.The protein central for systemic iron homeostasis is the hepatic peptide hormone hepcidin that regulates plasma iron concentrations and tissue iron distribution by inhibiting dietary iron absorption and mobilization. Hepcidin acts by controlling the number of iron channels through which cellular iron is delivered into plasma.40 In the small intestine, iron is transported out of the enterocyte into the blood via the basolateral iron export protein, ferroportin-1 (FP1).41,42 When plasma iron is in excess, hepcidin binds to FP1 receptor. This leads to internalisation and degradation of the FP1 receptor which in turn cause the blockage of iron efflux into plasma, resulting in iron retention within cells.40 Hepcidin follows an inverse relationship with body iron levels: low levels can potentially cause iron overload, whilst high levels cause anaemia.35,40 In turn, hepcidin concentration is regulated by a variety of influences. Iron loading increases hepcidin production (to block further iron absorption and maintain homeostasis), and so does inflammation (to decrease extracellular iron concentration and its accessibility to micro-organisms).43
Alterations in intracellular LIP are normally sensed by the cytosolic iron regulatory proteins 1 and 2 (IRPs) which function as post-transcriptional regulators of both iron uptake via the transferrin receptor 1 (TfR1) and iron sequestration by the iron storage protein, Ft. This regulation occurs via binding of IRPs to conserved iron responsive elements (IRE) in the untranslated region of Ft and TfR1 mRNAs.44 When iron is scarce in the LIP, Ft and TfR1 mRNAs are specifically recognized and bound by the active forms of IRPs, leading to stabilisation of the TfR1 mRNA and inhibition of Ft translation, both of which will lead to enhanced levels of LIP. Conversely, during an increase in iron supply, IRPs are converted to low affinity mRNA-binding proteins, leading to the induction of Ft mRNA translation and the degradation of TfR1 mRNA, which will ultimately lead to a reduction in the LIP.44
Iron regulatory protein 1 (IRP1) is the cytoplasmic counterpart of mitochondrial aconitase, the enzyme which converts citrate to isocitrate through a cis-aconitate intermediate in the tricarboxylic acid cycle by virtue of a catalytic iron-sulfur cluster.1 In iron-replete cells the cluster is assembled and IRP1 displays aconitase activity; in iron-depleted cells the cluster is lacking and IRP1 functions as an mRNA-binding protein. This reversible switch between a cluster-containing holoprotein and a cluster-deficient apoprotein therefore allows aconitase/IRP1 to constantly sense LI levels and to adapt them to cell requirements. Iron regulatory protein 2 (IRP2) is highly homologous to IRP1 but lacks aconitase activity, probably because of its inability to assemble an iron-sulfur cluster; the protein accumulates in iron-deficient cells and is rapidly targeted for proteasomal degradation in iron-replete cells.1 Unlike IRP1, IRP2 has a characteristic pattern of tissue expression44 and is also more sensitive to variations of iron in the diet.1 Moreover, when abundantly or uniquely expressed, it can act as the major or only modulator of intracellulariron metabolism.1
The involvement of H2O2 in numerous types of cell and tissue injury is well-documented.48–51 Although H2O2 itself has low reactivity towards cell constituents, it is capable of forming highly reactive ROS in the presence of trace amounts of catalytic LIviaFenton reaction. Under physiological conditions, cells protect themselves either by the H2O2-degrading enzymes catalase and GPx,52 or by minimising the intracellular level of potentially harmful LIPvia the cytosolic IRP1 and IRP2 regulatory network.44,53 However under pathological conditions, these conventional cellular defences are often insufficient, because the system is either overwhelmed by an increased H2O2 formation49,54–56 and/or by an excess presence of LI.57 The simultaneous presence of excess redox active chelatable LI and H2O2 can be potentially toxic for cells as it can catalyse the formation of oxygen free radicals such as OH˙ and can elicit irreversible oxidative damage. The pathological consequences of iron-catalyzed oxidative damage are recognized in diseases such as hepatitis, liver cirrhosis, hemochromatosis, cancer, chronic inflammatory disorders and neurodegenerative diseases.58–66 Excess iron may also aggravate diabetes, cancer, cardiovascular disease and alcoholic and non-alcoholic steatohepatitis.67–71
High levels of LI have also been identified as a risk factor for the development of cancer.72 Numerous studies across a variety of populations have found a positive correlation between iron stores in the body and risk of the development of a range of cancers including colorectal, liver, kidney, lung and stomach cancers.73 High dietary iron has also been reported to induce skin cancer in mice.74 Furthermore in both animals and humans, primary neoplasms develop at body sites of large iron deposits such as skin, which is a potential target for significant oxidative damage due to its constant exposure to high oxygen tensions and frequent exposure to ultraviolet (UV) light.32 During the process of carcinogenesis, intracellular iron homeostasis is altered, with neoplastic cells, poor in iron relative to their normal counterparts, internalizing iron from Tf at a tremendous rate, despite low levels of the iron storage protein, Ft.2
The presence of excess iron has also been demonstrated in a variety of skin disorders such as psoriasis,75 venous ulceration76 and atopic eczema,77 indicating the involvement of iron in the pathology of skin.
Iron accumulation in the body also occurs with age in males. In women, however, iron accumulation does not occur until after the menopause. Iron accumulation in post-menopausal women has been linked to increased incidence of heart disease.78 Increased levels of iron in post-menopausal women have also recently been considered as etiological agents potentially involved in the increased risk of oxidative damage in skin.79 There is also evidence for age-related accumulation of LIP associated with rheumatoid arthritis, atherosclerosis and Alzheimer's disease (AD).68 Interaction of iron and cholesterol in promoting oxidative damage has been suggested as causative of both atherosclerosis and neurodegeneration.80 Mutations in genes involved in controlling iron have also been shown to cause predisposition to AD.68Iron may also play a role in the carcinogenic process of other transition metals such as copper, or other types of carcinogens.72
We have also observed that exposure of skin cells to UVA (320–400 nm) inactivated the mRNA-binding activity of IRP114 and that this correlated with a reciprocal increase in the aconitase activity of the IRP-1 (which is known to occur in response to the increased availability of intracellular LIP1). The inactivation of IRP1 by UVA in skin fibroblasts is related to the excess LI and this was confirmed by measuring the LIP levels with the fluorescent Calcein assay.36 From these observations, it was concluded that UVA induces immediate release of LI in the skin fibroblast cells.14 Further studies from this laboratory revealed that in addition to UVA, H2O2 treatment of skin fibroblasts also promotes a dose-dependent increase in intracellular LIP.26,87,88 Furthermore our investigations revealed that the proteolytic degradation of Ft by UVA is a major contributor to the observed increase in LI in irradiated skin fibroblasts.14 Numerous other studies have also demonstrated that pro-oxidant stresses are capable of modulating iron homeostasis by elevating the intracellular LIPvia release of iron from sensitive sites, such as iron-sulfur clusters and Ft. Stress stimuli for which this has been observed include peroxides, nitrofurantoin, nitric oxide, phorone, ischemia-reperfusion and hypothermia injury.14,26,37,57,86,89
In addition to IRP1, IRP2 is also specifically modulated in response to oxidative stress in the form of enhanced production of ROS and reactive nitrogen intermediates.1
Oxidative stress can also affect iron homeostasis viaactivation of heme oxygenase 1 (HO-1). The strong transcriptional activation of HO-1 by UVA was first identified in human skin fibroblasts.90 Later studies demonstrated that this phenomenon also occurs in melanocytic and dendritic cells, and in certain pathologic states it is detected in epidermal keratinocytes.91 It is now recognised that all conditions that lead to perturbation of heme homeostasis (e.g. oxidative stress, tissue injury and inflammation) lead to rapid changes in heme synthesis (viaaminolevulinate synthases) and catabolism (viaHO). This adaptive response to restore heme homeostasis in oxidative stress and related pathological conditions must also take care of any perturbations in cellular LIP so that Ft is almost always induced, albeit more slowly, under the same conditions. The aberrant cellular and tissue iron distribution and severe iron-deficient anemias observed in HO-deficient rodents (and in the one human case recognised to date) indicate that heme oxygenases themselves also play a crucial role in controlling tissue iron distribution and homeostasis.92
Studies from Tyrrell's laboratory have shown that acute exposure to UVA or H2O2 promotes immediate heme release from microsomal hemoproteins in a human primary skin fibroblasts cell line, FEK4.15 Although heme itself is not a source of LIP, it is a substrate for the heme-catabolizing enzyme, HO which could release the heme iron. In the short term, HO activity can cause hypersensitivity to oxidative UVA radiation due to release of iron from heme.93 In the long term (i.e. 1–2 days) however, the UVA-mediated activation of HO-1 leads to HO-1-dependent increase in Ft94 and a consequent lowering of the pro-oxidant state in skin cells.95 Studies from Girotti and co-workers have also shown that short-term treatment of murine lymphocytic leukemic cells with hemin, causes an early increase in HO-1 and LI that sensitises the cells to acute oxidative killing, however long-term hemin treatment results in elevated H-Ft levels that sequesters the potentially harmful LI and enhances cellular resistance to acute exposure to oxidants.96 In contrast to acute conditions, repeated exposure of cells to oxidizing agents such as UVA or H2O2 has been shown to induce the HO-1 refractory response presumably as a safety mechanism to shut off the excess LI production as a result of repeated heme breakdown.16,97–99
Recent research aimed at understanding the mechanisms underlying the oxidant-induced alterations in iron homeostasis has revealed that the differential subcellular LI distribution plays a key role in promotion of iron-catalysed oxidative damage in cells. Furthermore translocation and trafficking of LI between subcellular compartments appears to be a hallmark of oxidative stress leading to propagation of iron-catalysed oxidative damage in cells. In order to understand the consequences of alteration of iron homeostasis by UVA, it is therefore necessary to understand how LI is distributed in subcellular compartments under physiological and oxidative conditions. The following sections provide an overview of the recent findings in this domain.
2.4 Labile iron distribution in subcellular compartments
Due to the high physiological and patho-physiological importance of LI distribution, great efforts have been made to develop suitable methods to investigate the intra- and sub-cellular LI levels. Most methods to characterize intracellular LI distribution seriously perturb the extent and nature of the LIP, rendering them inapplicable to living cells.100 In combination with non-invasive and high resolution confocal imaging, fluorescent chemosensors have proven to be highly sensitive tools to investigate the metabolism of LIP in individual cells with subcellular resolution. The methodology developed by Cabantchik and co-workers using Calcein-AM, a fluorescein derivative has so far been the most frequently applied fluorescent chemosensor to study intracellular LIP.4 However, due to hydrophilic nature of Calcein (CA) and lack of access to subcellular compartments, it has been demonstrated that the CA-based measurements reflect mostly the cytosolic LI rather than the overall intracellular LIP.101 Using this methodology, we observed that the cytosolic LIP levels of primary human skin keratinocytes were 2–3-fold lower than their matched primary human skin fibroblasts.28 The lower cytosolic LI content of skin keratinocytes correlated with their higher resistance to UVA-induced oxidative damage.28 Interestingly CA-assay also revealed that the cytosolic LIP content of highly proliferative human keratinocytes is much higher (i.e. 3–4-fold) than the low proliferating primary skin keratinocytes, as observed in a series of squameous cell carcinoma and psoriatic cell lines isolated from patients (unpublished data, this laboratory). The high requirement of neoplastic cells for iron that is required for rapid cell division has been shown to provoke an adaptive cellular response in the form of high Tf-bound iron uptake by increased TfR1 expression and low iron storage by decreased Ft content to maximise the availability of intracellular LIP for ribonucleotide reductase that requires iron for DNA synthesis.2To overcome the limitation of the CA-fluorescent assay, Hider and co-workers have recently developed a range of highly sensitive novel fluorescent iron chelators such that a fluorescent function (i.e.coumarin substitutes) is covalently linked to 3-hydroxypyridin-4-one (HPO). Among these, CP655 (7-diethylamino-N-[(5-hydroxy-6-methyl-4-oxo-1,4-dihydropyridin-3-yl)methyl]-N-methyl-2-oxo-2H-chromen-3-carboxamide), which is a moderately lipophilic fluorescent chelator, was found to be the most sensitive probe for monitoring intracellular LIP.102,103 The concentration of the intracellular chelatable iron pool in hepatocytes was determined by this probe to be 5.4 ± 1.3 micromolar.103
To measure the distribution of LI in subcellular compartments, Petrat and co-workers introduced a fluorescein-based iron sensor ‘Phen green SK’ incorporating a 1,10-phenanthrolin chelation unit.104,105 This iron sensor was capable of detecting much higher cytosolic LI concentrations in cultured hepatocytes as well as being able to detect LI in subcellular compartments in a number of cultured cell lines. The quantitative studies of chelatable LI with ‘Phen green SK’ revealed that both lysosomal and mitochondrial compartments have 2–3-fold higher levels of LI when compared to the cytosolic pool.105–107 These studies also highlighted the surprisingly high content of redox-active LI in the nucleus with clear implications for an increased probability of DNA damage.37
The presence of high levels of LI in mitochondria, lysosomes and nucleus makes these organelles highly vulnerable to oxidative damage upon exposure to oxidising agents such as UVA or H2O2.26 For example, we have observed that in skin fibroblasts, UVA promotes an immediate and dose-dependent damage to lysosomal, mitochondrial and nuclear membranes that can be prevented by organelle-permeable iron chelator pre-treatment.14,26,28,87,88,108
The development of real-time fluorimetry and flow cytometry techniques in conjunction with targeted fluorescent iron sensors capable of monitoring concurrently the cytosolic and mitochondrial LI changes,109 as well as lysosomal/endosomal-specific fluorescent iron sensors with high responsiveness towards alterations of endosomal/lysosomal LI100 have provided valuable insights into understating the mechanisms underlying the subcellular LI distribution and trafficking under physiological and patho-physiological conditions. Below, we provide a summary of the recent findings in this domain.
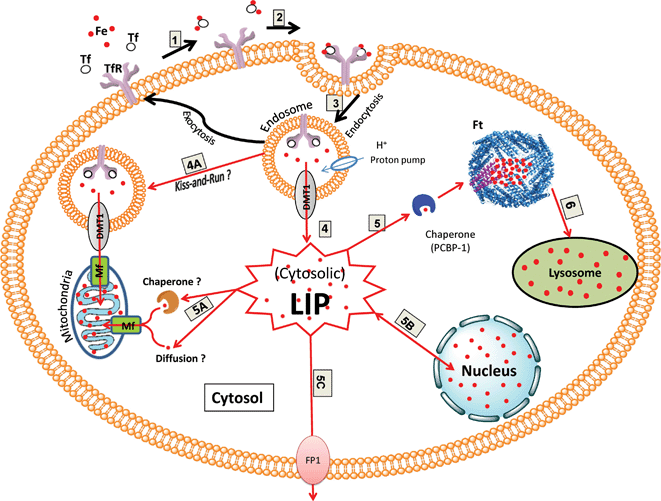 | ||
| Fig. 1 Schematic diagram illustrating the pathways of cytosolic LI uptake and distribution (modified from Breuer et al., 2008110). The main source of cytosolic LIP uptake is viareceptor-mediated endocytosis (1–3): In extracellular media, transferrin (Tf) binds two atoms of Fe3+ with high affinity (1). Two molecules of diferric-Tf bind to the transferrin receptor 1 (TfR1) on the cell surface (2). The Tf-TfR1 complex formed is internalized into an endosome. Within the endosome, iron is released from Tf following the decrease in intra-vesicular pH (3). Iron transfers from Tf to divalent metal transporter (DMT1) and is released in the Fe2+ form to the cytosol to join the pool of available cytosolic labile iron (LIP) (4). Endosomal iron may be directly delivered to mitochondria (i.e. without entering the cytosolic pool) via a hypothetical kiss-and-run mechanism involving DMT1 and mitoferrin (Mf) (4A). The cytosolic LIP can be distributed to various cellular targets (5–6): Cytosolic LI can be transported by a chaperone (PCBP-1) to be stored in ferritin (Ft) (5). Alternatively cytosolic LI may reach and replenish mitochondrial LI either via a hypothetical chaperone or possibly by diffusion (5A). Cytosolic LI can also establish a dynamic equilibrium with nuclear LI presumably vianuclear pores (5B). Cytosolic LI may also be exported from the cellsvia ferroportin-1 (FP1) (5C). The cytosolic iron stored in Ft may replenish the lysosomal LIviaautophagy and proteolytic degradation of Ft in lysosomes (6). | ||
The endosomes containing the Tf-TfR1 complex then undergo exocytosis to recycle TfR1 and return the apo-Tf to the bloodstream where it is able to bind more iron from the liver (see Fig. 1).114
The cytosolic LIP can reach several targets (see Fig. 1). It can either be stored in Ft for long term storage115 or be readily used in the synthesis of various proteins and enzymes such as ribonucleotide reductase.116 In general it is assumed that most of the cytosolic LI that is not metabolised is stored in Ft. Ferritin is an ubiquitously expressed cytosolic iron storage protein which forms a hetero-oligomeric protein shell composed of 24 Ft light (L-Ft, 19 kD) and heavy (H-Ft, 21 kD) chain subunits. Up to 4500 iron atoms can be sequestered in Ft as a crystalline core of ferric ions (Fe3+). Storing iron in Ft prevents LI from generating toxic radicals and allows the regulated release of iron.115 Studies with murine and rat models have shown that loss of cytosolic iron storage Ft by down-regulating or deleting H-Ft leads to an increase in LIP, oxidative stress, tissue damage and cell death.117,118 Recent work has identified a cytosolic iron chaperone, PCBP1 [poly(rC)-binding protein 1] that transports iron to Ft.119 Depletion of PCBP1 in human cells inhibited Ft iron loading and increased cytosolic LIP.119
The cytosolic LIP may also reach the mitochondria directly via the putative mitochondrial iron import protein, mitoferrin (Mf), for synthesis of heme and iron-sulfur clusters.110 The latter pathway was first supported by Lange et al. who measured Fe2+ ingress into isolated mitochondria.120 The recent development of real-time fluorimetry and flow cytometry techniques in conjunction with targeted fluorescent iron sensors capable of monitoring concurrently the cytosolic and mitochondrial changes in LI evoked by Tf-bound iron109 confirmed the initial observation by Lange et al.120 Using this methodology, Cabantchik and co-workers demonstrated that the bulk of Tf-bound iron initially delivered to cellsviaendocytosis can be quantitatively traced in the cytosol as cytosolic LIP, from where it is distributed to other cell compartments, notably mitochondria.109 At present it is not known whether cytosolic LI is delivered to mitochondria by high-affinity chaperone-like moieties, as proposed for Ft or simply by diffusion.109,110,119 The mitochondrial protein frataxin, that is deficient in individuals suffering from neuromuscular disease Friedreich's ataxia (FRDA), could conceivably fulfil the chaperone-like function, as frataxin is thought to deliver iron for the synthesis of iron-sulfur clusters to these organelles and has also been recently found in the cytosol.110 The presence of a high-affinity chaperone-type iron acquisition system to mitochondria is also supported by a study by Garrick et al., who demonstrated that high affinity iron chelates could donate iron for heme synthesis.121
It has been proposed that iron delivery to mitochondria may also occur via an alternative mechanism that bypasses the cytosolic LIP route. This includes the direct transfer of iron from endosomes (i.e.iron sequestered by Tf-TfR1 pathway) to the mitochondria as a ‘kiss-and-run’ mechanism, mediated by DMT1 (as endosomal iron exporter) and Mf (as mitochondrial iron importer) (see Fig. 1).122,123 This alternative mechanism is thought to apply primarily to erythroid cells which require a high rate of iron utilization for heme synthesis.122–125 The terminal step of heme biosynthesis requires the insertion of Fe2+ ion by ferrochelatase enzyme into heme precursor, protoporphyrin IX (PPIX) to produce heme. In hemoglobin-synthesizing cells, the vast majority of iron released from endosomes must cross both the outer and the inner mitochondrial membranes to reach ferrochelatase.126 It is remarkable that in these cells iron acquired from Tf continues to flow into mitochondria, even when the synthesis of PPIX is markedly compromised in vitro127–129 (by isonicotinic acid hydrazide or succinylacetone) or in vivo130 (patients with erythroid specific 5-aminolevulinate synthase deficiency). A significant proportion of non-heme iron that accumulates in mitochondria under these circumstances is in a form readily available for heme synthesis when PPIX formation is restored.127–129 The ‘kiss-and-run’ is defined as a transient endosome-mitochondrion interaction to mediate iron transfer between these organelles. This model proposes that, after iron is released from Tf in the endosome, it is passed directly from endosomal iron exporter DMT1 to mitochondrial iron importer Mf until it reaches the ferrochelatase in the mitochondrion. Interestingly, when heme synthesis is inhibited in definitive erythroid cells, very little128,129 or no131iron accumulates in cytosolic Ft. In contrast, it is well established that in normal non-erythroid cells, iron in excess of metabolic needs ends up in Ft. Thus, it seems highly likely that in erythroid cells the transport of iron into mitochondria is controlled differently than in non-erythroid cells.122,126
Metabolites can cross the mitochondrial outer membrane through a large diameter voltage dependent channel, also known as mitochondrial porin,132 while the inner mitochondrial barrier can only be crossed with the aid of specialised mitochondrial solute carrier family (MCF), localised in the inner mitochondrial membrane.133Mitoferrin-1 (Mf1, Slc25a37), is a member of the MCF that functions as an essential iron importer for the synthesis of heme and iron-sulfur clusters in erythroblasts.124 Recent work has shown that in erythroblasts, ferrochelatase forms an oligomeric complex with Mf1 and Abcb10 to synergistically integrate mitochondrial iron incorporation and use for heme biosynthesis.125 A similar protein, named mitoferrin-2 (Mf2) has also been identified in zebrafish and mammals that unlike erythroid Mf1, is expressed at low levels in all tissues.134 The latter may be involved in iron import into the mitochondria of non-erythroid cells.133 In skin fibroblasts, we have observed that growing cells under low serum concentration for several days could substantially decrease the basal intracellular level of LIP (as measured by IRP1 RNA-binding activity) and cause a related increase in the basal level concentrations of the mitochondrial heme precursor PPIX. The latter study suggests that the extent of heme biosynthesis in mitochondria of skin cells is directly related to the availability of intracellular LIP.135
Alternatively the cytosolic LIP may reach the plasma membrane where it is exported via the iron exporter protein FP1 (see Fig. 1).110 The divalent iron exporter FP1 (also called IREG1 and MPT1) has been found in the basolateral membrane of enterocytes.136 It has been shown that FP1 is only expressed in liver, spleen and kidney, so the mechanism of iron export from other cell types is unclear.37
Upon export from lysosomes, this rich source of LIP may contribute to the continued synthesis of new iron-containing proteins,142–144 although it is not known exactly how redox-active LI is transported from lysosomes to other subcellular compartments. The transport of lysosomal LI to cytosol may involve DMT1 similar to release of iron from late endosomes, although it is not known whether iron bound to DMT1 is redox-active.141 An alternative mechanism for the delivery of LI from late endosomes/lysosomes to mitochondria may involve the direct transport between these two organelles following temporary close contact (see ‘kiss-and-run’ in section 2.4.1).122,123
2.5 The role of subcellular LIP distribution in the cellular response to oxidative stress
Earlier studies aimed at understanding the role of intracellular LIP in potentiating oxidative damage in cells consisted of loading the cells with Tf-Fe or iron-salts followed by an oxidative challenge of cells with oxidizing agents such as t-butyl-hydroperoxide (TBHP), H2O2 or UVA radiation. These studies indicated that concomitantly with the rising of LIP levels in the cytosol (as measured by CA-assay), there was demonstrable rise in ROS production (possibly viaFenton reaction), lipid peroxidation and eventual cell death.2,146,147 Recent studies revealed however that the relationship between cytosolic LIP and intracellular ROS generation is more complex than previously thought. Indeed it is now known that intracellular LIP can not only act as a potent generator of ROS but its level can also be raised by oxidants or reductants.Direct measurements of intracellular LIP by electron paramagnetic resonance also indicated an expansion in intracellular LI with a concomitant drop in intracellular Ft levels in the liver of rats subjected to oxidative stress in the form of phorone (a GSH-depleting drug) or ischemia-reperfusion.57,86 The early drop in Ft in the latter studies was demonstrated to be a proteolytic degradation event leading to an increase in intracellular LIP.57,86 Studies from this laboratory have also demonstrated that the UVA-induced increase in cytosolic LIP is partly due to proteolytic degradation of Ft.14
Ferritin degradation in cytosol may occur via proteasomes upon oxidative damage, pathological conditions, FP1 overexpression or treatment with oral iron chelators such as deferiprone.115 However lysosomal proteases have also been shown to be involved in the oxidant-mediated degradation of Ft and the related increase in cytosolic LIP. Under physiological conditions, the predominant mechanism of iron release from Ft is through the constitutive degradation of the protein in lysosomes by specific lysosomal proteases, notably chymotrypsin.14 The half-life of Ft in the cytosol is estimated to be 24–72 h (depending on the cell type), after which it is transported into lysosomes where its iron is thought to be recycled for cellular requirements notably heme synthesis.141,158 The high turnover of both endocytosed and autophagocytosed iron-containing macromolecules (e.g. Ft and mitochondrial electron transport complexes) cause the accumulation of the potentially hazardous LI in lysosomal compartments that render these organelles highly vulnerable to oxidative damage upon exposure to oxidizing agents including UVA radiation.26 In skin fibroblasts, UVA has been shown to promote the proteolytic release of Ft iron in the cytosol. The UVA-induced proteolytic degradation of Ft occurs as a result of radiation-mediated damage to lysosomal membranes leading to leakage of lysosomal proteases to the cytosol.14 Pretreatment of skin fibroblasts with Leupeptin and Chymostatin (lysosomal protease inhibitors) prevented the UVA-induced Ft degradation and caused a substantial decrease in the level of cytosolic LI released by UVA.14 We have also previously reported a clear link between the lower basal level of Ft and UVA-induced release of cytosolic LI in keratinocytes that may be in part responsible for the resistance of primary skin keratinocyte cells to UVA-induced LI damage as compared to their UVA-sensitive matched primary skin fibroblasts that contain higher basal Ft level and release more cytosolic LI upon UVA exposure.28
So it appears that Ft iron can replenish the cytosolic LIP upon oxidative stress as it can be released either reductively or as a result of its proteolytic degradation.110 Therefore Ft plays a dual role in LIP homeostasis, acting on the one hand as an iron-sequestering protein and on the other hand as a potential source of LIP.26,37
The role of redox-active LI in compromising the stability of lysosomes under oxidative stress is well-demonstrated in studies involving simultaneous exposure of cells to an oxidising agent and a potent iron-specific chelator. For example, Brunk and co-workers have reported that the combined treatment of cells with H2O2 and the highly lipophilic iron chelator salicaldehyde isonicotinoyl hydrazone (SIH) almost fully prevents both lysosomal rupture and cell death. The lack of oxidative effect of H2O2 in presence of SIH strongly suggests that H2O2per se is not particularly toxic but rather must work in concert with iron in order to damage cells.141 Similarly the pre-treatment of skin cells with lysosomotrophic strong iron chelator desferrioxamine (DFO) or the highly lipophilic iron chelator pyridoxal isonicotinoyl hydrazone (PIH) significantly protected the cells against UVA-mediated lysosomal damage (unpublished data, this laboratory) and the ensuing necrotic cell death87 by virtue of their strong iron-chelating properties.141,159,160 It is noteworthy that H2O2 and O21 are recognised as the predominant intracellular ROS generated by UVA promoting biological damage in exposed tissues viairon-catalysed oxidative reactions.17–20
It has been suggested that the concomitant release of lysosomal proteases and LIP following oxidative damage and the consequent destabilisation of the lysosomal membranes might activate feedback processes that would cause further lysosomal rupture. Such feedback processes may be either LIP-mediated and/or due to activation of lytic cytosolic pro-enzymes such as caspases.14,161,162Lysosomal proteases such as cathepsins B/L/D and chymotrypsin have been specifically detected in the cytosol of cells treated with oxidising agents, indicating their translocation from lysosomal compartments to the cytosol as a result of oxidative damage to lysosomal organelles.14,27,161 The level of chymotrypsin, the lysosomal protease responsible for the degradation of Ft, was found to increase in a dose-dependent manner in the cytosol of skin fibroblasts subjected to a series of doses of UVA at natural exposure levels.14 The latter provided a rational explanation for the observed rapid proteolytic degradation of Ft in the cytosol of skin cells subjected to UVA radiation.14Iron loading of cells prior to UVA radiation exacerbated both the lysosomal damage (unpublished data, this laboratory) and the ensuing cell death in skin cells.28
Lysosomal organelle rupture resulting from intra-lysosomal Fenton-type reactions upon exposure to oxidative agents has also been associated with enhanced DNA damage due to sudden release of lysosomal LI into the cytoplasm.162 It has also been suggested that permeabilization of mitochondrial membranes by release of lysosomal proteases may also induce a loss of mitochondrial membrane potential, irreversibly leading to cell death.100,142,163 The pre-treatment of cells with lysosomotrophic iron chelator DFO prevented the oxidant-induced lysosomal membrane damage, DNA damage, loss of mitochondrial membrane potential and cell death,26,164,165 implying the important role of lysosomal LI in catalysing harmful oxidative damage.
The consequences of a pathological rise in mitochondrial LIP have been well documented in a series of genetic disorders with defective cellular iron utilization (i.e. trafficking and incorporation into proteins) whereby LI concentrations rise to toxic levels in mitochondria of excitable cells often leaving the cytosol iron-depleted. These inherited disorders include FRDA, myopathy with iron-sulfur cluster scaffold protein deficiency, X-linked sideroblastic anemias and neurodegeneration with brain iron accumulation.169 The studies in this field have highlighted a positive correlation between preferential accumulation of redox-active LI in mitochondria and oxidative damage in sensory neurons, the myocardium and endocrine glands.169 Wong et al. have also demonstrated that cultured fibroblasts from FRDA patients that contain high mitochondrial iron level are highly sensitive to iron stress and significantly more sensitive to H2O2-mediated cell death than controls.170
In addition to its involvement in pathological conditions, mitochondrial LI level has also been recently identified as the key determinant of susceptibility of cells to oxidative stress. Studies from this laboratory have demonstrated that exposure of cultured human skin fibroblasts to UVA promotes immediate damage to mitochondrial membranes and that this plays a key role in UVA-induced necrotic cell death.28 The mitochondrial damage leads to abrupt interruption of electron chain reactions within the mitochondrial membrane and production of ROS as well as depletion of mitochondrial ATP that leads to necrotic cell death of UVA-irradiated skin cells.28 Pretreatment of skin cells with highly lipophilic iron chelators SIH, PIH and their caged-photolabile derivative prevented damage to mitochondrial membranes, ATP depletion and the ensuing necrotic cell death, as these compounds were capable of accessing and sequestering the loosely available LI in these organelles.26,87 Similarly Wong et al. observed that FRDA fibroblasts treated with DFO were rescued from H2O2-induced death to a greater extent than controls.170 Chelating of intracellular iron with the iron chelator DFO has also provided some protection against the formation of H2O2-induced mitochondrial DNA breaks.171 Richardson and co-workers have also demonstrated that the mitochondria permeable iron chelator 2-pyridylcarboxaldehyde 2-thiophenecarboxyl hydrazone (PCTH) was more effective than DFO in preventing the H2O2-induced cytotoxicity in cultured FRDA fibroblasts.172
The critical role of lysosomal and mitochondrial LI in oxidative injury has also been well demonstrated in a series of studies by Lemasters and co-workers showing that translocation of LI from lysosomes into mitochondria is a key event during oxidative stress-induced hepatocellular injury and involves the synergistic action of oxidative stress and LI to promote toxic radical formation, mitochondrial dysfunction and cell death.173
The nuclear redox-active LI is also thought to be involved in DNA damage induced by H2O2 and other oxidising compounds.37 This assumption is strengthened by studies showing a significant correlation between cellular LIP and the yield of 7,8-dihydro-8-oxoguanine base damage (i.e. a typical marker of ROS-mediated DNA damage) and DNA breaks in cells treated with oxidising agents such as H2O2.37 Furthermore, pre-treatment of cells with the iron chelator DFO has been shown to prevent the formation of DNA breaks and cytotoxicity by H2O2 to nuclear DNA.37 Interestingly UVA doses that promote a substantial increase in cytosolic LIP, have also been shown to induce 7,8-dihydro-8-oxoguanine base damage in human skin fibroblasts, a classical marker of oxidised DNA.175
3 UVA-induced iron-mediated damage
3.1 Mechanisms underlying the phenomenon of UVA-induced LI release
Fig. 2 summarises potential pathways by which UVA promotes an increase in cytosolic LIP. Our research aimed at understanding the source of UVA-induced increase in cytosolic LIP has revealed that preventing UVA-induced Ft degradation with specific protease inhibitors only partially decreased the observed increase in cytosolic LI following UVA radiation.14 The latter strongly suggested that other sources in addition to Ft's iron contribute to UVA-induced immediate increase in cytosolic LIP.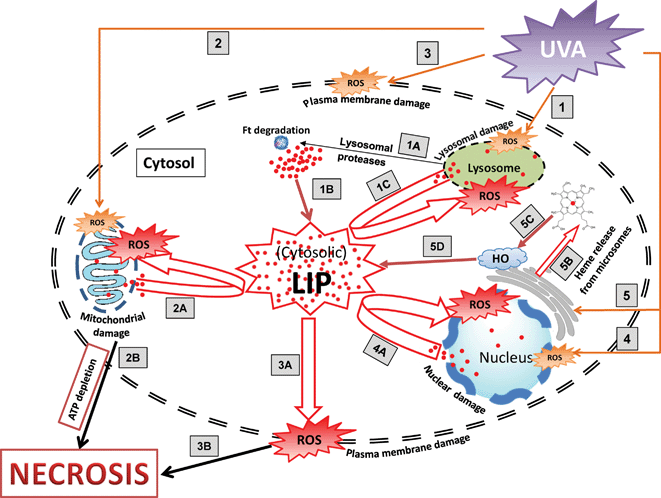 | ||
| Fig. 2 Schematic diagram illustrating the pathways involved in UVA-induced cytosolic LI release and necrotic cell death in human skin fibroblasts. Exposure of skin fibroblasts to UVA generates ROS (orange colour) that promote oxidative damage in lysosomal (1), mitochondrial (2), plasma (3) and nuclear (4) membranes. UVA also promotes the immediate degradation of microsomal hemoproteins (5) that leads to release of free heme (5B) leading to increase in cytosolic LIviaheme oxygenase (HO)-mediated breakdown of heme (5C–5D). Damage to lysosomal membrane (1) leads to release of lysosomal proteases (1A) which in turn degrade the cytosolic iron storage protein ferritin (Ft) and release its iron in the labile form (1B). Damage to mitochondrial membrane (2) leads to interruption of electron chain transport in mitochondrial membrane causing the generation of ROS, loss of the electrochemical gradient across the inner membrane and ATP depletion (2B). The release of potentially harmful LI in cytosolvia routes (1–5), along with the pre-existing pool of cytosolic LI contribute to a massive increase in cytosolic pool of LI (LIP) that catalyses the formation of more harmful ROS (in red) that is thought to further exacerbate the peroxidative damage in the lysosomal (1C), mitochondrial (2A), plasma (3A) and nuclear (4A) membranes leading to the loss of organelles' and plasma membrane's integrity. The loss of plasma membrane integrity (3A) together with mitochondrial ATP depletion (2B) results in necrotic cell death. | ||
Clearly the presence of high level of redox active LI in lysosomes, mitochondria and nucleus of skin cells sensitises these organelles to UVA-induced oxidative damage, so it is not surprising that exposure of skin cells to UVA at natural exposure levels promotes dose-dependent damage to these organelles.28,88,108 The rapid release of LI from these organelles is likely to contribute to the measurable increase in cytosolic LIP which in turn will exacerbate the oxidative damage to cell constituents. While the cytosolic LIP is physiologically kept low in cells by storage in Ft, the UVA-induced degradation of Ft by lysosomal proteases, further contribute to the expansion of cytosolic LIP. The lack of the cytosolic iron storage protein, Ft within the critical first hours after irradiation further exacerbates the iron-catalyzed damage in irradiated skin cells, since a potentially harmful excess of cytosolic LI can not be safely sequestered. Indeed increase in cytosolic LI along with ROS generated by UVA radiation promotes further peroxidative damage in exposed skin cells notably in plasma membranes and this results in turn in a loss of cell membrane integrity. The latter predisposes the cells to necrotic cell death as a result of the influx of extracellular media to the intracellular environment leading to swelling and rupture of subcellular compartments and cell lysis.26
Concomitant with destabilisation of cell membrane, UVA also promotes an immediate depletion in mitochondrial ATP, that is a hallmark of necrotic cell death.28,87 The UVA-induced mitochondrial ATP depletion is thought to be triggered by several simultaneous events. Firstly the presence of a high level of LI in mitochondria may initiate the destabilisation of mitochondrial membranes upon UVA treatment as a result of LI-induced oxidative damage. Secondly the UVA-induced expansion of cytosolic LI by lysosomal and nuclear damage as well as Ft degradation may exacerbate the iron-catalysed oxidative damage that already occurs in the mitochondrial membrane. Finally, the rapid release of lysosomal proteases after UVA radiation might further attack the mitochondrial membrane leading to additional permeabilisation of the mitochondrial organelles. All these events will almost certainly contribute to the observed UVA-induced rapid depletion of mitochondrial ATP that leads to the demise of cells in the form of necrosis. Replenishing the cellular ATP with glucose led to a substantial decrease in the extent of UVA-induced necrotic cell death in skin fibroblasts.28 Most importantly, pre-treatment of skin fibroblasts with iron chelators dramatically decreased the level of mitochondrial ATP depletion, plasma membrane damage and necrotic cell death.28,87
Previous observations from this laboratory have shown that although UVA promotes mitochondrial damage and cytochrome c release in fibroblast cell lines such as FEK4, these cell lines are particularly resistant to UVA-induced apoptotic cell death.27 As the presence of ATP is essential for the activation of apoptosis protease activating factor-1 (Apaf-1) and subsequent activation of caspases that induce apoptosis,27 it appears that the UVA-induced immediate depletion of ATP in skin fibroblasts provides a rational explanation for the predominance of necrotic cell death induced by UVA.
In all these damaging processes, LI is clearly involved as pre-treatment of cells with both the lysosomotropic iron chelator DFO and the highly lipophilic/cell organelle-permeable chelators SIH and PIH fully protected the skin cells against UVA-induced lysosomal, mitochondrial and nuclear membrane damage.28,87,108 In contrast pre-treatment of skin cells with membrane antioxidants such as α-tocopherol succinate (α-Toco), Trolox and butyryl hydroxytoluene (BHT) only partially protected the cells against UVA-induced peroxidative damage presumably because of the high amount of LI still present in these organelles. Indeed the CA-assay revealed that UVA still promotes an increase in LIP in cytosolic compartments of cells pre-treated with α-Toco or BHT (unpublished data, this laboratory). Numerous other studies in this field have also demonstrated that ‘conventional’ antioxidants have had a very modest protective effect, presumably because the investigators have overlooked the phenomenon of UVA-induced excess LI release in cells that could still contribute to generation of ROS and oxidative damage.26 Conventional radical scavengers can often not neutralise the overall ROS production upon exposure of cells to strong oxidising agents such as UVA which also promote redox-active LI release in cells. Because of this dual action, only bifunctional antioxidants targeting both LI and ROS have been effective in protecting skin cells against UVA-induced oxidative damage and cell death.26,88,176,177
UVA not only promotes the early degradation of Ft, but also the degradation of a series of hemoproteins notably catalase and microsomal hemoproteins. Microsomal hemoproteins are found to be immediately degraded in human skin fibroblasts following relatively low doses of UVA with the consequent release of free heme.15,16 The UVA-induced heme release provides another likely source of LI that may contribute to UVA-induced increase in cytosolic LIP, as it has been shown that it can be readily broken down to release LI by both the constitutive HO-2 as well as the inducible HO-1 enzymes. The UVA-induced heme release has been identified as a key factor in the upregulation of the HO-1 gene in skin fibroblasts.16
3.2 Mechanisms underlying the resistance of keratinocytes to UVA radiation
Compared to skin fibroblasts, skin keratinocytes are more resistant to UVA-mediated membrane damage178 and cytotoxicity,13 although the underlying mechanism is yet to be determined. The evaluation of intracellular LIP in a series of matched human primary skin fibroblasts and keratinocytes revealed that the basal level concentrations of both cytosolic LIP and Ft in keratinocytes is several-fold lower than in fibroblasts.28Keratinocytes also possess less lysosomal organelles than fibroblasts. The quantification of lysosomal membrane damage by neutral red dye uptake assay revealed that the overall UVA-induced damage to lysosomal organelles in keratinocytes is also much lower than in skin fibroblasts (unpublished data, this laboratory). Moreover, the quantification of lysosomal cysteine proteases such as cathepsin-B, -L and -D with ELISA assays revealed that keratinocytes possess up to 20-fold lower cathepsin levels than skin fibroblasts. Accordingly following UVA irradiation, the level of lysosomal cathepsins delocalised to cytosol was found to be much higher in skin fibroblasts than keratinocytes. This was also confirmed by in situdetection of cathepsin-B release by epifluorescence microscopy after immunostaining of cells with a polyclonal rabbit anti-human cathepsin-B antibody (unpublished data, this laboratory).So it appears that although UVA triggers lysosomal damage, Ft degradation and cytosolic LI release in keratinocytes, the absolute level of UVA-induced LI release remains several-fold lower in these cells than in fibroblasts and this almost certainly contributes to the lower level of mitochondrial damage, ATP depletion and necrotic cell death following radiation treatment.28 We propose that a combination of low basal and UVA-induced increase in cytosolic LI, low basal level of Ft and low lysosomal organelle and low lysosomal cathepsin content will all contribute to the higher resistance of keratinocytes to UVA-induced membrane damage and cell death. The level of lactate dehydrogenase release from UVA-irradiated keratinocytes was found to be 4-fold lower than from UVA-irradiated fibroblasts, consistent with the notion that keratinocytes are more resistant to UVA-induced membrane damage (unpublished data, this laboratory). Furthermore the UVA dose to give an equivalent level of necrosis was found to be several-fold lower in fibroblasts than in keratinocytes.28 An artificial increase in intracellular Ft levels in keratinocytes by overnight treatment of cells with hemin or iron-citrate caused a massive increase in cytosolic LI following UVA and a strong sensitisation of the cells to very low doses of UVA, implying the importance of cytosolic LI concentrations in increased susceptibility of skin cells to UVA-induced oxidative damage and cell death.28,87
Given that the upper layer of skin (epidermis) receives high levels of UV radiation on sunlight exposure, it appears that nature has considerably protected the keratinocytes against UVA-induced LI damage. Interestingly in keratinocytes HO-1 is not inducible by UVA, however these cells exhibit a high level of constitutive HO-2 protein. In addition to HO-2, keratinocytes also possess other strong constitutive antioxidant defense molecules and enzymes7,52 that together should be effective in protecting keratinocytes against the deleterious effect of UVA-induced oxidative iron damage, as these cells release very low amount of cytosolic LI after UVA irradiation.28
3.3 The long term effects of UVA-induced LI release
The kinetics of LI mobilisation in both fibroblast and keratinocyte cell lines revealed that in both cell types, the immediate increase in cytosolic LIP is sustained up to 2 h after irradiation and returns to around control value 4–6 h after UVA. The cytosolic LIP remains then unchanged as monitored up to 48 h after UVA irradiation.28 The recovery of cytosolic LIP after UVA coincides with the reciprocal de novo synthesis of degraded Ft that occurs within the first 2 h after irradiation. The full recovery of Ft content to control levels was observed between 4–6 h after irradiation.28 The kinetics of Ft synthesis after UVA irradiation further revealed that the rate of Ft synthesis increases 8–10 h after irradiation and as a result the level of cytosolic Ft becomes 2-fold higher 24 h after irradiation when compared to unirradiated control cells.14 This phenomenon was previously thought to occur solely as a result of HO-1 induction by UVA.94,95 However in the light of our recent findings, it appears that the UVA-induced long-term increase in Ft is a general response of UVA-irradiated cells to UVA-induced increase in cytosolic LI, where HO-1 also appears to be involved as a potential enhancer of cytosolic LI. This argument is strengthened by the observations that pre-treatment of skin fibroblasts with DFO not only blocked the UVA-induced overnight increase in Ft attributed to HO-1 induction94 but also the UVA-induced increase in cytosolic LI.26,283.4 Concluding remarks
Overall, our findings demonstrate that UVA-mediated increase in cytosolic LI and heme release plays a key role in the increased susceptibility of cells to UVA-induced damage and necrotic cell death. Although cells have mechanisms to remove iron (i.e. long-term increase in Ft) and heme (HO-1 activation), these defence mechanisms develop over several hours and days. Therefore it appears that high intensity short-term exposures to UVA radiation are the most likely to be damaging. This is precisely the situation for exposure to sunlamps where people tend to expose themselves habitually for short high intensity periods. A clear role for iron in exacerbating UVA damage suggests potential pathways for protection through iron chelation. However prolonged exposure to strong iron chelators leads to severe side-effects as a result of the removal of essential iron from various iron-containing proteins, including IRPs and hypoxia-inducing factor-1.26,179,180 As a result, topical formulations of pro-chelators that are either activated by UVA light87 or UVA-induced ROS179 should provide a safer and more efficient alternative for skin photoprotection.26List of abbreviations
| α-Toco | alpha-Tocopherol |
| AD | Alzheimer's disease |
| Apaf-1 | Apoptosis protease activating factor 1 |
| BHT | Butyryl hydroxytoluene |
| CA | Calcein |
| CP655 | 7-Diethylamino-N-[(5-hydroxy-6-methyl-4-oxo-1,4-dihydropyridin-3-yl)methyl]-N-methyl-2-oxo-2H-chromen-3-carboxamide |
| DFO | Desferrioxamine |
| DMT1 | Divalent metal transporter 1 |
| Fe2+ | Ferrous ion |
| Fe3+ | Ferric ion |
| FP1 | Ferroportin 1 |
| FRDA | Friedreich's ataxia |
| Ft | Ferritin |
| g | Gram |
| GSH | Glutathione |
| GPx | Glutathione peroxidase |
| h | Hour |
| H-Ft | Heavy chain of ferritin |
| H2O2 | Hydrogen peroxide |
| HO | Heme oxygenase |
| HO-1 | Heme oxygenase 1 |
| HO-2 | Heme oxygenase 2 |
| HPO | 3-Hydroxypyridin-4-one |
| IRE | Iron responsive element(s) |
| IRP1 | Iron regulatory protein 1 |
| IRP2 | Iron regulatory protein 2 |
| IRPs | Iron regulatory proteins |
| L-Ft | Light chain of ferritin |
| LI | Labile iron |
| LIP | Labile iron pool |
| mg | Milligram |
| MCF | Mitochondrial solute carrier family |
| Mf | Mitoferrin |
| Mf1 | Mitoferrin 1 |
| Mf2 | Mitoferrin 2 |
| NF-kappaB | Nuclear factor kappa-B |
| 1O2 | Singlet oxygen |
| O2˙ − | Superoxide anion |
| OH˙ | Hydroxyl radical |
| PCBP1 | Poly(rC)-binding protein 1 |
| PCTH | 2-Pyridylcarboxaldehyde 2-thiophenecarboxyl hydrazone |
| PIH | Pyridoxal isonicotinoyl hydrazone |
| PPIX | Protoporphyrin IX |
| ROS | Reactive oxygen species |
| SIH | Salicaldehyde isonicotinoyl hydrazone |
| SOD | Superoxide dismutase |
| Steap3 | Six-transmembrane epithelial antigen of the prostate 3 |
| TBHP | t-Butyl-hydroperoxide |
| Tf | Transferrin |
| TfR1 | Transferrin receptor 1 |
| UV | Ultraviolet |
| UVA | Ultraviolet A |
| UVB | Ultraviolet B |
Acknowledgements
We thank Mrs Patricia Holley and Mr Dave Vinicombe for their excellent technical assistance to work presented here. This research was supported by a Wellcome Trust Showcase Award (Contract n°067653/Z/02/Z) and core grants from the Association for International Cancer Research (UK) and the UK Department of Health (Contract n°121/6378). AA was a recipient of PhD scholarship from Algerian ministration of education. JLZ was a recipient of overseas ORS PhD scholarship from the University of Bath.References
- G. Cairo, F. Bernuzzi and S. Recalcati, A precious metal: Iron, an essential nutrient for all cells, Genes Nutr., 2006, 1, 25–39 CrossRef CAS.
- N. T. Le and D. R. Richardson, The role of iron in cell cycle progression and the proliferation of neoplastic cells, Biochim. Biophys. Acta, 2002, 1603, 31–46 CAS.
- D. R. Richardson and P. Ponka, The molecular mechanisms of the metabolism and transport of iron in normal and neoplastic cells, Biochim. Biophys. Acta, 1997, 1331, 1–40 CAS.
- O. Kakhlon and Z. I. Cabantchik, The labile iron pool: Characterization, measurement and participation in cellular processes, Free Radical Biol. Med., 2002, 33, 1037–1046 CrossRef CAS.
- B. Halliwell and J. M. C. Gutteridge, Biologically relevant metal ion-dependent hydroxyl radical generation: An update, FEBS Lett., 1992, 307, 108–112 CrossRef CAS.
- R. M. Tyrrell, The molecular and cellular pathology of solar ultraviolet radiation, Mol. Aspects Med., 1994, 15, 1–77 CrossRef CAS.
- R. M. Tyrrell, Activation of mammalian gene expression by the UV component of sunlight-from models to reality, BioEssays, 1996, 18, 139–148 CrossRef CAS.
- H. J. Danpure and R. M. Tyrrell, Oxygen dependence of near-UV (365 nm) lethality and the interaction of near-UV and X-rays in two mammalian cell lines, Photochem. Photobiol., 1976, 23, 171–177 CrossRef CAS.
- R. M. Tyrrell, in Oxidative Stress: Oxidants and Antioxidants, ed. H. Sies, Academic Press, London, 1991, pp 57–83 Search PubMed.
- C. Beauchamp and I. A. Fridovich, A mechanism for the production of ethylene from methional. The generation of the hydroxyl radical by xanthine oxidase, J. Biol. Chem., 1970, 245, 4641–4646 CAS.
- J. A. Badwey and M. L. Karnovsky, Active oxygen species and the functions of phagocytic leukocytes, Annu. Rev. Biochem., 1980, 49, 695–726 CrossRef CAS.
- A. Valencia and I. E. Kochevar, Nox1-based NADPH oxidase is the major source of UVA-induced reactive oxygen species in human keratinocytes, J. Invest. Dermatol., 2007, 128, 214–222 CrossRef.
- R. M. Tyrrell and M. Pidoux, Correlation between endogenous glutathione content and sensitivity of cultured human skin cells to radiation at defined wavelengths in the solar UV range, Photochem. Photobiol., 1988, 47, 405–412 CrossRef CAS.
- C. Pourzand, R. D. Watkin, J. E. Brown and R. M. Tyrrell, Ultraviolet A radiation induces immediate release of iron in human primary skin fibroblasts: The role of ferritin, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 6751–6756 CrossRef CAS.
- E. Kvam, A. Noel, S. Basu-Modak and R. M. Tyrrell, Cyclooxygenase dependent release of heme from microsomal hemeproteins correlates with induction of heme oxygenase 1 transcription in human fibroblasts, Free Radical Biol. Med., 1999, 26, 511–517 CrossRef CAS.
- R. M. Tyrrell, Solar ultraviolet A radiation: An oxidizing skin carcinogen that activates heme oxygenase-1, Antioxid. Redox Signal., 2004, 6, 835–840 CAS.
- G. F. Vile and R. M. Tyrrell, UVA radiation-induced oxidative damage to lipids and proteins in vitro and in human skin fibroblasts is dependent on iron and singlet oxygen, Free Radical Biol. Med., 1995, 18, 721–730 CrossRef CAS.
- R. M. Tyrrell and V. E. Reeve, Potential protection of skin by acute UVA irradiation-from cellular to animal models, Prog. Biophys. Mol. Biol., 2006, 92, 86–91 CrossRef CAS.
- C. J. Bertling, F. Lin and A. W. Girotti, Role of hydrogen peroxide in the cytotoxic effects of UVA/B radiation on mammalian cells, Photochem. Photobiol., 1996, 64, 137–142 CrossRef CAS.
- G. T. Wondrak, M. K. Jacobson and E. L. Jacobson, Endogenous UVA-photosensitizers: mediators of skin photodamage and novel targets for skin photoprotection, Photochem. Photobiol. Sci., 2006, 5, 215–237 CAS.
- P. Morliere, A. Moysan, R. Santus, G. Huppe, J-C Maziere and L. Ertret, UVA-induced lipid peroxidation in cultured human fibroblasts, Biochim. Biophys. Acta, 1991, 1084, 261–268 CAS.
- K. Punnonen, C. T. Jansen, A. Puntala and M. Ahotoupa, Effects of ultraviolet A and B irradiation on lipid peroxidation and activity of the antioxidant enzymes in keratinocytes in culture, J. Invest. Dermatol., 1991, 96, 255–259 CAS.
- J. Dissemond, L. A. Schneider, P. Brenneisen, K. Briviba, J. Wenk, M. Wlaschek and K. Scharfetter-Kochanek, Protective and determining factors for the overall lipid peroxidation in ultraviolet A1-irradiated fibroblasts: in vitro and in vivo investigations, Br. J. Dermatol., 2003, 149, 341–349 CrossRef CAS.
- S. D. Aust, L. A. Morehouse and C. E. Thomas, Role of metals in oxygen radical reactions, J. Free Radicals Biol. Med., 1985, 1, 3–25 CrossRef CAS.
- A. W. Girotti, Photosensitized oxidation of membrane lipids: reaction pathways, cytotoxic effects, and cytoprotective mechanisms, J. Photochem. Photobiol., B, 2001, 63, 103–113 CrossRef CAS.
- O. Reelfs, I. M. Eggleston and C. Pourzand, Skin protection against UVA-induced iron damage by multiantioxidants and iron chelating drugs/prodrug, Curr. Drug Metab., 2010, 11, 242–249 CrossRef CAS.
- C. Pourzand and R. M. Tyrrell, Apoptosis, the role of oxidative stress and the example of solar ultraviolet radiation, Photochem. Photobiol., 1999, 70, 380–391 CrossRef CAS.
- J. L. Zhong, A. Yiakouvaki, P. Holley, R. M. Tyrrell and C. Pourzand, Susceptibility of skin cells to UVA-induced necrotic cell death reflects the intracellular level of labile iron, J. Invest. Dermatol., 2004, 123, 771–780 CrossRef CAS.
- K. Scharfetter-Kochanek, M. Wlaschek, P. Brenneisen, M. Scauen, R. Blaudschun and J. Wenk, UV-induced reactive oxygen species in photocarcinogenesis and photoaging, Biol. Chem., 1997, 378, 1247–1257 Search PubMed.
- M. Shayeghi, G. O. Latunde-Dada, J. S. Oakhill, A. H. Laftah, K. Takeuchi, N. Halliday, Y. Khan, A. Warley, F. E. McCann, R. C. Hider, D. M. Frazer, G. J. Anderson, C. D. Vulpe, R. J. Simpson and A. T. McKie, Identification of an intestinal heme transporter, Cell, 2005, 122, 789–801 CrossRef CAS.
- N. C. Andrews, Molecular control of iron metabolism, Best Pract. Res. Clin. Haematol., 2005, 18, 159–169 CrossRef CAS.
- C. W. Trenam, D. R. Blake and C. J. Morris, Skin inflammation: Reactive oxygen species and the role of iron, J. Invest. Dermatol., 1992, 99, 675–682 CAS.
- L. C. Kuhn, Molecular regulation of iron proteins, Bailliere's Clin. Haematol., 1994, 7, 763–785 CrossRef CAS.
- R. Crichton, B. Danielson and P. Geisser, in Iron therapy with special emphasis on intravenous administration (4th Edition), UNI-MED, Bremen, Germany, 2008, pp. 14–16 Search PubMed.
- T. Ganz, Molecular control of iron transport, J. Am. Soc. Nephrol., 2007, 18, 394–400 CrossRef CAS.
- S. Epsztejn, O. Kakhlon, H. Glickstein, W. Breuer and Z. I. Cabantchik, Fluorescence analysis of the labile iron pool of mammalian cells, Anal. Biochem., 1997, 248, 31–40 CrossRef CAS.
- M. Kruszewski, Labile iron pool: The main determinant of cellular response to oxidative stress, Mut. Res., 2003, 29, 81–92 CrossRef.
- B. P. Espósito, S. Epsztejn, W. Breuer and Z. I. Cabantchik, A review of fluorescence methods for assessing labile iron in cells and biological fluids, Anal. Biochem., 2002, 304, 1–18 CrossRef.
- C. Hershko, G. Graham, G. W. Bates and E. A. Rachmilewitz, Non-specific serum iron in thalassaemia: an abnormal serum iron fraction of potential toxicity, Br. J. Haematol., 1978, 40, 255–263 CrossRef CAS.
- T. Ganz and E. Nemeth, Hepcidin and disorders of iron metabolism, Annu. Rev. Med., 2011, 62, 347–360 CrossRef CAS.
- E. Nemeth, M. S. Tuttle, J. Powelson, M. B. Vaughn, A. Donovan, D. M. Ward, T. Ganz and J. Kaplan, Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization, Science, 2004, 306, 2090–2093 CrossRef CAS.
- B. A. Syed, P. J. Sargent, S. Farnaud and R. W. Evans, An overview of molecular aspects of iron metabolism, Hemoglobin, 2006, 30, 69–80 CrossRef CAS.
- L. Klautz and E. Nemeth, Novel tools for the evaluation of iron metabolism, Haematologica, 2010, 95, 1989–1991 CrossRef.
- M. W. Hentze and L. C. Kuhn, Molecular control of vertebrate iron metabolism: mRNA-based regulatory circuits operated by iron, nitric oxide, and oxidative stress, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 8175–8182 CrossRef CAS.
- N. C. Andrews, Forging a field: the golden age of iron biology, Blood, 2008, 112, 219–230 CrossRef CAS.
- I. De Domenico, D. McVey Ward and J. Kaplan, Regulation of iron acquisition and storage: consequences for iron-linked disorders, Nat. Rev. Mol. Cell Biol., 2008, 9, 72–81 CrossRef CAS.
- Z. D. Liu and R. C. Hider, Design of clinically useful iron(III)-selective chelators., Med. Res. Rev., 2002, 22, 26–64 CrossRef CAS.
- B. Halliwell and J. M. C. Gutteridge, Free radicals, ageing, and disease, in Free Radicals in Biology and Medicine, second edition, Clarendon, Oxford, 1989, pp 416–493 Search PubMed.
- H. De Groot, Reactive oxygen species in tissue injury., Hepato-Gastroenterology, 1994, 41, 328–332 CAS.
- S. J. Stohs and D. Bagchi, Oxidative mechanisms in the toxicity of metal ions, Free Radical Biol. Med., 1995, 18, 321–336 CrossRef CAS.
- M. Tenopoulo, P. T. Doulis, A. Barbouti, U. Brunk and D. Galaris, Role of compartmentalized redox active iron in hydrogen peroxide-induced DNA damage and apoptosis, Biochem. J., 2005, 387, 703–710 CrossRef.
- C. Pourzand, O. Reelfs and R. M. Tyrrell, Approaches to define the involvement of reactive oxygen species and iron in ultraviolet-A inducible gene expression, Methods Mol. Biol., 2000, 99, 257–276 CAS.
- G. Cairo and A. Pietrangelo, Iron regulatory proteins in pathobiology., Biochem. J., 2000, 352, 241–250 CrossRef CAS.
- B. G. Rosser and G. J. Gores, Liver cell necrosis: cellular mechanisms and clinical implications., Gastroenterology, 1995, 108, 252–275 CrossRef CAS.
- M. S. Sussman and G. B. Bulkley, Oxygen-derived free radicals in reperfusion injury, Methods Enzymol., 1990, 186, 711–722 CAS.
- H. De Groot and M. Brecht, Reoxygenation injury in rat hepatocytes: mediation by O2−/H2O2 liberated by sources other than xanthine oxidase, Biol. Chem. Hoppe-Seyler, 1991, 372, 35–41 CrossRef CAS.
- L. Tacchini, S. Recalcati, A. Bernelli-Zazzera and G. Cairo, Induction of ferritin synthesis in ischemic-reperfused rat liver: analysis of the molecular mechanisms, Gastroenterology, 1997, 113, 946–953 CrossRef CAS.
- N. C. Andrews, Disorders of iron metabolism, N. Engl. J. Med., 1999, 341, 1986–1995 CrossRef CAS.
- A. Gaeta and R. C. Hider, The crucial role of metal ions in neurodegeneration: the basis for a promising therapeutic strategy, Br. J. Pharmacol., 2009, 146, 1041–1059 CrossRef.
- S. Kalinowski and D. R. Richardson, The evolution of iron chelators for the treatment of iron overload disease and cancer, Pharmacol. Rev., 2005, 57, 547–583 CrossRef.
- M. Molina-Holgado, R. C. Hider, A. Gaeta, R. Williams and P. Francis, Metals ions and neurodegeneration, BioMetals, 2007, 20, 639–654 CrossRef.
- B. R. Bacon and R. S. Britton, The pathology of hepatic iron overload: a free radical-mediated process?, Hepatology, 1990, 11, 127–137 CrossRef CAS.
- J. P. Kehrer, The Haber-Weiss reaction and mechanisms of toxicity, Toxicology, 2000, 149, 43–50 CrossRef CAS.
- K. V. Kowdley, Iron, hemochromatosis, and hepatocellular carcinoma, Gastroenterology, 2004, 127, S79–S86 CrossRef CAS.
- M. Valko, C. J. Rhodes, J. Moncol, M. Izakovic and M. Mazur, Free radicals, metals and antioxidants in oxidative stress-induced cancer, Chem.-Biol. Interact., 2006, 160, 1–40 CrossRef CAS.
- M. Valko, D. Leibfritz, J. Moncol, M. T. D. Cronin and M. Mazur, Free radicals and antioxidants in normal physiological functions and human diseases, Int. J. Biochem. Cell Biol., 2007, 39, 44–84 CrossRef CAS.
- C. A. Swanson, Iron intake and regulation: implications for iron deficiency and iron overload, Alcohol, 2003, 30, 99–102 CrossRef CAS.
- G. J. Brewer, Risks of copper and iron toxicity during aging in humans, Chem. Res. Toxicol., 2010, 23, 319–326 CrossRef CAS.
- Y. Kongho, T. Ohtake, K. Ikuta, Y. Suzuki, Y. Hosoki and H. Saito, Iron accumulation in alcoholic liver diseases, Alcohol.: Clin. Exp. Res., 2005, 29, S189–S193 CrossRef.
- D. R. Petersen, Alcohol, iron-associated oxidative stress, and cancer, Alcohol, 2005, 35, 243–249 CrossRef CAS.
- N. Imeryuz, V. Tahan, A. Sonsuz, F. Eren, S. Uraz and M Yuksel, Iron preloading aggravates nutritional steatohepatitis in rats by increasing apopotic cell death, J. Hepatol., 2007, 47, 851–859 CrossRef CAS.
- S. Toyokuni, Iron-induced carcinogenesis: the role of redox regulation, Free Radical Biol. Med., 1996, 20, 553–566 CrossRef CAS.
- D. R. Richardson, D. S. Kalinowski, S. Lau, P. J. Jansson and D. B. Lovejoy, Cancer cell iron metabolism and the development of potent iron chelators as anti-tumour agents, Biochim. Biophys. Acta, 2009, 1790, 702–717 CrossRef CAS.
- H. W. Hann, M. W. Stahlhut and B. S. Blumberg, Iron nutrition and tumor growth: decreased tumor growth in iron-deficient mice, Cancer Res., 1988, 48, 4168–4170 CAS.
- L. Molin and P. O. Wester, Iron content in normal and psoriatic epidermis, Acta Derm. Venereol., 1973, 53, 473–476 CAS.
- Z. Ackerman, M. Seidenbaum, E. Loewenthal and A. Rubinow, Overload of iron in the skin of patients with varicose ulcers. Possible contributing role of iron accumulation in progression of the disease, Arch. Dermatol., 1988, 124, 1376–1378 CAS.
- T. J. David, F. E. Wells, T. C. Sharpe, A. C. Gibbs and J. Devlin, Serum levels of trace metals in children with atopic eczema, Br. J. Dermatol., 1990, 122, 485–489 CrossRef CAS.
- J. L. Sullivan, Iron and the sex difference in heart disease risk, Lancet, 1981, 317, 1293–1294 CrossRef.
- J. Jian, E. Pelle and X. Huang, Iron and menopause: does increased iron affect the health of postmenopausal women?, Antioxid. Redox Signaling, 2009, 11, 2939–2943 CrossRef CAS.
- W. Y. Ong and B. Halliwell, Iron, atherosclerosis, and neurodegeneration: a key role for cholesterol in promoting iron-dependent oxidative damage?, Ann. N. Y. Acad. Sci., 2004, 1012, 51–64 CrossRef CAS.
- M. W. Hentze, T. A. Rouault, J. B. Harford and R. D. Klausner, Oxidation-reduction and the molecular mechanism of a regulatory RNA-protein interaction, Science, 1989, 244, 357–359 CAS.
- E. W. Müllner, S. Rothenberger, A. M. Müller and L. C. Kühn, In vivo and in vitro modulation of the mRNA-binding activity of iron-regulatory factor. Tissue distribution and effects of cell proliferation, iron levels and redox state, Eur. J. Biochem., 1992, 208, 597–605 CrossRef.
- E. A. Martins, R. L. Robalinho and R. Meneghini, Oxidative stress induces activation of a cytosolic protein responsible for control of iron uptake, Arch. Biochem. Biophys., 1995, 316, 128–134 CrossRef CAS.
- K. Pantopoulos and M. W. Hentze, Rapid responses to oxidative stress mediated by iron regulatory protein, EMBO J., 1995, 14, 2917–2924 CAS.
- G. Cairo, E. Castrusini, G. Minotti and A. Bernelli-Zazzera, Superoxide and hydrogen peroxide-dependent inhibition of iron regulatory prtein activity : A protective stratagen against oxidative injury, FASEB J., 1996, 10, 1326–1335 CAS.
- G. Cairo, L. Tacchini, G. Pogliaghi, E. Anzon, A. Tomasi and A. Bernelli-Zazzera, Induction of ferritin synthesis by oxidative stress. Transcriptional and post-transcriptional regulation by expansion of the ‘free’ iron pool, J. Biol. Chem., 1995, 270, 700–703 CrossRef CAS.
- A. Yiakouvaki, J. Savovic, A. Al-Qenaei, J. Dowden and C. Pourzand, Caged-Iron Chelators a novel approach towards protecting skin cells against UVA-induced necrotic cell death, J. Invest. Dermatol., 2006, 126, 2287–2295 CrossRef CAS.
- S. Basu-Modak, D. Ali, M. Gordon, T. Polte, A. Yiakouvaki, C. Pourzand, C. Rice-Evans and R. M. Tyrrell, Suppression of UVA-mediated release of labile iron by epicathecin-A link to lysosomal protection, Free Radical Biol. Med., 2006, 41, 1197–1204 CrossRef CAS.
- W. Breuer, M. Shvartsman and Z. I. Cabantchik, Intracellular labile iron, Int. J. Biochem. Cell Biol., 2008, 40, 350–354 CrossRef CAS.
- S. M. Keyse and R. M. Tyrrell, Heme oxygenase is the major 32 kDA stress protein induced in human skin fibroblasts by UVA radiation, hydrogen peroxide, and sodium arsenite., Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 99–103 CrossRef CAS.
- C. Hanselmann, C. Mauch and S. Werner, Haem oxygenase-1: a novel player in cutaneous wound repair and psoriasis?, Biochem. J., 2001, 353, 459–466 CrossRef CAS.
- R. M. Tyrrell, in Heme-Oxygenase - The Elegant Orchestration of its Products in Medicine, ed. L. E. Otterbein and B. S. Zucherbraun, Nova, Science Publishers Inc, New York, 2005, pp. 333–350 Search PubMed.
- E. Kvam, V. Hejmadi, S. Ryter, C. Pourzand and R. M. Tyrrell, Heme oxygenase activity causes transient hypersensitivity to oxidative ultraviolet A radiation that depends on release of iron from heme, Free Radical Biol. Med., 2000, 28, 1191–1196 CrossRef CAS.
- G. F. Vile and R. M. Tyrrell, Oxidative stress resulting from ultraviolet A irradiation of human skin fibroblasts leads to a heme oxygenase-dependent increase in ferritin, J. Biol. Chem., 1993, 268, 14678–14681 CAS.
- G. F. Vile, S. Basu-Modak, C. Waltner and R. M. Tyrrell, Heme-oxygenase 1 mediates an adaptive response to oxidative stress in human skin fibroblasts, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 2607–2610 CrossRef CAS.
- F. Lin and A. W. Girotti, Elevated ferritin production, iron containment, and oxidant resistance in hemin-treated leukemia cells, Biochem. Pharmacol., 1997, 14, 1361–1363 Search PubMed.
- V. E. Reeve and D. Domanski, Refractoriness of UVA-induced protection from photo-immunosuppression correlates with heme oxygenase response to repeated UVA exposure, Photochem. Photobiol., 2002, 76, 401–405 CrossRef CAS.
- W. K. McCourbey and M. D. Maines, The structure, organisation and differential expression of the gene encoding rat heme-oxygenase-2, Gene, 1994, 139, 155–161 CrossRef.
- A. Noël and R. M. Tyrrell, Development of refractoriness of induced human heme oxygenase-1 gene expression to reinduction by UVA irradiation and hemin, Photochem. Photobiol., 1997, 66, 456–463 CrossRef.
- S. Fakih, M. Podinovskaia, X. Kong, H. L. Collins, U. E. Schaible and R. C. Hider, Targeting the lysosome: Fluorescent iron (III) chelators to selectively monitor endosomal/lysosomal labile iron pools, J. Med. Chem., 2008, 51, 4539–4552 CrossRef CAS.
- M. Tenopoulou, T. Kurz, P. T. Doulias, D. Galaris and U. T. Brunk, Does the calcein-AM method assay the total cellular ‘labile iron pool’ or only a fraction of it?, Biochem. J., 2007, 403, 261–266 CrossRef CAS.
- Y. Ma, H. de Groot, Z. Liu, R. C. Hider and F. Petrat, Chelation and determination of labile iron in primary hepatocytes by pyridinone fluorescent probes, Biochem. J., 2006, 395, 49–55 CrossRef CAS.
- Y. Ma, W. Luo, P. J. Quinn, Z. Liu and R. C. Hider, Design, Synthesis, Physicochemical properties and evaluation of novel iron chelators with fluorescent sensors, J. Med. Chem., 2004, 47, 6349–6362 CrossRef CAS.
- F. Petrat, U U. Rauen and H. de Groot, Determination of the chelatable iron pool of isolated rat hepatocytes by digital fluorescence microscopy using the fluorescent probe, phen green SK, Hepatology, 1999, 29, 1171–1179 CrossRef CAS.
- F. Petrat, H. de Groot and U. Rauen, Determination of the chelatable iron pool of single intact cells by laser scanning microscopy, Arch. Biochem. Biophys., 2000, 376, 74–81 CrossRef CAS.
- F. Petrat, H. de Groot and U. Rauen, Subcellular distribution of chelatable iron: a laser scanning microscopic study in isolated hepatocytes and liver endothelial cells, Biochem. J., 2001, 356, 61–69 CrossRef CAS.
- F. Petrat, D. Weisheit, M. Lensen, H. de Groot, R. Sustmann and U. Rauen, Selective determination of mitochondrial chelatable iron in viable cells with a new fluorescent sensor, Biochem. J., 2002, 362, 137–147 CrossRef CAS.
- O. Reelfs, R. M. Tyrrell and C. Pourzand, Ultraviolet A radiation-induced immediate iron release is a key modulator of the activation of NF-kappaB in human skin fibroblasts, J. Invest. Dermatol., 2004, 122, 1440–1447 CrossRef CAS.
- M. Shvartsman, E. Fibach and Z. I. Cabantchik, Transferrin iron routing to the cytosol and mitochondria as studied by live and real-time fluorescence, Biochem. J., 2010, 429, 185–193 CrossRef CAS.
- W. Breuer, M. Shvartsman and Z. I. Cabantchik, Intracellular labile iron, Int. J. Biochem. Cell Biol., 2008, 40, 350–354 CrossRef CAS.
- Y. Yu, Z. Kovacevic and D. R. Richardson, Tuning cell cycle regulation with an iron key, Cell Cycle, 2007, 6, 1982–1994 CrossRef CAS.
- R. S. Ohgami, D. R. Campagna, E. L. Greer, B. Antiochos, A. McDonald, J. Chen, J. J. Sharp, Y. Fujiwara, J. E. Barker and M. D. Fleming, Identification of a ferrireductase required for efficient transferrin-dependent iron uptake in erythroid cells, Nat. Genet., 2005, 37, 1264–1269 CrossRef CAS.
- H. Gunshin, B. Mackenzie, U. V. Berger, Y. Gunshin, M. F. Romero, W. F. Boron, S. Nussberger, J. L. Gollan and M. A. Hediger, Cloning and characterization of a mammalian proton-coupled metal-ion transporter, Nature, 1997, 388, 482–488 CrossRef CAS.
- R. S. Eisenstein, Iron regulatory proteins and the molecular control of mammalian iron metabolism, Annu. Rev. Nutr., 2000, 20, 627–662 CrossRef CAS.
- P. Arosio and S. Levi, Cytosolic and mitochondrial ferritins in the regulation of cellular iron homeostasis and oxidative damage, Biochim. Biophys. Acta, 2010, 1800, 783–792 CrossRef CAS.
- Y. Yu, J. Wong, D. B. Lovejoy, D. S. Kalinowski and D. R. Richardson, Chelators at the cancer coalface: desferrioxamine to Triapine and beyond, Clin. Cancer Res., 2006, 12, 6876–6883 CrossRef CAS.
- D. Darshan, L. Vanoaica, L. Richman, F. Beerman and L. C. Kühn, Conditional deletion of ferritin H in mice induces loss of iron storage and liver damage, Hepatology, 2009, 50, 852–860 CrossRef CAS.
- S. Omiya, S. Hikoso, Y. Imanishi, A. Saito, O. Yamaguchi, T. Takeda, I. Mizote, T. Oka, M. Taneike, Y. Nakano, Y. Matsumura, K. Nishida, Y. Sawa, M. Hori and K. Otsu, Downregulation of ferritin heavy chain decreases labile iron pool, oxidative stress and cell death in cardiomyocytes, J. Mol. Cell. Cardiol., 2009, 46, 59–66 CrossRef CAS.
- H. Shi, K. Z. Bencze, T. L. Stemmler and C. C. Philpott, A cytosolic iron chaperone that delivers iron to ferritin, Science, 2008, 320, 1207–1210 CrossRef CAS.
- H. Lange, G. Kispal and R. Lill, Mechanism of iron transport to the site of heme synthesis inside yeast mitochondria, J. Biol. Chem., 1999, 274, 18989–18996 CrossRef CAS.
- L. M. Garrick, K. Gniecko, J. E. Hoke, A. al-Nakeeb, P. Ponka and M. D. Garrick, Ferric-salicylaldehyde isonicotinoyl hydrazone, a synthetic iron chelate, alleviates defective iron utilization by reticulocytes of the Belgrade rat, J. Cell. Physiol., 1991, 146, 460–465 CrossRef CAS.
- A. S. Zhang, A. D. Sheftel and P. Ponka, Intracellular kinetics of iron in reticulocytes: evidence for endosome involvement in iron targeting to mitochondria, Blood, 2005, 105, 368–375 CrossRef CAS.
- A. D. Sheftel, A. S. Zhang, C. Brown, O. S. Shirihai and P. Ponka, Direct intraorganellar transfer of iron from endosomes to mitochondria, Blood, 2007, 110, 125–132 CrossRef CAS.
- G. C. Shaw, J. J. Cope, I. Li, K. Corson, C. Hersey, G. E. Ackermann, B. Gwynn, A. J. Lambert, R. A. Wingert, D. Traver, N. S. Trede, B. A. Barut, Y. Zhou, E. Minet, A. Donovan, A. Brownlie, R. Balzan, M. J. Weiss, L. I. Peters, J. Kaplan, L. I. Zon and B. H. Paw, Mitoferrin is essential for erythroid iron assimilation, Nature, 2006, 440, 96–100 CrossRef CAS.
- W. Chen, H. A. Dailey and B. H. Paw, Ferrochelatase forms an oligomeric complex with mitoferrin-1 and Abcb10 for erythroid heme synthesis, Blood, 2010, 116, 628–630 CrossRef CAS.
- P. Ponka, Tissue-specific regulation of iron metabolism and heme synthesis: distinct control mechanisms in erythroid cells, Blood, 1997, 89, 1–25 CAS.
- J. Borova, P. Ponka and J. Neuwirt, Study of intracellular iron distribution in rabbit reticulocytes with normal and inhibited heme synthesis, Biochim. Biophys. Acta, 1973, 320, 143–156 CrossRef CAS.
- P. Ponka, A. Wilczynska and H. M. Schulman, Iron utilization in rabbit reticulocytes: a study using succinylacetone as an inhibitor of heme synthesis, Biochim. Biophys. Acta, Mol. Cell Res., 1982, 720, 96–105 CrossRef CAS.
- D. R. Richardson, P. Ponka and D. Vyoral, Distribution of iron in reticulocytes after inhibition of heme synthesis with succinylacetone: examination of the intermediates involved in iron metabolism., Blood, 1996, 87, 3477–3488 CAS.
- A. May and E. Fitzsimons, Sideroblastic anaemia, Bailliere's Clin. Haematol., 1994, 7, 851–879 CrossRef CAS.
- M. L. Adams, I. Ostapiuk and J. A. Grasso, The effects of inhibition of heme synthesis on the intracellular localization of iron in rat reticulocytes, Biochim. Biophys. Acta, Mol. Cell Res., 1989, 1012, 243–253 CrossRef CAS.
- T. K. Rostovtseva and S. M. Bezrukov, ATP transport through a single mitochondrial channel, VDAC, studied by current fluctuation analysis, Biophys. J., 1998, 74, 2365–2373 CrossRef CAS.
- S. Levi and E. Rovida, The role of iron in mitochondria function, Biochim. Biophys. Acta, 2009, 1790, 629–636 CrossRef CAS.
- F. Y. Li, B. Leibiger, I. Leibiger and C. Larsson, Characerisation of a putative murine mitochondrial transporter homology of hMRS3/4, Mamm. Genome, 2002, 13, 20–23 CrossRef CAS.
- C. Pourzand, O. Reelfs, E. Kvam and R. M. Tyrrell, IRP can determine the effectiveness of δ-aminolevulinic acid in inducing protoporphyrin IX (PPIX) in primary human skin fibroblasts, J. Invest. Dermatol., 1999, 112, 101–107 CrossRef.
- A. T. McKie, P. Marciani, A. Relfs, K. Brennan, K. Wehr, D. Barrow, S. Miret, T. J. Bomford, T. J. Peters, F. Farzaneh, M. A. Hediger, M. W. Hentze and R. J. Simpson, A novel duodenal iron-regulated transporter, IREG1, implicated in the basolateral transfer of iron to the circulation, Mol. Cell, 2000, 5, 299–309 CrossRef CAS.
- R. Lill, Functions and biogenesis of iron sulfur proteins, Nature, 2009, 460, 831–888 CrossRef CAS.
- I. J. Schultz, C. Chen, B. H. Paw and I Hamza, Iron and porphyrin trafficking in heme biogenesis, J. Biol. Chem., 2010, 285, 26753–26759 CrossRef CAS.
- U. Rauen, A. Springer, D. Weisheit, F. Petrat, H. G. Korth, H. de Groot and R. Sustmann, Assessment of chelatable mitochondrial iron by using mitochondrion-selective fluorescent iron indicators with different iron-binding affinities, ChemBioChem, 2007, 8, 341–352 CrossRef CAS.
- B. Sturm, U. Bistrich, M. Schranzhofer, J. P. Sarsero, U. Rauen, B. Scheiber-Mojdehkar, H. de Groot, P. Ioannou and F. Petrat, Friedreich's ataxia, no changes in mitochondrial labile iron in human lymphoblasts and fibroblasts: a decrease in antioxidative capacity?, J. Biol. Chem., 2005, 280, 6701–6708 CrossRef CAS.
- T. Kurz, A. Terman, B. Gustafsson and U. T. Brunk, Lysosomes and oxidative stress in aging and apoptosis, Biochim. Biophys. Acta, 2008, 1780, 1291–1303 CrossRef CAS.
- Z. Yu, H. L. Persson, J. W. Eaton and U. T. Brunk, Intralysosomal iron: a major determinant of oxidant-induced cell death, Free Radical Biol. Med., 2003, 34, 1243–1252 CrossRef CAS.
- K. Kislyov, J. J. Jennings Jr, Y. Rbaibi and C. T. Chu, Autophagy, mitochondria and cell death in lysosomal storage diseases, Autophagy, 2007, 3, 259–262 Search PubMed.
- C. Settembre, A. Fraldi, L. Jahreiss, C. Spampanato, C. Venturi, D. Medina, R. D. Pablo, C. Tacchetti, D. C. Rubinsztein and A. Ballabio, A block of autophagy in lysosomal storage disorders, Hum. Mol. Genet., 2007, 17, 119–129 CrossRef.
- H. Glickstein, R. B. El, G. Link, W. Breuer, A. M. Konijn, C. Hershko, H. Nick and Z. I. Cabantchik, Action of chelators in iron-loaded cardiac cells: Accessibility to intracellular labile iron and functional consequences,, Blood, 2006, 108, 3195–3203 CrossRef CAS.
- W. Breuer, E. Greenberg and Z. I. Cabantchik, Newly delivered transferrin iron and oxidative cell injury, FEBS Lett., 1997, 403, 213–219 CrossRef CAS.
- P. Morliere, S. Salmon, M. Aubailly, A. Rister and R. Santus, Sensitization of skin fibroblasts to UVA by excess iron, Biochim. Biophys. Acta, 1997, 1334, 283–290 CrossRef CAS.
- A. M. Konijn, H. Glickstein, B. Vaisman, E. G. Meyron-Holtz, I. N. Slotki and Z. I. Cabantchik, The cellular labile iron pool and intracellular ferritin in K562 cells, Blood, 1999, 94, 2128–2134 CAS.
- W. Breuer, S. Epsztejn and Z. I. Cabantchik, Dynamics of the cytosolic chelatable iron pool of K562 cells, FEBS Lett., 1996, 382, 304–308 CrossRef CAS.
- C. E. Thomas and S. D. Aust, Reductive release of iron from ferritin by cation free radicals of paraquat and other bipyridyls, J. Biol. Chem., 1986, 261, 13064–13070 CAS.
- B. J. Bolann and R. J. Ulvik, On the ability of superoxide to release iron from ferritin, Eur. J. Biochem., 1990, 193, 899–904 CrossRef CAS.
- Y. Bando and K. Aki, Superoxide-mediated release of iron from feritin by some flavoenzymes, Biochem. Biophys. Res. Commun., 1990, 168, 389–395 CrossRef CAS.
- F. Funk, J. P. Lenders, R. R. Crichton and W. Schneider, Reductive mobilisation of ferritin iron, Eur. J. Biochem., 1985, 152, 167–172 CrossRef CAS.
- D. W. Reif, Ferritin as a source of iron for oxidative damage, Free Radical Biol. Med., 1992, 12, 417–427 CrossRef CAS.
- S. D. Aust, C. F. Chignell, T. M. Bray, B. Kalyanaraman and R. P. Mason, Free radicals in toxicology, Toxicol. Appl. Pharmacol., 1993, 120, 168–178 CrossRef CAS.
- M. Aubailly, R. Santus and S. Salmon, Ferrous iron release from ferritin by ultraviolet A radiation, Photochem. Photobiol., 1991, 54, 769–773 CrossRef CAS.
- P. I. Oteiza, C. G. Kleinman, M. Demasi and E. J. H. Bechara, 5-aminolevulinic acid induces iron release from ferritin, Arch. Biochem. Biophys., 1995, 316, 607–611 CrossRef CAS.
- B. Vaisman, E. Fibach and A. M. Konijn, Utilization of intracellular ferritin iron for hemoglobin synthesis in developing human erythroid precursors, Blood, 1997, 90, 831–838 CAS.
- M. Horackova, P. Ponka and Z. Byczko, The antioxidant effects of a novel iron chelator salicaldehyde isonicotinoyl hydrazone in the prevention of H2O2 injury in adult cardiomyocytes, Cardiovasc. Res., 2000, 47, 529–536 CrossRef CAS.
- T. Simunek, C. Boer, R. A. Bouwman, R. Vasblom, A. M. Versteilen, M. Sterba, V. Gersl, R. Hrdina, P. Ponka, J. J. de Lange, W. J. Paulus and R. J. Musters, SIH, a novel lipophilic iron chelator, protects H9c2 cardiomyoblasts from oxidative stress-induced mitochondrial injury and cell death, J. Mol. Cell. Cardiol., 2005, 39, 345–354 CrossRef CAS.
- T. Kurz, A. Terman and U. T. Brunk, Autophagy, ageing and apoptosis: the role of oxidative stress and lysosomal iron, Arch. Biochem. Biophys., 2007, 462, 220–230 CrossRef CAS.
- T. Kurz, A. Leake, T. Von Zglinicki and U. T. Brunk, Relocalized redox active lysosomal iron is an important mediator of oxidative-stress-induced DNA damage, Biochem. J., 2004, 378, 1039–1045 CrossRef CAS.
- M. Zhao, F. Antunes, J. W. Eaton and U. T. Brunk, Lysosomal enzymes promote mitochondrial oxidant production, cytochrome c release and apoptosis, Eur. J. Biochem., 2003, 270, 3778–3786 CrossRef CAS.
- T. Kurz, B. Gustafsson and U. T. Brunk, Intralysosomal iron chelation protects against oxidative stress-induced cellular damage, FEBS J., 2006, 273, 3106–3117 CrossRef CAS.
- H. L. Persson, Z. Yu, O. Tirosh, J. W. Eaton and U. T. Brunk, Prevention of oxidant-induced cell death by lysosomotropic iron chelators, Free Radical Biol. Med., 2003, 34, 1295–1305 CrossRef CAS.
- A. Cozzi, B. Corsi, s. Levi, P. Santambrogio, A. Albertini and P. Arosio, Overexpression of wild type and mutated human ferritin H-chain in Hela cels: in vivo role of ferroxidase activity, J. Biol. Chem., 2000, 275, 25122–25129 CrossRef CAS.
- S. Epsztejn, H. Glickstein, V. Piccard, I. N. Slotki, W. Breuer, C. Beaumont and Z. I. Cabantchik, H-ferritin subunit Overexpression in erythroid cells reduces the oxidative stress response and induces multidrug resistance properties, Blood, 1999, 94, 3593–3603 CAS.
- H. Glickstein, R. B. El, M. Shvartsman and Z. I. Cabantchik, Intracellular labile iron pools as direct targets of iron chelators: a fluorescence study of chelator action in living cells, Blood, 2005, 106, 3242–3250 CrossRef CAS.
- O. Kakhlon, W. Breuer, A. Munnich and Z. I. Cabantchik, Iron redistribution as a therapeutic strategy for treating diseases of localized iron accumulation, Can. J. Physiol. Pharmacol., 2010, 88, 187–196 CrossRef CAS.
- A. Wong, J. Yang, P. Cavadini, C. Gellera, B. Lonnerdal, F. Taroni and G. Cortopassi, The Friedreich's ataxia mutation confers cellular sensitivity to oxidant stress which is rescued by chelators of iron and calcium and inhibitors of apoptosis, Hum. Mol. Genet., 1999, 8, 425–430 CrossRef CAS.
- H. Itoh, T. Shioda, T. Matsura, S. Koyama, T. Nakanishi, G. Kajiyama and T. Kawasaki, Iron ions induces mitochondrial DNA damage in HTC rat hepatoma cell culture- role of antioxidants in mitochondrial DNA protection from oxidative stresses, Arch. Biochem. Biophys., 1994, 313, 120–125 CrossRef CAS.
- C. K. Lim, D. S. Kalinowski and D. R. Richardson, Protection against hydrogen peroxide-mediated cytotoxicity in Friedreich's ataxia fibroblasts using novel iron chelators of the 2-pyridylcarboxaldehyde isonicotinoyl hydrazone class, Mol. Pharmacol., 2008, 74, 225–235 CrossRef CAS.
- A. Uchiyama, J-S. Kim, K. Kon, H. Jaeschke, K. Ikejima, S. Watanabe and J. J. Lemasters, Translocation of iron from lysosome into mitochondria is a key event during oxidative stress-induced hepatocellular injury, Hepatology, 2008, 48, 1644–1654 CrossRef CAS.
- R. A. Swerlick and N. J. Korman, UVA and NF-kappaB activity: ironing out the details, Comment on: J. Invest. Dermatol. 2004 122:1440–1447, J. Invest. Dermatol., 2004, 122, xi–xii CrossRef CAS.
- E. Kvam and R. M. Tyrrell, Induction of oxidative DNA base damage in human skin cells by UV and near visible radiation, Carcinogenesis, 1997, 18, 2379–2384 CrossRef CAS.
- S. Mecklenburg, C. A. Collins, M. Döring, T. Burkholz, M. Abbas, F. H. Fry, C. Pourzand and C. Jacob, The design of multifunctional antioxidants against the damaging ingredients of oxidative stress, Phosphorus, Sulfur Silicon Relat. Elem., 2008, 183, 863–888 CrossRef CAS.
- C. A. Collins, F. H. Fry, A. L. Holme, S. Pariagh, A. Yiakouvaki, A. Al-Qenaei, C. Pourzand and C. Jacob, Towards Multifunctional Antioxidants: Synthesis, Electrochemistry, in vitro and Cell Culture Evaluation of Compounds with Ligand/Catalytic Properties, Org. Biomol. Chem., 2005, 3, 1541–1546 CAS.
- L. A. Applegate, A. Noel, G. Vile, E. Frenk and R. M. Tyrrell, Two genes contribute to different extents to the heme oxygenase enzyme activity measured in cultured human skin fibroblasts and keratinocytes: implications for protection against oxidant stress, Photochem. Photobiol., 1995, 61, 285–291 CrossRef CAS.
- M. J. Pygmalion, L. Ruiz, E. Popovic, J. Gizard, P. Portes, X. Marat, K. Lucet-Levannier, B. Muller, J. B. Galey, MJ Pygmalion, L Ruiz, E Popovic, J Gizard, P Portes, X Marat, K Lucet-Levannier, B Muller and JB Galey, Skin cell protection against UVA by Sideroxyl, a new antioxidant complementary to sunscreens, Free Radical Biol. Med., 2010, 49, 1629–1637 CrossRef CAS.
- M. Creighton-Gutteridge and R. M. Tyrrell, A novel iron chelator that does not induce HIF-1 activity, Free Radical Biol. Med., 2002, 33, 356–363 CrossRef CAS.
Footnotes |
| † Contribution to the themed issue on the biology of UVA. |
| ‡ Equal contribution. |
| § Current Address: College of Bioengineering, Chongqing University, Chongqing, China. |
| This journal is © The Royal Society of Chemistry and Owner Societies 2012 |