N-Substituted 2-aminoimidazole inhibitors of MRSA biofilm formation accessed through direct 1,3-bis(tert-butoxycarbonyl)guanidine cyclization†
Andrew A.
Yeagley
,
Zhaoming
Su
,
Kára D.
McCullough
,
Roberta J.
Worthington
and
Christian
Melander
*
Department of Chemistry, North Carolina State University, Raleigh, USA. E-mail: Christian_Melander@ncsu.edu; Tel: +1-919-513-2960
First published on 9th October 2012
Abstract
Antibiotic resistance is a significant problem and is compounded by the ability of many pathogenic bacteria to form biofilms. A library of N-substituted derivatives of a previously reported 2-aminoimidazole/triazole (2-AIT) biofilm modulator was constructed via α-bromoketone cyclization with 1,3-bis(tert-butoxycarbonyl)guanidine, followed by selective substitution. Several compounds exhibited the ability to inhibit biofilm formation by three strong biofilm forming strains of methicillin resistant Staphylococcus aureus (MRSA). Additionally, a number of members of this library exhibited synergistic activity with oxacillin against planktonic MRSA. Compounds with this type of dual activity have the potential to be used as adjuvants with conventional antibiotics.
Introduction
A significant factor contributing to the antibiotic/host immune resistance of many pathogenic bacteria is the ability of the bacteria to form a biofilm. Biofilms, defined as communities of surface-attached bacteria encased in an extracellular polymeric substance, are now accepted as being ubiquitous in nature and comprising the major portion of the lifecycle of many bacteria.1 The National Institutes of Health has estimated that over 80% of microbial infections involve biofilms,2 and examples include: lung infections in cystic fibrosis patients, ear infections, periodontitis, infections in burn patients, infections of surgical implants, endocarditis, and urinary tract infections.3–9 Within a biofilm state, bacteria are upwards of 1000-fold more resistant to antibiotics and the host immune response than their free-floating counterparts.10 Novel approaches to treating bacterial infections that involve the targeting of: bacterial communication, virulence, biofilm formation, and suppression of resistance to current antibiotics are being increasingly recognized as a viable alternative approach in the fight against infectious disease.11–15Natural products such as oroidin 1 and bromoageliferin 2 (Fig. 1), which contain a 2-aminoimidazole (2-AI) moiety, have been reported to possess the ability to inhibit and disperse bacterial biofilms.16 We have developed synthetically accessible 2-aminoimidazole containing scaffolds based upon these natural products that exhibit increased ability to inhibit and disperse bacterial biofilms at non-microbicidal concentrations.17–23 The 2-aminoimidazole/triazole (2-AIT) family,20–23 to which 3 and 4 belong, represents some of the most potent anti-biofilm compounds developed to date. The ability of these compounds to eradicate biofilms at concentrations that do not kill bacteria has the advantage of avoiding evolutionary pressures on the bacteria to develop resistance.24
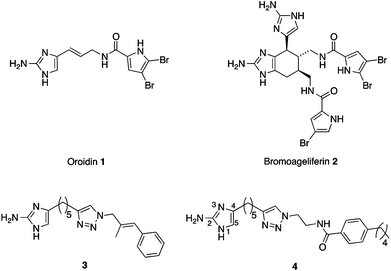 | ||
| Fig. 1 Biofilm inhibitors: natural products oroidin 1 and bromoageliferin 2 and synthetic 2-AIT derivatives 3 and 4. | ||
Some 2-AI derivatives also possess the ability to suppress resistance of several strains of drug resistant bacteria to conventional antibiotics.23–25 Examples include suppression of resistance of methicillin-resistant Staphylococcus aureus (MRSA) to penicillin G and methicillin, and of multidrug-resistant Acinetobacter baumannii (MDRAB) to imipenem by compound 3,24 and suppression of resistance of a NDM-1 producing strain of Klebsiella pneumoniae to imipenem and meropenem by a 2-AI derivative.25 We have also recently shown that compound 4 is able to suppress the resistance of MRSA to oxacillin.26 We previously reported that modification of the 2-AI moiety of 3 by substitution at the 5-position (Fig. 1) resulted in compounds with greater ability to inhibit and disperse MDRAB biofilms (the lead compound displayed IC50 values for inhibition of biofilm formation by MRSA and MDRAB of 1.4 μM and 11.3 μM respectively), and to suppress oxacillin resistance in MRSA.23 We therefore posited that modification of the 2-amino functionality may also lead to compounds with improved ability to modulate MRSA biofilms and suppress oxacillin resistance. However, selective substitution of this functionality has proven synthetically challenging and thus eluded our endeavours until recently.
To this end, we have elaborated a method primarily used to prepare substituted guanidine derivatives27 to obtain analogues of 2-AIT 4 that are selectively substituted at the 2-amino position. We constructed a library of analogues and investigated the effect of 2-amino substitution on the anti-biofilm and resistance suppression properties of this scaffold. Several of these compounds exhibited an increased ability to inhibit biofilms of three different MRSA strains compared to the parent compound 4, in addition to the ability to decrease the minimum inhibitory concentration (MIC) of oxacillin against MRSA.
Results and discussion
Synthesis of 2-AIT library
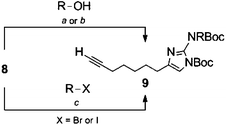
Selective substitution of the 2-amino position of the 2-AI is difficult due to the presence of the more nucleophilic imidazole nitrogens. Functionalization of the 2-position of the unprotected heterocycle has been limited to acylations and reductive aminations, made possible by reversible attack on these electrophiles by the endocyclic nitrogen. Amide functionalized 2-aminoimidazoles can be reduced to the corresponding alkyl analogues, though this requires harsh conditions which are not always compatible with the remainder of the scaffold. There are few examples of reductive aminations with aliphatic aldehydes and even reactions with aromatic aldehydes suffer from low yields as a result of the poor nucleophilicity of the 2-amino group.28 Examples in which the 2-amino position is derivatized via alkylation of a preformed carbamate have been reported,29,30 and this approach appears the most amenable to the preparation of diverse analogues.In order to construct a library of 2-amino substituted analogues of 2-AIT 4 we first set out to develop an efficient preparation of the di-tert-butoxycarbonyl (Boc) protected 2-AI 8 (Scheme 1). The known α-bromoketone 622 was prepared by isomerization of 2-octyn-1-ol 5 under “zipper”31 conditions, followed by Jones oxidation to yield oct-7-ynoic acid. The crude carboxylic acid was converted to the corresponding acyl chloride by treatment with oxalyl chloride, which was followed by addition of diazomethane and subsequent quenching of the intermediate diazoketone with aqueous HBr to provide α-bromoketone 6. Typically, the 2-aminoimidazole is obtained from condensation of the α-bromoketone with Boc-guanidine over several days and we therefore envisioned the possibility of using 1,3-di-Boc-guanidine as a precursor to 8. However, 1,3-di-Boc-guanidine exhibits decreased nucleophilicity and does not readily condense with α-bromoketones in a similar fashion to Boc-guanidine (Scheme 1). To overcome the lack of nucleophilicity, the sodium salt of 1,3-di-Boc-guanidine was prepared by treatment with sodium hydride, and the salt was then allowed to react with 6, this resulted in a mixture of di-Boc protected 2-aminoimidazole 8 and the intermediate hydrate, which was readily converted to 8 by treatment with mesyl chloride.
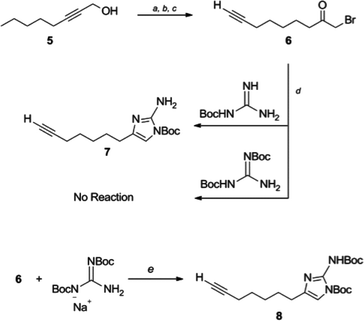 | ||
| Scheme 1 Synthetic approach to di-Boc 2-amino imidazole 8. Reaction conditions: (a) ethylenediamine, NaH, 86%, (b) CrO3, H2SO4, H2O, acetone, 92%, (c) i. CH2N2, Et2O-CH2Cl2 0 °C; ii. aq HBr 67%, (d) DMF, 3 days, 59%, (e) i. di-Boc guanidine, NaH, DMF, 4 h 0 °C to rt; ii. MsCl, Et3N, CH2Cl2, 1 h 82%. | ||
The use of 1,3-di-Boc-guanidine provides the desired selective protection of the 1-imidazole and 2-amino positions without necessitating purification of a mixture of regioisomers or multiple carbamate containing products. The carbamate functionality renders the 2-amino group unreactive to direct acylations or reductive aminations. However, the di-Boc protected 2-AI 8 can be activated by deprotonation of the carbamate with sodium hydride, which following treatment with alkyl halides or acyl chlorides, delivers the 2-amino alkylated or acylated imidazole derivatives. Mitsunobu conditions were also investigated to obtain the desired products from the corresponding alcohols. Both methods proved equally viable, providing similar yields. In both cases, alkylation with primary alkyl derivatives was most efficient, while yields suffered in the case of secondary halides or secondary alcohols. No product was obtained upon reaction with either tertiary halides or tertiary alcohols. Acylation of 8 (to give compound 9i) was also low yielding.
Following substitution of the 2-amino group, alkynes 9a–p were subjected to the copper catalyzed azide-alkyne Huisgen cycloaddition (click reaction) with azide 1126 to produce triazoles 12a–p in excellent yields (75–97%, Scheme 2). Removal of the Boc groups from 12a–p was achieved with TFA/CH2Cl2. In a few examples, most notably 9n and 9o, a large excess of TFA resulted in decomposition; however this issue could simply be resolved by reducing the concentration of TFA. The resulting products were treated with HCl in MeOH and concentrated to provide the hydrochloride salts for biological screening.
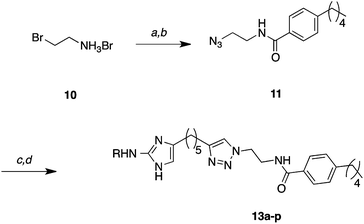 | ||
| Scheme 2 General cycloaddition and carbamate deprotection. Reaction conditions: (a) NaN3, H2O 99% by NMR, (b) 4-pentylbenzoyl chloride, Et3N, CH2Cl2 99%, (c) 9a–p, CuSO4, sodium ascorbate, tBuOH/H2O/CH2Cl2 (75–97%), (d) 10–30% TFA-CH2Cl2 (89–100%). | ||
A disubstituted derivative 13p (Scheme 3) was also prepared through a 1,3-di-Boc-guanidine cyclization. Guanidine derivative 15 was prepared by a known displacement32 of the corresponding N,N′-di-Boc-S-methylthiourea followed by purification and generation of the sodium salt via treatment with sodium hydride. Reaction of 15 with α-bromoketone 6 only yielded the corresponding 2-aminoimidazole 16 upon treatment with TFA. 2-AI 16 was isolated by column chromatography, but quickly oxidized and proved unstable under “click” cyclization conditions. Faced with this situation, the 2-AI heterocycle was Boc protected by treatment with di-tert-butyl dicarbonate to provide 17. This species was found to undergo cycloaddition in good yield that following treatment with TFA and conversion to the HCl salt yielded 13p for biological screening (Table 1).
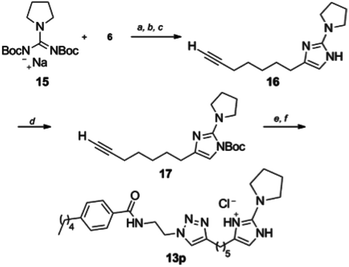 | ||
| Scheme 3 Synthesis of pyrrolidine derivative 13p. Reagents and conditions (a) NaH, DMF, (b) 1-bromonon-8-yl-one, (c) 30% TFA/CH2Cl2, (d) di-tert-butyl dicarbonate, Et3N, DMAP, DMF (58%), (e) 11, CuSO4, sodium ascorbate, tBuOH/H2O/CH2Cl2, (f) 10–30% TFA-CH2Cl2 (89–100%). | ||
|
|
|||
|---|---|---|---|
| R | Conditions | Product | Yield |
| a Yield determined after azide-alkyne Huisgen cycloaddition and represents isolated material after two steps. Reaction conditions: (a) DIAD, PPh3, R-OH, THF 0 °C to 70 °C; (b) DEAD, PPh3, R-OH, THF 0 °C to 70 °C; (c) NaH, R-X, DMF 0 °C to 70 °C; (d) i. NaH, DMF ii. TFA, CH2Cl2, iii. Boc2O, Et3N, DMAP, DMF. | |||
| Me | C | 9a | 47% |
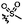
|
C | 9b | 24% |
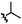
|
A | 9c | 36% |
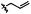
|
A | 9d | 38%a |

|
C | 9e | 27% |
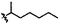
|
C | 9f | 9% |

|
C | 9g | 28% |
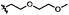
|
B | 9h | 23%a |
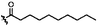
|
C | 9i | 10%a |
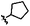
|
A | 9j | 14% |
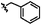
|
C | 9k | 37% |
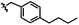
|
C | 9l | 70% |
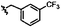
|
C | 9m | 54% |
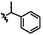
|
C | 9n | 23%a |
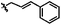
|
A | 9o | 39% |
|
D | 9p | 58% |
Inhibition of MRSA biofilm formation
The parent compound 4 originated from a library of previously prepared 2-AITs that exhibit potent biofilm inhibition activity, including against one strain of MRSA (ATCC BAA-44).22 We chose to investigate the activity of this library of 2-amino substituted derivatives against three strains of MRSA that we found to form considerably robust biofilms in comparison to BAA-44. Several ATCC MRSA strains at our disposal were grown under typical conditions (in tryptic soy broth supplemented with 0.5% glucose at 37 °C for 24 h in polyvinylchloride 96 well plates)22 and analysed by a crystal violet reporter assay.33 Biofilms formed by strains BAA-1770, BAA-1765 and 43![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300 contained considerably greater bacterial mass than those formed by the other strains investigated. The IC50 values of 4 for inhibition of biofilm formation are 34 μM and 22 μM for BAA-1770 and BAA-1685 respectively, while the compound was microbicidal to strain 43300. These values are considerably higher than the previously reported IC50 of 4.3 μM22 against the weaker biofilm forming strain BAA-44 (Table 2).
300 contained considerably greater bacterial mass than those formed by the other strains investigated. The IC50 values of 4 for inhibition of biofilm formation are 34 μM and 22 μM for BAA-1770 and BAA-1685 respectively, while the compound was microbicidal to strain 43300. These values are considerably higher than the previously reported IC50 of 4.3 μM22 against the weaker biofilm forming strain BAA-44 (Table 2).
| Compound | IC50 (μM) | ||
|---|---|---|---|
| BAA-1770 | BAA-1685 | 43300 |
|
| NA = not active up to 200 μM (highest concentration tested). Toxic = dose response curve displayed a precipitous drop in activity over a narrow concentration range indicative of a toxic mechanism of action, therefore an IC50 value was not calculated. | |||
| 13a | 42 ± 4.0 | 20 ± 1.6 | 36 ± 6.4 |
| 13b | NA | NA | NA |
| 13c | 6 ± 9.5 | 48 ± 14 | 53 ± 9.2 |
| 13d | 36 ± 2.3 | 25 ± 2.0 | 30 ± 5.7 |
| 13e | 11 ± 1.3 | 5.7 ± 3.7 | 10 ± 1.4 |
| 13f | 11 ± 0.83 | 9.2 ± 1.1 | 8.6 ± 2.1 |
| 13g | 7.4 ± 1.4 | 3.7 ± 0.92 | 5.3 ± 1.5 |
| 13h | Toxic | Toxic | 76 ± 4.0 |
| 13i | NA | NA | NA |
| 13j | 16 ± 1.3 | 18 ± 1.0 | 16 ± 0.55 |
| 13k | 15 ± 0.54 | 8.1 ± 1.1 | 12 ± 1.9 |
| 13l | 5.9 ± 0.66 | 5.5 ± 0.77 | 4.4 ± 0.76 |
| 13m | 8.9 ± 1.1 | 4.5 ± 1.1 | 7.4 ± 0.72 |
| 13n | 8.3 ± 1.0 | 7.5 ± 0.06 | 8.3 ± 3.3 |
| 13o | NA | NA | NA |
| 13p | Toxic | Toxic | 67 ± 7.8 |
| 4 | 34 ± 2.8 | 22 ± 1.3 | Toxic |
The library of 2-amino substituted derivatives was initially tested against these three MRSA strains using the crystal violet reporter assay at 200 μM. Analogues possessing alkyl chain substituents of less than four carbons (13a, 13c, and 13d) inhibited biofilm formation with similar efficiency to the parent compound 4. Analogues possessing longer alkyl chain or benzyl substituents exhibited an increased ability to inhibit biofilm formation in comparison to the parent compound. The 4-butyl-benzyl derivative 13l exhibited the highest activity across the three strains with IC50 values of 5.9, 5.5 and 4.4 μM against BAA-1770, BAA-1765 and 43300 respectively. All compounds in this library, and the parent compound 4, exhibited some effect on the growth curves of planktonic bacteria of these three strains at their IC50 concentrations (ESI†), indicating that these higher substituted 2-AI derivatives are not modulating biofilms for these strains through an entirely non-microbicidal mechanism, similar to what we have observed for other highly substituted 2-AI compounds.34
Suppression of oxacillin resistance
Compound 4 is able to suppress MRSA resistance to oxacillin, effecting a four-fold reduction in MIC for strain ATCC BAA-44 when co-dosed at a quarter of its MIC (12.5 μM).26 The 2-amino substituted library was therefore screened against this MRSA strain for oxacillin resistance suppression activity. The MIC for each analogue was first determined using a standard broth microdilution protocol.35 MICs of analogues possessing alkyl substituents were, on the whole, lower than that of the parent compound while analogues with sulfonyl or acyl substituents were much weaker microbicides than compound 4. Following determination of the MIC of each analogue, the MIC of oxacillin was determined in the presence of each compound at a quarter of its MIC (up to a limit of 50 μM) using a previously reported protocol.23 Similar to the parent compound, the majority of the 2-amino substituted derivatives effected two-four fold reductions in the oxacillin MIC (Table 3). Compounds 13c and 13d caused an eight-fold drop in the oxacillin MIC while the deactivated amino derivatives 13b and 13i did not elicit any change in MIC.| Compound | MIC (μg mL−1) | Conc. tested (μg mL−1) | Fold reduction in oxacillin MIC | Compound | MIC (μg mL−1) | Conc. Tested (μg mL−1) | Fold reduction in oxacillin MIC |
|---|---|---|---|---|---|---|---|
| 13a | 32 | 8 | 4 | 13i | >256 | 31 | 0 |
| 13b | >256 | 28 | 0 | 13j | 16 | 4 | 2 |
| 13c | 64 | 16 | 8 | 13k | 16 | 4 | 2 |
| 13d | 32 | 8 | 8 | 13l | 16 | 4 | 4 |
| 13e | 8 | 2 | 2 | 13m | 8 | 2 | 2 |
| 13f | 8 | 2 | 2 | 13n | 16 | 4 | 2 |
| 13g | 8 | 2 | 2 | 13o | >256 | 29 | 2 |
| 13h | 128 | 32 | 4 | 13p | 32 | 8 | 4 |
| 4 | 23 | 6 | 4 |
To determine whether these compounds were suppressing resistance to oxacillin through a non-microbicidal mechanism, growth curves were constructed for bacteria in the presence of each compound at the concentrations used in the resistance suppression assay (see ESI†). The most active compounds 13c and 13d were shown to be acting as antibiotics at these concentrations, and exhibited considerable effects on bacterial growth at a quarter of their MIC, even up to the 24 h time point. Due to this antibiotic activity, checkerboard assays were performed to determine whether the active compounds were acting synergistically, or simply additively, with oxacillin.36 Fractional inhibitory concentrations (FICs) for each antibiotic were determined by dividing the MIC of the antibiotic in the combination by the MIC of the antibiotic alone. A FIC index (ΣFIC) was then determined for each combination. Combinations exhibiting ΣFIC values of −0.5 are considered synergistic. The parent compound 4 was shown to be acting synergistically with oxacillin, displaying an ΣFIC index of 0.5 (Table 4). A number of 2-amino alkyl derivatives exhibited greater synergy, with compounds 13c and 13d, which possess short alkyl substituents, both exhibiting ΣFIC indexes of 0.38.
Conclusions
A procedure utilizing 1,3-di-Boc-guanidine for the preparation of 2-aminoimidazoles has been developed to enable the selective preparation of 2-amino substituted 2-AI derivatives. These derivatives exhibited improved biofilm inhibition activities against several MRSA strains relative to the unsubstituted parent compound, with aliphatic and benzyl substitution providing the most potent compounds. Substitution with short aliphatic chains led to an increase in synergy with oxacillin against MRSA. The mechanism of suppression of oxacillin resistance suppression of compound 4, and related analogues, is currently being investigated and will be reported in due course.Experimental
All reagents used for chemical synthesis were purchased from commercially available sources and used without further purification. Chromatography was performed using 60 Å mesh standard grade silica gel from Sorbtech. Deuterated NMR solvents were obtained from Cambridge Isotope Labs and used as is. All 1H NMR (300 MHz or 400 MHz) and 13C NMR (75 MHz or 100 MHz) spectra were recorded at 25 °C on Varian Mercury spectrometers. Chemical shifts (δ) are given in ppm relative to the respective NMR solvent; coupling constants (J) are in hertz (Hz). Abbreviations used are s = singlet, bs = broad singlet, d = doublet, dd = doublet of doublets, t = triplet, dt = doublet of triplets, bt = broad triplet, qt = quartet, m = multiplet, bm = broad multiplet, p = pentet, sep = septet, and br = broad. Mass spectra were obtained at the NCSU Department of Chemistry Mass Spectrometry Facility. Funding was obtained from the North Carolina Biotechnology Center and the NCSU Department of Chemistry. Infrared spectra were obtained on a FT/IR-4100 spectrophotometer (νmax in cm−1). UV absorbance was recorded on a Genesys 10 scanning UV/visible spectrophotometer (λmax in nm).MRSA strains (BAA-44, BAA-1685, BAA-1770 and 43300) were obtained from the ATCC. Oxacillin sodium salt was purchased from TCI (# O0353). Mueller–Hinton broth was made based on the following procedure. To 1 L deionized water was added 2 g BBL™ beef extract (# 212303) and 1.5 g Difco™ soluble starch (# 217820) which were obtained from BD, casein hydrolysate (17.5 g) purchased from MP Biomedicals (# 101290) was then added and the pH was adjusted to 7.4 at ambient temperature. The resulting solution was autoclaved at 120 °C for 15 min.
tert-Butyl 2-((tert-butoxycarbonyl)amino)-4-(hept-6-yn-1-yl)-1H-imidazole-1-carboxylate (8)
To a 0 °C slurry of 60% sodium hydride dispersion in mineral oil (596 mg, 14.9 mmol) in THF (5 mL) was added 1,3-di-tert-butylcarboxy guanidine (3.96 g, 15.3 mmol). After evolution of gas and all solids had dissolved (∼30 min) the 1-bromonon-8-yl-one (2.93 g, 13.47 mmol) dissolved in DMF (25 mL) was added dropwise while maintaining the 0 °C temperature. After complete addition the cold bath was removed and the temperature allowed to reach 25 °C. TLC confirmed complete conversion of the ketone (visualized by KMnO4) after four hours. Reaction was quenched with water and diluted with EtOAc. Organic layer was then washed with water (3 × 30 mL) and brine (3 × 30 mL) to remove DMF. The organic layer was then dried (MgSO4), filtered, and concentrated in vacuo. The crude material was then submitted to flash chromatography (gradient from 10 to 40% EtOAc in hexanes) to isolate a mixture of 2-aminoimidazole and the more polar hydrate (5.072 g). This mixture was then dissolved in dry CH2Cl2 (125 mL) with Et3N (2.0 mL, 14.4 mmol) and cooled to −78 °C. To this solution was added mesyl chloride (990 μL, 12.8 mmol) and the cold bath removed. TLC showed complete conversion of the hydrate to the imidazole product after one hour. The reaction then diluted with CH2Cl2 then washed with saturated NH4Cl (3 × 25 mL) and saturated NaHCO3 (3 × 25 mL). The organic layer was then dried (MgSO4), filtered, and concentrated in vacuo. The crude material was then purified by flash chromatography (10–20% EtOAc in hexanes) to provide a white solid (3.972 g, 10.5 mmol, 82%). 1H NMR (CDCl3, 400 MHz) δ 9.02 (brs, 1H), 6.57 (t, J = 0.89 Hz, 1H), 2.42 (t, J = 7.2 Hz, 2H), 2.07 (dt, J = 7.0, 2.6 Hz, 2H), 1.82 (t, J = 2.6 Hz, 1H), 1.59–1.31 (m, 6H), 1.49 (s, 9H), 1.41 (s, 9H); 13C NMR (CDCl3, 100 MHz) δ 149.5, 149.0, 141.3, 140.2, 107.7, 85.6, 84.2, 81.1, 67.9, 28.1, 28.0, 27.9, 27.8 27.6, 27.5, 18.0; IR (CDCl3) 2980, 2939, 2862, 2332, 1758, 1731, 1576, 1536, 1370, 1253, 1140, 1048; UV (λmax nm) 249, HRMS (ESI+) m/z 400.2202 [(M + Na)+; calculated mass for C20H31N3NaO4+: 400.2207 amu].General procedure for alkylation of tert-butyl 2-((tert-butoxycarbonyl)amino)-4-(hept-6-yn-1-yl)-1H-imidazole-1-carboxylate 8
From alkyl halides: To a 0 °C solution of tert-butyl 2-((tert-butoxycarbonyl)amino)-4-(hept-6-yn-1-yl)-1H-imidazole-1-carboxylate 8 (∼0.3 or ∼0.6 mmol) in DMF (4 mL) was added 60% sodium hydride dispersion in mineral oil (1 eq) and stirred until complete dissolution. To this solution was then added the alkyl halide (1.1 eq) dropwise. The reactions were then heated to 70 °C for four hours. Note that longer heating lead to increased decomposition or loss of the protected 2-aminoimidazole material. The reactions were then diluted with EtOAc and washed with water and brine. The organic layer was then dried (MgSO4), filtered, and concentrated in vacuo. The crude material was then purified by flash chromatography (typically 5–10% EtOAc in hexanes).From alkyl alcohols: To a 0 °C solution of tert-butyl 2-((tert-butoxycarbonyl)amino)-4-(hept-6-yn-1-yl)-1H-imidazole-1-carboxylate 8 (∼0.3 mmol), triphenylphosphine (2 eq), and alcohol (3 eq) in THF (2.5 mL) was added DIAD or DEAD (1.5 eq) dropwise and allowed cold bath to warm to room temperature. DEAD or DIAD were used interchangeably depending on the retention factor of the final product and thus aiding in final purification. After 16 h the reaction was concentrated in vacuo and purified by flash chromatography (typically 5–10% EtOAc in hexanes).
Note: Many alkylated di-Boc 2-aminoimidazoles exhibited rotomers in both the 1H and 13C NMR. An attempt has been made to single out the minor rotamer peaks if and when possible. The observed minor rotamer frequencies have been denoted by “rotamer” following the observed frequencies for the 1H NMR. For clarity the proton count for these frequencies corresponds to the expected proton count for the molecule rather than the fraction or ratio of the two rotamers. For 13C NMR, when possible the carbon peak pairs corresponding to the different rotamers have been singled out but no 2D experiments were performed to distinguish their identities.
General procedure for the azide-alkyne Huisgen cycloaddition between 9a–o or 17 and N-(2-azidoethyl)-4-pentylbenzamide 11
To a solution of alkyne 9a–o or 17 in tBuOH (2 mL), water (2 mL), and CH2Cl2 (1 mL) was added N-(2-azidoethyl)-4-pentylbenzamide 11 (1.1 eq), CuSO4 (0.2 eq), and sodium ascorbate (0.4 eq). The above mixture was stirred vigorously until TLC indicated complete conversion of the alkyne starting material (1–5 h). The reaction was then diluted with water and extracted with CH2Cl2. The combined organic layers were then dried (MgSO4), filtered, and concentrated in vacuo to afford the crude material. The crude material was then purified by flash chromatography (typically 70–100% EtOAc in hexanes or 0–5% MeOH in CH2Cl2). In cases were copper color was still present in final material (typical of MeOH/CH2Cl2 purification) the final product was dissolved in CH2Cl2 and washed with a 0.01 M EDTA solution until organic layer was free of blue-green color.General procedure for Boc-deprotection of 12a–p
Di-tert-butoxycarbonyl 12a–p is dissolved in 30% TFA/CH2Cl2 (2 mL) or 10% for branched derivatives prone to loss of the substitution as a stable cation. This solution was then monitored by TLC for completion (approximately 3 h for 30% TFA/CH2Cl2 and overnight for 10%). The reaction is then concentrated in vacuo to afford the TFA salt then dissolved in MeOH (2 mL) with 1–2 drops of concentrated HCl. Any solid particulates are filtered through a cotton plug and the solution is then concentrated in vacuo to afford the HCl salt. The majority of samples are pure by 1H NMR. Impure products were purified by flash chromatography (1–10% concentrated ammonia in MeOH/CH2Cl2). The ammonia free dried samples are then dissolved in MeOH with concentrated HCL as above and concentrated to afford the HCl salt.Biological screening
Checkerboard assay. HB was inoculated with MRSA (5 × 105 CFU mL−1) and 100 μL was distributed into each wells of a 96-well plate except well 1A. Inoculated MHB (200 μL) containing compound (at 2 × the highest concentration being tested) was added to well 1A, 100 μL of the same sample was placed in each of wells 2A–12A. Column A wells were mixed 6 to 8 times then 100 μL was withdrawn and transferred to column B. Column B wells were mixed 6 to 8 times, followed by a 100 μL transfer to column C. This procedure was repeated to serially dilute the rest of the columns of the plate up to column G (column H was not mixed to allow the MIC of antibiotic alone to be determined). Inoculated media (100 μL) containing antibiotic at 2 × the highest concentration being tested was placed in wells A1–H1 and serially diluted in the same manner to row 11 (row 12 was not mixed to allow the MIC of compound alone to be determined). The plates were incubated for 16 h at 37 °C. The MICs of both compound and antibiotic in the combination were recorded, as well as the MICs of compound alone (from row 12) and antibiotic alone (from column H). The ΣFICs were calculated as follows: ΣFIC = FIC (compound) + FIC (antibiotic), where FIC (compound) is the MIC of the compound in the combination/MIC of the compound alone, and FIC (antibiotic) is the MIC of the antibiotic in the combination/MIC of the antibiotic. The combination is considered synergistic when the ΣFIC is ≤0.5, indifferent when the ΣFIC is >0.5 to <2, and antagonistic when the ΣFIC is ≥2.
Acknowledgements
The authors thank the DOD DMRDP program (W81XWH-11-2-0115) for support of this work. The DMRDP program is administered by the Department of Army, The U.S. Army Medical Research Acquisition Activity, 820 Chandler Street, Fort Detrick, MD 21702-5014 is the awarding and administering office. The content of this manuscript does not necessarily reflect the position or the policy of the Government, and no official endorsement should be inferred.Notes and references
- R. J. Worthington, J. J. Richards and C. Melander, Org. Biomol. Chem., 2012, 10, 7457–7474 CAS.
- D. Davies, Nat. Rev. Drug Discov., 2003, 2, 114 CrossRef CAS.
- J. R. W. Govan and V. Deretic, Microbiol. Rev., 1996, 60, 539 CAS.
- C. G. Gourin and R. N. Hubbel, Arch. Otolaryngol. Head Neck Surg., 1999, 125, 446 CAS.
- J. Biedlingmaier, R. Samaranayake and P. Whelan, Otolaryngol. Head Neck Surg., 1998, 118, 444 CAS.
- C. von Eiff, B. Jansen, W. Kohnen and K. Becker, Drugs, 2005, 65, 179 CrossRef CAS.
- M. R. Paresk and P. K. Singh, Annu. Rev. Microbiol., 2003, 57, 677 CrossRef.
- J. C. Nickel, J. W. Costerton, R. J. McLean and M. Olson, J. Antimicrob. Chemother., 1994, 33, 31 CrossRef.
- J. W. Costerton, Z. Lewandowski, D. Caldwell, D. Korber and H. M. Lapin-Scott, Annu. Rev. Microbiol., 1995, 49, 711 CrossRef CAS.
- T. B. Rasmussen and M. Givskov, Int. J. Med. Microbiol., 2006, 296, 149 CrossRef CAS.
- L. Kalan and G. D. Wright, Expert Rev. Mol. Med., 2011, 13, 1–17 CrossRef.
- A. E. Clatworthy, E. Pierson and D. T. Hung, Nat. Chem. Biol., 2007, 3, 541 CrossRef CAS.
- S. Muschiol, L. Bailey, Å. Gylfe, C. Sundin, K. hultenby, S. Bergström, M. Elofsson, H. Wolf-Watz, S. Normark and B. Henriques-Normark, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 14566 CrossRef CAS.
- J. S. Pinker, H. Remaut, F. Buelens, E. Miller, V. Åberg, N. Pemberton, M. Hedenström, A. Larsson, P. Seed, G. Waksman, S. J. Hultgren and F. Almqvist, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 17897 CrossRef.
- Y. M. Lee, F. Almqvist and S. J. Hultgren, Curr. Opin. Pharmacol., 2003, 3, 513 CrossRef CAS.
- A. Yamada, H. Kitamura, K. Yamaguchi, S. Fukuzawa, C. Kamijima, K. Yazawa, M. Kuramoto, G. Y. S. Wang, Y. Fujitani and D. Uemura, Bull. Chem. Soc. Jpn., 1997, 70, 3061–3069 CrossRef CAS.
- J. J. Richards, R. W. Huigens, T. E. Ballard, A. Basso, J. Cavanagh and C. Melander, Chem. Commun., 2008, 1698–1700 RSC.
- J. J. Richards, T. E. Ballard and C. Melander, Org. Biomol. Chem., 2008, 6, 1356–1363 CAS.
- R. W. Huigens, G. Parise, J. J. Richards, T. E. Ballard, W. Zeng, R. Deora and C. Melander, J. Am. Chem. Soc., 2007, 129, 6966–6967 CrossRef CAS.
- S. A. Rogers and C. Melander, Angew. Chem., Int. Ed., 2008, 47, 5229–5231 CrossRef CAS.
- S. Reyes, R. W. Huigens, Z. Su, M. L. Simon and C. Melander, Org. Biomol. Chem., 2011, 9, 3041–3049 CAS.
- S. A. Rogers, J. D. Bero and C. Melander, ChemBioChem, 2010, 11, 396–410 CrossRef CAS.
- Z. Su, L. Peng, R. J. Worthington and C. Melander, ChemMedChem, 2011, 6, 2243–2251 CrossRef CAS.
- S. A. Rogers, R. W. Huigins III, J. Cavanagh and C. Melander, Antimicrob. Agents Chemother., 2010, 54, 2112–2118 CrossRef CAS.
- R. J. Worthington, C. A. Bunders, C. S. Reed and C. Melander, ACS Med. Chem. Lett., 2012, 3, 357–361 CrossRef CAS.
- Z. Su, A. A. Yeagley, R. Su, L. Peng and C. Melander, ChemMedChem, 2012 DOI:10.1002/cmdc.201200350.
- G. Vaidyanathan and M. R. Zalutsky, J. Org. Chem., 1997, 62, 4867 CrossRef CAS.
- G. Galley, Z. K. G. Zbinden, R. Norcross and H. Stalder, PCT Int. Appl., 2008071574, 2008 Search PubMed.
- P. Ciapetti, F. Chery-Mozziconacci, C. G. Wermuth, F. Leblanc, M. Schneider, S. Ropp, C. Morice and B. Giethlen, PCT Int. Appl., 2009106967, 2009 Search PubMed.
- F. Perron-Sierra, D. S. Dizier, M. Betrand, A. Genton, G. C. Tucker and P. Casara, Bioorg. Med. Chem. Lett., 2002, 12, 3291 CrossRef CAS.
- C. A. Brown and A. Yamashita, J. Am. Chem. Soc., 1975, 97, 891 CrossRef CAS.
- H. Ube, D. Uraguchi and M. Terada, J. Org. Chem., 2007, 692, 545–549 CrossRef CAS.
- E. Burton, N. Yakandawala, K. LoVetri and M. S. Madhyastha, J. Ind. Microbiol. Biotechnol., 2007, 34, 1 CrossRef CAS.
- Z. Su, L. Peng and C. Melander, Tetrahedron Lett., 2012, 53, 1204–1206 CrossRef CAS.
- CSLI, Performance Standards for Antimicrobial Susceptibility Testing; Twentieth Informational Supplement, Clinical and Laboratory Standards Institute, Wayne, PA, January 2010, M100-S20 Vol. 30 No. 1 Search PubMed.
- G. Orhan, A. Bayram, Y. Zer and I. Balci, J. Clin. Microbiol., 2005, 43, 140–143 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2ob26469b |
| This journal is © The Royal Society of Chemistry 2013 |