N-Heterocyclic carbene catalyzed synthesis of oxime esters†
Dieter
Enders
*,
André
Grossmann
and
David
Van Craen
Institute of Organic Chemistry, RWTH Aachen University, Landoltweg 1, 52074 Aachen, Germany. E-mail: enders@rwth-aachen.de; Fax: (+49) 241-809-2127
First published on 16th October 2012
Abstract
A triazolium salt derived N-heterocyclic carbene catalyzes the redox esterification reaction between α–β-unsaturated aldehydes and oximes. The resulting saturated oxime esters were obtained in very good yields for a broad range of aliphatic, aromatic and heteroaromatic substrates.
When designing a synthesis strategy for a complex target molecule through retrosynthetic analysis, the concept of umpolung has turned out to be very useful allowing non-traditional disconnections.1 In this context N-heterocyclic carbenes have evolved into a very successful class of catalysts for the umpolung of aldehydes over the past few decades.2 Among the vast number of publications on NHC organocatalysis, Chow and Bode3a and Rovis et al.3b independently reported on the conjugate umpolung of α-reducible aldehydes in 2004 (Scheme 1). Thereby, N-heterocyclic carbenes react with aldehydes bearing a double bond equivalent or a leaving group in the alpha position to the carbonyl group forming an acyl azolium salt, which subsequently reacts with an alcohol leading to saturated esters via internal redox esterification.3,4
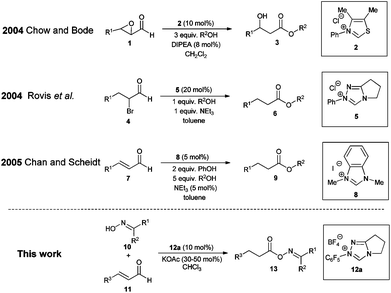 | ||
| Scheme 1 Examples of NHC catalyzed redox esterifications; an alcohol is the nucleophile in all three literature cases. | ||
In comparison with the classic preparation methods of esters via substitution reaction of carboxylic acid derivatives5 this catalytic or sub-stoichiometric approach has, theoretically, the advantage of avoiding stoichiometric amounts of salt waste and/or coupling reagents, therefore being atom6 and redox economical.7 In reality, all reports on intermolecular redox esterifications so far do not fulfill these advantages by applying stoichiometric amounts of base, protic additives or large excesses of the O-nucleophile (Scheme 1). We now would like to report a highly efficient redox esterification reaction between α–β-unsaturated aldehydes and oximes under very mild, nearly equimolar conditions where no protic additives are necessary. All starting materials and reagents are commercially available and the resulting oxime esters are very useful biologically active molecules for fragrance,8 medical9 and agricultural industries.10
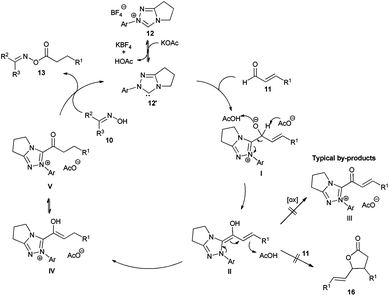
We started our investigations by utilizing crotonaldehyde (11a) and p-tolylaldoxime (10a) in the presence of the triazolium salt 12a (5 mol%) and potassium acetate as a base (30 mol%) in chloroform for 24 h. Although crotonaldehyde is thought to be a poor substrate for the redox esterification,3j we could successfully isolate the corresponding oxime ester 13a in 69% yield with 72% converted starting material (Table 1, entry 1). It is noteworthy that no typical by-products via the benzoin reaction or dimerization of the aldehyde were observed in the crude reaction mixture. From the range of the tested bases (Table 1, entries 2–8) strong bases such as KOtBu or DBU showed no activity in this reaction while weaker ones, such as amino or acetate bases, could promote the reaction albeit less effectively as compared to potassium acetate. Therefore, in agreement with the literature, only bases whose corresponding acid is strong enough for the protonation of the extended Breslow intermediate II (Scheme 2) led to a measurable conversion of the starting material.3d
|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Base | Base (mol%) | Precatalyst (mol%) | Conversionb (%) |
| a Reaction conditions: 10a (1.0 mmol), 11a (1.0 mmol), solvent (2.0 mL), 24 h, rt, under air. b Conversion of the oxime 10a in the crude reaction mixture was determined via1H NMR spectroscopy; values in brackets are yields of isolated product 13a. c Excess aldehyde 11a (1.1 equiv.) was used. | |||||
| 1 | CHCl3 | KOAc | 30 | 12a (5) | 72 (69) |
| 2 | CHCl3 | NaOAc | 30 | 12a (5) | 53 |
| 3 | CHCl3 | CsOAc | 30 | 12a (5) | 51 |
| 4 | CHCl3 | DBU | 30 | 12a (5) | Trace |
| 5 | CHCl3 | KOtBu | 30 | 12a (5) | Trace |
| 6 | CHCl3 | (iPr)2NEt | 30 | 12a (5) | 43 |
| 7 | CHCl3 | NEt3 | 30 | 12a (5) | 32 |
| 8 | CHCl3 | Cs2CO3 | 30 | 12a (5) | Trace |
| 9 | CHCl3 | KOAc | 30 | 12a (10) | 86 |
| 10 | CH2Cl2 | KOAc | 30 | 12a (10) | 83 |
| 11 | Toluene | KOAc | 30 | 12a (10) | 73 |
| 12 | Et2O | KOAc | 30 | 12a (10) | 65 |
| 13 | Acetone | KOAc | 30 | 12a (10) | 34 |
| 14 | iPrOH | KOAc | 30 | 12a (10) | 43 |
| 15 | MeOH | KOAc | 30 | 12a (10) | 13 |
| 16 | EtOAc | KOAc | 30 | 12a (10) | 55 |
| 17 | CHCl3 | KOAc | 30 | 12b (10) | 13 |
| 18 | CHCl3 | KOAc | 30 | 12c (10) | 13 |
| 19 | CHCl3 | KOAc | 30 | 12d (10) | 10 |
| 20 | CHCl3 | KOAc | 30 | 12e (10) | 52 |
| 21 | CHCl3 | KOAc | 30 | 14 (10) | Trace |
| 22 | CHCl3 | KOAc | 30 | 15 (10) | Trace |
| 23c | CHCl3 | KOAc | 50 | 12a (10) | 91 (90) |
In order to improve the reaction rate we increased the catalyst loading to 10 mol% and tested different solvents (Table 1, entries 9–16). Aprotic solvents and especially chlorinated ones gave the best results, while ethers, esters and primary alcohols led to poor conversion rates. Interestingly, the catalytic system was highly selective concerning the choice of the nucleophile. Even when the reaction was performed in methanol only the desired oxime ester was observed without any by-products such as methylesters resulting from the redox esterification reaction with the solvent. Subsequently, we tested different azolium salts for their catalytic activity (Table 1, entries 9, 17–22). While thiazolium (14) and imidazolium salts 15 were catalytically inactive, the triazolium catalyst precursors 12 gave varying results strongly dependent on the aromatic N-substituent. Thereby, only the azolium salts 12a and 12e bearing ortho-substituents on the N-phenyl moiety produced reasonable oxime conversions with triazolium salt 12a being far superior to 12e. Finally, using 12a as a catalyst precursor and increasing the potassium acetate loading to 0.5 equivalents raised the conversion of oxime to 91% and the yield of isolated product to 90% (Table 1, entry 23).
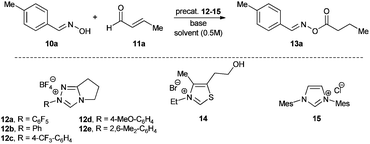
With the optimized conditions in hand we turned our attention to the scope of the oxime and aldehyde substrates. Aliphatic esters of aromatic aldoximes have recently attracted special attention due to their distinct and characteristic aroma of berries making them interesting to the fragrance and food industries.8 Therefore we prepared a range of these compounds varying the aromatic moiety and utilizing our protocol (Table 2, entries 1–10). Indeed, irrespective of their electronic or steric properties, all these aldoximes performed well with high yields (78–90%). Sole limitations were the solubility and/or the basicity of the aldoxime leading to slightly lower yields for 3,4-dimethoxyphenyl and 4-dimethylaminophenyl substituted aldoximes (Table 2, entries 8, 10). Similarly, heteroaromatic aldoximes gave good results (Table 2, entries 11–13) although their low solubility required a change of solvent in the case of R1 = 2-pyridinyl and 3-indolyloxime (Table 2, entries 11–12). Since even ortho-substituted aldoximes do not inhibit the reaction (Table 2, entry 9), we envisaged that ketoximes could be good substrates as well (Table 2, entries 14–19). In fact, ketoximes irrespective of their aromatic or aliphatic substituents reacted to give the corresponding oxime esters in synthetically useful yields (56–95%). Only some methyl-substituted ketoximes gave moderate results (Table 2, entries 14, 16).
|
|
|||||
|---|---|---|---|---|---|
| Entry | R1 | R2 | R3 | 13 | Yieldb (%) |
a Reaction conditions: 10 (2.0 mmol), 11 (2.2 mmol), solvent (4.0 mL), 24 h, rt, under air.
b Yields of isolated product 13.
c CH2Cl2 (4.0 mL) was used as solvent.
d A mixture of CHCl3–CH2Cl2–Et2O 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 (8.0 mL) was used as solvent.
e A mixture of CHCl3–CH2Cl2 1:1 (8 mL) was used as solvent. :1:1 (8.0 mL) was used as solvent.
e A mixture of CHCl3–CH2Cl2 1:1 (8 mL) was used as solvent.
|
|||||
| 1 | 4-MeC6H4 | H | Me | a | 90 |
| 2 | Ph | H | Me | b | 81 |
| 3 | 1-Naphthyl | H | Me | c | 93 |
| 4 | 4-BrC6H4 | H | Me | d | 90 |
| 5 | 3-ClC6H4 | H | Me | e | 90 |
| 6 | 4-(CF3)C6H4 | H | Me | f | 83 |
| 7 | 3,4,5-F3C6H2 | H | Me | g | 90 |
| 8 | 4-(Me2N)C6H4 | H | Me | h | 79 |
| 9 | 2-(MeO)C6H4 | H | Me | i | 88 |
| 10c | 3,4-(MeO)2C6H3 | H | Me | j | 85 |
| 11c | 2-Pyridinyl | H | Me | k | 73 |
| 12d | 3-Indolyl | H | Me | l | 84 |
| 13 | 2-Furanyl | H | Me | m | 87 |
| 14 | Me | Me | Me | n | 56 |
| 15 | Et | Me | Me | o | 92 |
| 16 | Ph | Me | Me | p | 68 |
| 17 | Ph | Ph | Me | q | 92 |
| 18e | 1-Indanonyl | Me | r | 95 | |
| 19 | –(CH2)5– | Me | s | 83 | |
| 20 | 4-Me-C6H4 | H | Et | t | 82 |
| 21 | 4-Me-C6H4 | H | nPr | u | 80 |
| 22 | 4-Me-C6H4 | H | iPr | v | 90 |
| 23 | 4-Me-C6H4 | H | CH2OTIPS | w | 74 |
| 24 | 4-Me-C6H4 | H | Bn | x | 79 |
| 25 | 4-Me-C6H4 | H | Ph | y | 85 |
Finally, different α–β-unsaturated aldehydes were tested in this reaction. In comparison to crotonaldehyde the longer chained or bulkier aliphatic aldehydes were all equally active resulting in high yields of 74–90% (Table 2, entries 20–24). Similarly, aromatic aldehydes such as cinnamaldehyde are good substrates as well. The oxime ester formed from cinnamaldehyde and p-tolyloxime was isolated in 85% yield (Table 2, entry 25).
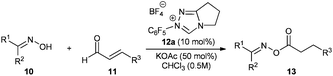
The proposed mechanism of the reaction is shown in Scheme 2. First, the triazolium salt 12 is deprotonated by potassium acetate. Considering the low basicity of acetate bases and previous labeling experiments by Sohn and Bode3d the concentration of the free carbene 12′ is probably low and the protonation is reversible. Subsequently, the resulting carbene 12′ attacks the aldehyde 11 forming the tetrahedral intermediate I,11 which rearranges to the Breslow intermediate II. Recent DFT calculations showed that such proton transfers are protonation–deprotonation processes more than symmetry forbidden intramolecular [1,2]-H-shifts.12 In the current case, the acetate base and the corresponding acetic acid predominantly take the role of the proton shuttle agent. Indeed, when the reaction was performed with triethylamine as a base, addition of 30 mol% acetic acid improved the conversion of oxime significantly, up to around 50% (previously 32%, see Table 1 entry 7). The important regioselective step is the subsequent protonation of the Breslow intermediate II to the azolium-enol IV and its tautomeric acyl-azolium V. Taking into account that no significant amounts of by-products were observed, the protonation of the Breslow intermediate occurs faster than the reaction with an additional molecule of the aldehyde 11 preventing C–C bond formation leading to lactones 1613 or the oxidation leading to α–β-unsaturated acyl-azolium III.3j,14,15 Finally, the oxime 10 performs a nucleophilic substitution on the carboxyl surrogate V yielding the oxime ester 13 and liberating the carbene catalyst 12′ together with acetic acid.
In conclusion we developed a practical, efficient and highly selective redox esterification reaction between enals and oximes. The reaction worked well for all tested aliphatic, aromatic and heteroaromatic substrates with the only limitation being substrate solubility and basicity. In fact, the presented methodology is a good alternative to the classical transesterification processes of carboxylic acid derivatives leading to industrially valuable oxime esters in good to excellent yields.
General procedure for the NHC catalyzed redox esterification
Oxime 10 (2 mmol) and aldehyde 11 (2.2 mmol) were mixed in a flask and subsequently dissolved in chloroform (8 mL). The clear solution was treated with triazolium salt 12a (0.2 mmol) and potassium acetate (1 mmol). The heterogeneous reaction mixture was stirred at ambient temperature.16 After 24 h the reaction mixture was transferred onto a small amount of silica gel and purified by column chromatography.Acknowledgements
We thank the German Research Foundation for support (scholarship for A. G., International Research Training Group “Selectivity in Chemo- and Biocatalysis” – SeleCa). The donation of chemicals by the former Degussa AG and BASF SE is gratefully acknowledged.Notes and references
- D. Seebach, Angew. Chem., 1979, 91, 259–278 ( Angew. Chem., Int. Ed. Engl. , 1979 , 18 , 239–258 ) CrossRef CAS.
- For selected reviews on NHC catalyzed reactions, see: (a) K. Zeitler, Angew. Chem., 2005, 117, 7674–7678 ( Angew. Chem., Int. Ed. , 2005 , 44 , 7506–7510 ) CrossRef; (b) D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606–5655 CrossRef CAS; (c) V. Nair, S. Vellalath and B. P. Babu, Chem. Soc. Rev., 2008, 37, 2691–2698 RSC; (d) J. Read de Alaniz and T. Rovis, Synlett, 2009, 1189–1207; (e) E. M. Phillips, A. Chan and K. A. Scheidt, Aldrichimica Acta, 2009, 42, 55–66 CAS; (f) J. L. Moore and T. Rovis, Top. Curr. Chem., 2010, 291, 77–144 CAS; (g) V. Nair, R. S. Menon, A. T. Biju, C. R. Sinu, R. R. Paul, A. Jose and V. Sreekumar, Chem. Soc. Rev., 2011, 40, 5336–5346 RSC; (h) P.-C. Chiang and J. W. Bode, in N-Heterocyclic Carbenes, The Royal Society of Chemistry, Cambridge, 2011, pp. 399–435 Search PubMed; (i) A. T. Biju, N. Kuhl and F. Glorius, Acc. Chem. Res., 2011, 44, 1182–1195 CrossRef CAS; (j) K. Hirano, I. Piel and F. Glorius, Chem. Lett., 2011, 40, 786–791 CrossRef CAS; (k) D. T. Cohen and K. A. Scheidt, Chem. Sci., 2012, 53–57 Search PubMed; (l) P.-C. Chiang and J. W. Bode, in Asymmetric Organocatalysis, Georg Thieme Verlag, Stuttgart, 2012, vol. 1, pp. 639–672 Search PubMed; (m) H. U. Vora, P. Wheeler and T. Rovis, Adv. Synth. Catal., 2012, 354, 1617–1639 Search PubMed.
- For selected examples of intermolecular NHC catalyzed redox esterifications, see: (a) K. Y.-K. Chow and J. W. Bode, J. Am. Chem. Soc., 2004, 126, 8126–8127 CrossRef CAS; (b) N. T. Reynolds, J. Read de Alaniz and T. Rovis, J. Am. Chem. Soc., 2004, 126, 9518–9519 CrossRef CAS; (c) A. Chan and K. A. Scheidt, Org. Lett., 2005, 7, 905–908 CrossRef CAS; (d) S. S. Sohn and J. W. Bode, Org. Lett., 2005, 7, 3873–3876 CrossRef CAS; (e) N. T. Reynolds and T. Rovis, J. Am. Chem. Soc., 2005, 127, 16406–16407 CrossRef; (f) S. S. Sohn and J. W. Bode, Angew. Chem., 2006, 118, 6167–6170 ( Angew. Chem., Int. Ed. , 2006 , 45 , 6021–6024 ) Search PubMed; (g) K. Zeitler, Org. Lett., 2006, 8, 637–640 CrossRef CAS; (h) G.-L. Zhao and A. Córdova, Tetrahedron Lett., 2007, 48, 5976 CrossRef CAS; (i) B. E. Maki, A. Chan and K. A. Scheidt, Synthesis, 2008, 1306–1315 CAS; (j) B. E. Maki, E. V. Patterson, C. J. Cramer and K. A. Scheidt, Org. Lett., 2009, 11, 3942–3945 CrossRef CAS; (k) H. Jiang, B. Gschwend, L. Albrecht and K. A. Jørgensen, Org. Lett., 2010, 12, 5052 CrossRef CAS; (l) J. Kaeobamrung, J. Mahatthananchai, P. Zheng and J. W. Bode, J. Am. Chem. Soc., 2010, 132, 8810–8812 CrossRef CAS.
- For selected examples of intramolecular NHC catalyzed redox esterifications, see: (a) K. Zeitler and C. A. Rose, J. Org. Chem., 2009, 74, 1759–1762 CrossRef CAS; (b) G.-Q. Li, L.-X. Dai and S.-L. You, Org. Lett., 2009, 11, 1623–1625 CrossRef CAS; (c) L. Wang, K. Thai and M. Gravel, Org. Lett., 2009, 11, 891–893 CrossRef CAS; (d) A. D. Smith and K. B. Ling, Chem. Commun., 2011, 47, 373–375 RSC.
- J. Otera, Esterification: Methods, Reactions, and Applications, Wiley & Sons, New York, 2003 Search PubMed.
- B. M. Trost, Science, 1991, 254, 1471–1477 CrossRef CAS.
- N. Z. Burns, P. S. Baran and R. W. Hoffmann, Angew. Chem., 2009, 121, 2896–2910 ( Angew. Chem., Int. Ed. , 2009 , 48 , 2854–2867 ) CrossRef.
- (a) N. A. Zhukovskaya, E. A. Dikusar and O. G. Vyglazov, Chem. Nat. Compd., 2008, 44, 688–691 Search PubMed; (b) N. A. Zhukovskaya, E. A. Dikusar, V. I. Potkin and O. G. Vyglazov, Chem. Nat. Compd., 2009, 45, 148–151 Search PubMed.
- (a) A. Lukaszczyk, H. Martin, P. J. Diel, W. Föry, K. Gätzi, H. Kristinsson, B. Müller, R. Muntwyler, J. P. Pachlatko, H. Rempfler, R. Schurter and H. Szczepanski (CIBA GEIGY A(G)), Eur. Pat. Appl., EP0012158A2, June 25, 1980 Search PubMed; (b) S. Ke, Z. Zhang, Z. Yang, K. Wang, Y. Liang, Y. Zhang, T. Long and A. Jiang (Hubei Engineering Research Center of Bio-pesticid(e)), CN102030680A, Oct. 20, 2010 Search PubMed.
- G. V. Crichlow, K. F. Cheng, D. Dabideen, M. Ochani, B. Aljabari, V. A. Pavlov, E. J. Miller, E. Lolis and Y. Al-Abed, J. Biol. Chem., 2007, 285, 23089–23095 Search PubMed.
- For NHC–aldehyde adducts, see: (a) R. Breslow and E. McNelis, J. Am. Chem. Soc., 1959, 81, 3080–3082 CrossRef CAS; (b) R. Breslow and R. Kim, Tetrahedron Lett., 1994, 35, 699–702 CrossRef CAS; (c) W. Schrader, P. P. Handayani, C. Burstein and F. Glorius, Chem. Commun., 2007, 716–718 RSC.
- K. J. Hawkes and B. F. Yates, Eur. J. Org. Chem., 2008, 5563–5570 CrossRef CAS.
- For selected examples of NHC catalyzed lactonizations, see: (a) C. Burstein and F. Glorius, Angew. Chem., 2004, 116, 6331–6334 ( Angew. Chem., Int. Ed. , 2004 , 43 , 6205–6208 ) CrossRef; (b) S. S. Sohn, E. L. Rosen and J. W. Bode, J. Am. Chem. Soc., 2004, 126, 14370–14371 CrossRef CAS; (c) K. Hirano, I. Piel and F. Glorius, Adv. Synth. Catal., 2008, 350, 984–988 CrossRef CAS; (d) L.-H. Sun, L.-T. Shen and S. Ye, Chem. Commun., 2011, 47, 10136–10138 RSC; (e) D. T. Cohen, C. C. Eichman, E. M. Phillips, E. R. Zarefsky and K. A. Scheidt, Angew. Chem., 2012, 124, 7421–7425 ( Angew. Chem., Int. Ed. , 2012 , 51 , 7309–7313 ) Search PubMed.
- For an early example of the oxidation of the Breslow intermediate and subsequent esterification, see: J. Guin, S. De Sarkar, S. Grimme and A. Studer, Angew. Chem., 2008, 120, 8855–8858 ( Angew. Chem., Int. Ed. , 2008 , 47 , 8727–8730 ) Search PubMed.
- For a recent review on the oxidation of the Breslow intermediate and subsequent esterification, see: C. E. I. Knappke, A. Imami and A. Jacobi von Wangelin, ChemCatChem, 2012, 4, 937–941 Search PubMed.
- It is important to prevent the precipitates from agglomeration by applying moderate stirring. Agglomeration of the undissolved salts leads to a strong decrease in yield.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and full spectroscopic data for all isolated compounds. See DOI: 10.1039/c2ob26974k |
| This journal is © The Royal Society of Chemistry 2013 |